
Blood–Brain Barrier and Neurodegenerative Diseases—Modeling with iPSC-Derived Brain Cells

 ,
,
Abstract
:1. Introduction
2. The Blood–Brain Barrier
2.1. Endothelial Cells
2.2. Astrocytes
2.3. Pericytes
3. Blood–Brain Barrier Dysfunction in Neurodegenerative Diseases
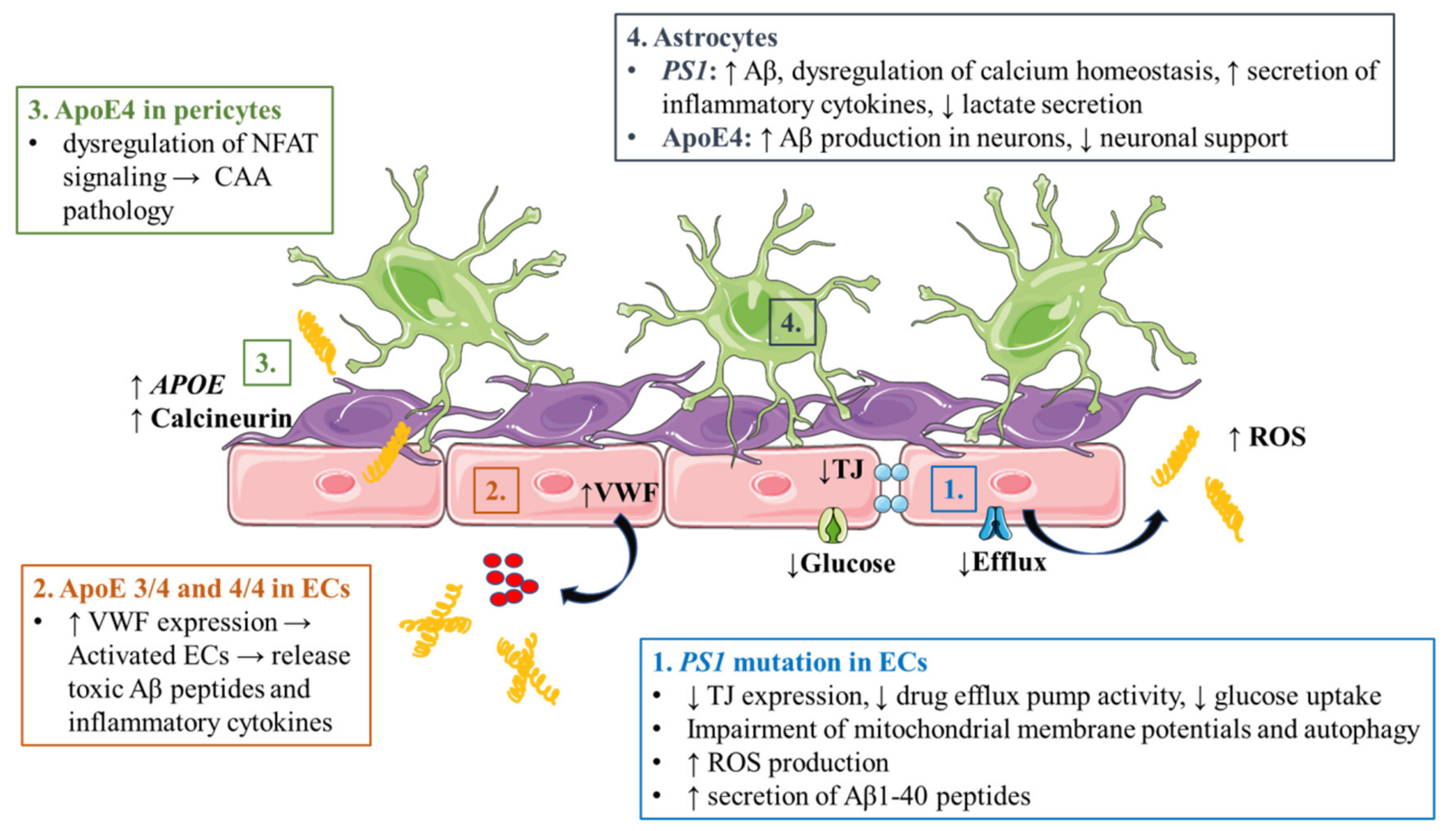
3.1. Alzheimer’s Disease
3.1.1. Accumulation of Aβ and Phosphorylated Tau in Cerebrovasculature
3.1.2. Endothelial Cell Dysfunction
3.1.3. Astrocyte Dysfunction
3.1.4. Pericyte Dysfunction
3.1.5. Peripheral Immune Cell Infiltration
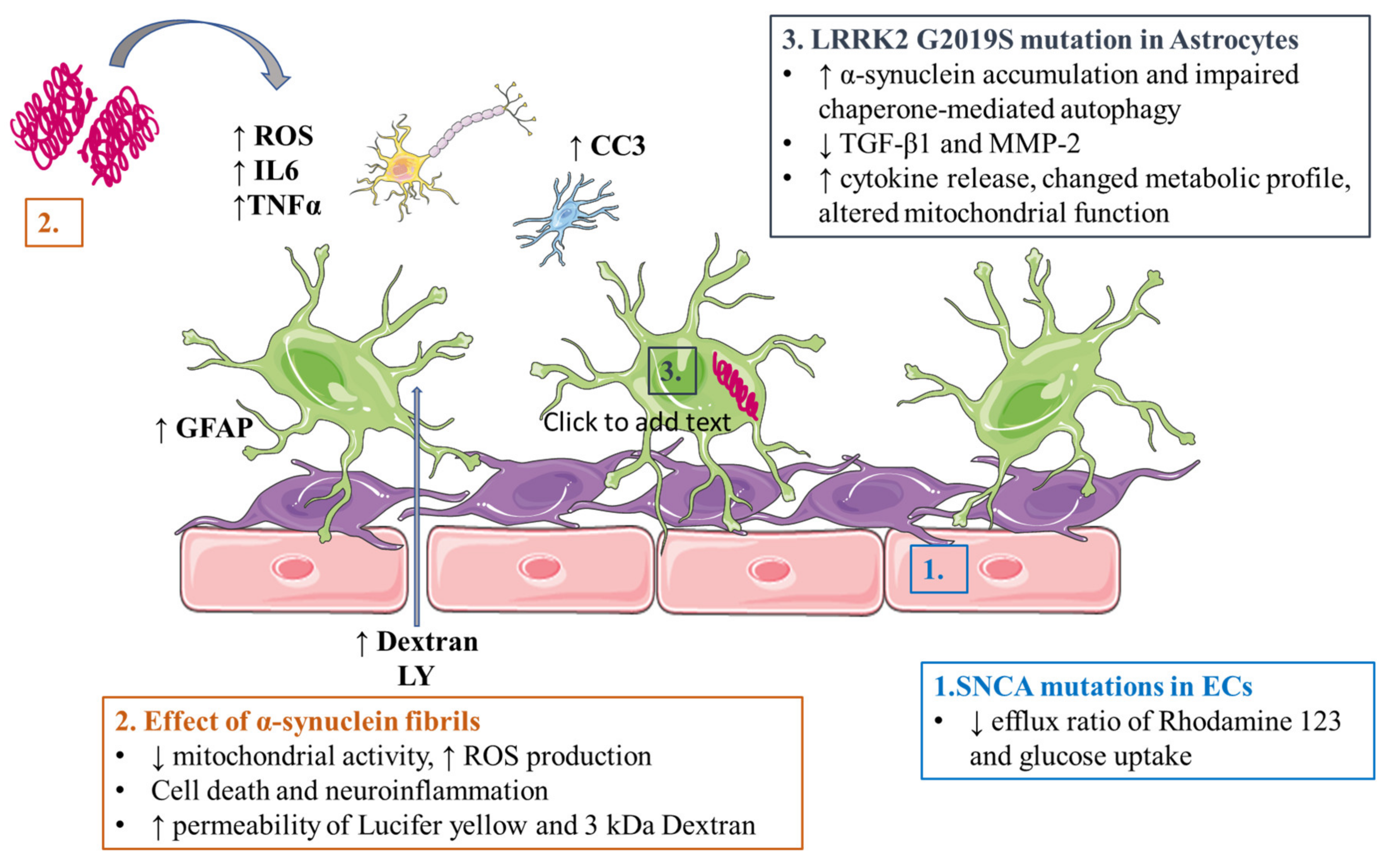
3.2. Parkinson’s Disease
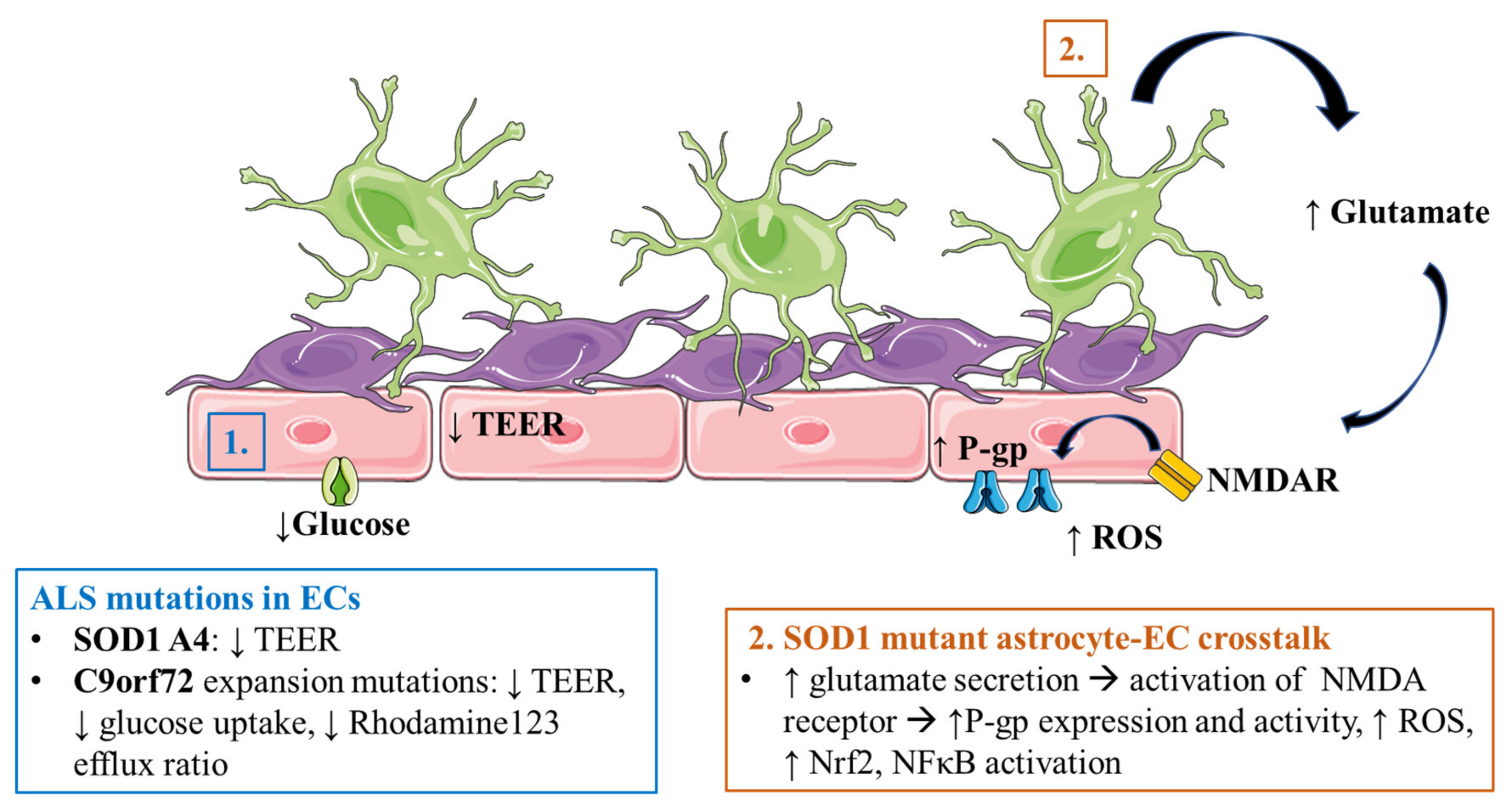
3.3. Amyotrophic Lateral Sclerosis
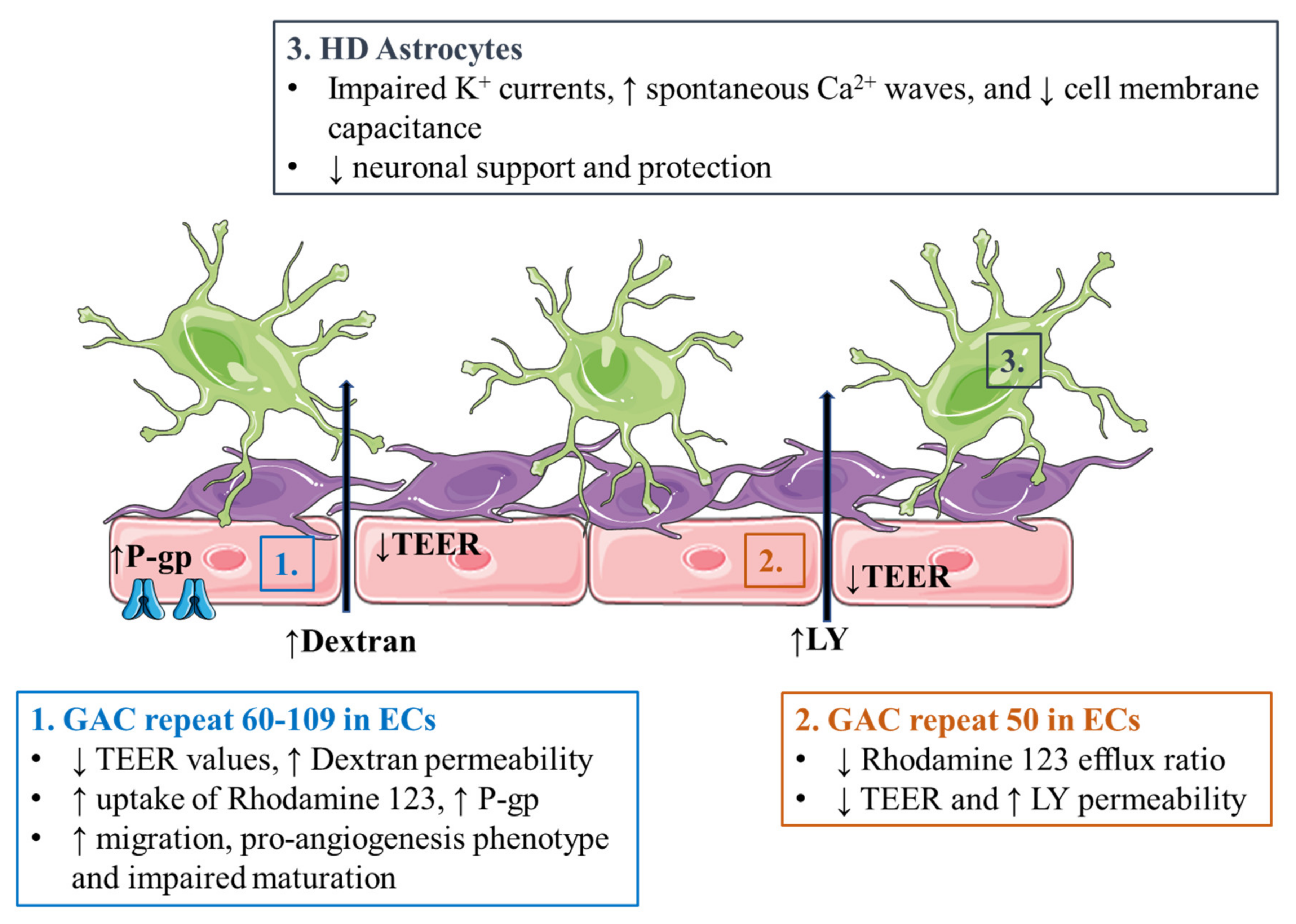
3.4. Huntington’s Disease
4. Human-Induced Pluripotent Stem Cells in Blood–Brain Barrier Modeling
4.1. Differentiation of BBB-Related Cells
4.1.1. Brain Endothelial Cells
4.1.2. Astrocytes
4.1.3. Pericytes
4.2. Blood–Brain Barrier In Vitro Models
4.2.1. Transwell
4.2.2. Spheroid
4.2.3. Organ-on-Chip
4.2.4. Hydrogel Models
4.2.5. Vascularized Brain Organoids
5. iPSC–BBB Models of Neurodegenerative Diseases
5.1. Alzheimer’s Disease
5.2. Parkinson’s Disease
5.3. Amyotrophic Lateral Sclerosis
5.4. Huntington’s Disease
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Xie, A.; Gao, J.; Xu, L.; Meng, D. Shared Mechanisms of Neurodegeneration in Alzheimer’s Disease and Parkinson’s Disease. Biomed. Res. Int. 2014, 2014, 648740. [Google Scholar] [CrossRef]
- Jellinger, K.A. Basic Mechanisms of Neurodegeneration: A Critical Update. J. Cell Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.B.; Egefjord, L.; Angleys, H.; Mouridsen, K.; Gejl, M.; Møller, A.; Brock, B.; Brændgaard, H.; Gottrup, H.; Rungby, J.; et al. Capillary Dysfunction Is Associated with Symptom Severity and Neurodegeneration in Alzheimer’s Disease. Alzheimers Dement. 2017, 13, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Erdener, Ş.E.; Dalkara, T. Small Vessels Are a Big Problem in Neurodegeneration and Neuroprotection. Front. Neurol. 2019, 10, 889. [Google Scholar] [CrossRef] [PubMed]
- Korte, N.; Nortley, R.; Attwell, D. Cerebral Blood Flow Decrease as an Early Pathological Mechanism in Alzheimer’s Disease. Acta Neuropathol. 2020, 140, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Kim, C. Disease Modeling and Cell Based Therapy with IPSC: Future Therapeutic Option with Fast and Safe Application. Blood Res. 2014, 49, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and Function of the Blood-Brain Barrier. NeuroBiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R. The Blood-Brain Barrier in Health and Disease. Ann. Neurol. 2012, 72, 648–672. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Balda, M.S.; Whitney, J.A.; Flores, C.; González, S.; Cereijido, M.; Matter, K. Functional Dissociation of Paracellular Permeability and Transepithelial Electrical Resistance and Disruption of the Apical-Basolateral Intramembrane Diffusion Barrier by Expression of a Mutant Tight Junction Membrane Protein. J. Cell Biol. 1996, 134, 1031–1049. [Google Scholar] [CrossRef]
- Luissint, A.-C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O. Tight Junctions at the Blood Brain Barrier: Physiological Architecture and Disease-Associated Dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyahi, A.; Nik, A.M.; Ghiami, M.; Gritli-Linde, A.; Pontén, F.; Johansson, B.R.; Carlsson, P. Foxf2 Is Required for Brain Pericyte Differentiation and Development and Maintenance of the Blood-Brain Barrier. Dev. Cell 2015, 34, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a Is Critical for the Formation and Function of the Blood-Brain Barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, C.-H.; Huang, S.; Fu, Z.; Tomita, Y.; Britton, W.R.; Cho, S.S.; Chen, C.T.; Sun, Y.; Ma, J.-X.; et al. Wnt Signaling Activates MFSD2A to Suppress Vascular Endothelial Transcytosis and Maintain Blood-Retinal Barrier. Sci. Adv. 2020, 6, 7457. [Google Scholar] [CrossRef]
- Sabbagh, M.F.; Heng, J.S.; Luo, C.; Castanon, R.G.; Nery, J.R.; Rattner, A.; Goff, L.A.; Ecker, J.R.; Nathans, J. Transcriptional and Epigenomic Landscapes of CNS and Non-CNS Vascular Endothelial Cells. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K.M. L-Type Amino Acid Transporter 1 as a Target for Drug Delivery. Pharm. Res. 2020, 37, 88. [Google Scholar] [CrossRef]
- Veys, K.; Fan, Z.; Ghobrial, M.; Bouché, A.; García-Caballero, M.; Vriens, K.; Conchinha, N.V.; Seuwen, A.; Schlegel, F.; Gorski, T.; et al. Role of the GLUT1 Glucose Transporter in Postnatal CNS Angiogenesis and Blood-Brain Barrier Integrity. Circ. Res. 2020, 127, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Rössler, K.; Neuchrist, C.; Kitz, K.; Scheiner, O.; Kraft, D.; Lassmann, H. Expression of Leucocyte Adhesion Molecules at the Human Blood-Brain Barrier (BBB). J. Neurosci. Res. 1992, 31, 365–374. [Google Scholar] [CrossRef]
- Henninger, D.D.; Panés, J.; Eppihimer, M.; Russell, J.; Gerritsen, M.; Anderson, D.C.; Granger, D.N. Cytokine-Induced VCAM-1 and ICAM-1 Expression in Different Organs of the Mouse. J. Immunol. 1997, 158, 1825–1832. [Google Scholar]
- Bergersen, L.H. Lactate Transport and Signaling in the Brain: Potential Therapeutic Targets and Roles in Body-Brain Interaction. J. Cereb. Blood Flow Metab. 2015, 35, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Vijay, N.; Morris, M.E. Role of Monocarboxylate Transporters in Drug Delivery to the Brain. Curr. Pharm. Des. 2014, 20, 1487–1498. [Google Scholar] [CrossRef] [Green Version]
- Leino, R.L.; Gerhart, D.Z.; Drewes, L.R. Monocarboxylate Transporter (MCT1) Abundance in Brains of Suckling and Adult Rats: A Quantitative Electron Microscopic Immunogold Study. Brain Res. Dev. Brain Res. 1999, 113, 47–54. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s Amyloid-Ss(1-40) Peptide from Brain by LDL Receptor-Related Protein-1 at the Blood-Brain Barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE Mediates Amyloid-Beta Peptide Transport across the Blood-Brain Barrier and Accumulation in Brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-Endothelial Interactions at the Blood-Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Higashi, K.; Fujita, A.; Inanobe, A.; Tanemoto, M.; Doi, K.; Kubo, T.; Kurachi, Y. An Inwardly Rectifying K(+) Channel, Kir4.1, Expressed in Astrocytes Surrounds Synapses and Blood Vessels in Brain. Am. J. Physiol. Cell Physiol. 2001, 281, C922–C931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, C.F.; Mohanta, S.K.; Frontczak-Baniewicz, M.; Swinny, J.D.; Zablocka, B.; Górecki, D.C. Absence of Glial α-Dystrobrevin Causes Abnormalities of the Blood-Brain Barrier and Progressive Brain Edema. J. Biol. Chem. 2012, 287, 41374–41385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial Influence on the Blood Brain Barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michinaga, S.; Koyama, Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int. J. Mol. Sci. 2019, 20, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, K.-J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.-F.; Turk, J.; et al. Matrix Metalloproteinases Expressed by Astrocytes Mediate Extracellular Amyloid-Beta Peptide Catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels Is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. J. Cereb. Blood Flow. Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. Signals for the Induction of Nitric Oxide Synthase in Astrocytes. Neurochem. Int. 2006, 49, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Li, C.; Arrick, D.M.; Yang, S.; Baluna, A.E.; Sun, H. Role of Nitric Oxide Synthases in Early Blood-Brain Barrier Disruption Following Transient Focal Cerebral Ischemia. PLoS ONE 2014, 9, e93134. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.R.; Mayanil, C.S.; Kim, K.S.; Tomita, T. Angiopoietin-1 Regulates Brain Endothelial Permeability through PTPN-2 Mediated Tyrosine Dephosphorylation of Occludin. PLoS ONE 2015, 10, e0130857. [Google Scholar] [CrossRef]
- Utsumi, H.; Chiba, H.; Kamimura, Y.; Osanai, M.; Igarashi, Y.; Tobioka, H.; Mori, M.; Sawada, N. Expression of GFRalpha-1, Receptor for GDNF, in Rat Brain Capillary during Postnatal Development of the BBB. Am. J. Physiol. Cell Physiol. 2000, 279, C361–C368. [Google Scholar] [CrossRef] [Green Version]
- Yosef, N.; Ubogu, E.E. GDNF Restores Human Blood-Nerve Barrier Function via RET Tyrosine Kinase-Mediated Cytoskeletal Reorganization. Microvasc. Res. 2012, 83, 298–310. [Google Scholar] [CrossRef]
- Muller, W.A. How Endothelial Cells Regulate Transmigration of Leukocytes in the Inflammatory Response. Am. J. Pathol. 2014, 184, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Derivation of Blood-Brain Barrier Endothelial Cells from Human Pluripotent Stem Cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef]
- Heithoff, B.P.; George, K.K.; Phares, A.N.; Zuidhoek, I.A.; Munoz-Ballester, C.; Robel, S. Astrocytes Are Necessary for Blood-Brain Barrier Maintenance in the Adult Mouse Brain. Glia 2021, 69, 436–472. [Google Scholar] [CrossRef]
- Trost, A.; Lange, S.; Schroedl, F.; Bruckner, D.; Motloch, K.A.; Bogner, B.; Kaser-Eichberger, A.; Strohmaier, C.; Runge, C.; Aigner, L.; et al. Brain and Retinal Pericytes: Origin, Function and Role. Front. Cell Neurosci. 2016, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, K.Y.; Brekken, R.A. Recruitment and Retention: Factors That Affect Pericyte Migration. Cell Mol. Life Sci. 2014, 71, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes Are Required for Blood-Brain Barrier Integrity during Embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood-Brain Barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Durham, J.T.; Surks, H.K.; Dulmovits, B.M.; Herman, I.M. Pericyte Contractility Controls Endothelial Cell Cycle Progression and Sprouting: Insights into Angiogenic Switch Mechanics. Am. J. Physiol. Cell Physiol. 2014, 307, C878–C892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, R.; Kawahara, M.; Nakano-Doi, A.; Takahashi, A.; Tanaka, Y.; Narita, A.; Kuwahara-Otani, S.; Hayakawa, T.; Yagi, H.; Matsuyama, T.; et al. Brain Pericytes Serve as Microglia-Generating Multipotent Vascular Stem Cells Following Ischemic Stroke. J. Neuroinflamm. 2016, 13, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagomi, T.; Kubo, S.; Nakano-Doi, A.; Sakuma, R.; Lu, S.; Narita, A.; Kawahara, M.; Taguchi, A.; Matsuyama, T. Brain Vascular Pericytes Following Ischemia Have Multipotential Stem Cell Activity to Differentiate into Neural and Vascular Lineage Cells. Stem Cells 2015, 33, 1962–1974. [Google Scholar] [CrossRef]
- Özen, I.; Deierborg, T.; Miharada, K.; Padel, T.; Englund, E.; Genové, G.; Paul, G. Brain Pericytes Acquire a Microglial Phenotype after Stroke. Acta Neuropathol. 2014, 128, 381–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, R.; Compte, M.; Álvarez-Vallina, L.; Sanz, L. Immune Regulation by Pericytes: Modulating Innate and Adaptive Immunity. Front. Immunol. 2016, 7, 480. [Google Scholar] [CrossRef] [Green Version]
- Rustenhoven, J.; Jansson, D.; Smyth, L.C.; Dragunow, M. Brain Pericytes As Mediators of Neuroinflammation. Trends Pharm. Sci. 2017, 38, 291–304. [Google Scholar] [CrossRef]
- Jansson, D.; Rustenhoven, J.; Feng, S.; Hurley, D.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Faull, R.L.M.; Dragunow, M. A Role for Human Brain Pericytes in Neuroinflammation. J. Neuroinflamm. 2014, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Kovac, A.; Erickson, M.A.; Banks, W.A. Brain Microvascular Pericytes Are Immunoactive in Culture: Cytokine, Chemokine, Nitric Oxide, and LRP-1 Expression in Response to Lipopolysaccharide. J. Neuroinflamm. 2011, 8, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocahan, S.; Doğan, Z. Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-Methyl-D-Aspartate Receptors, Tau Protein and Other Risk Factors. Clin. Psychopharmacol. Neurosci. 2017, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Moraes, C.H.; Oliveira-Pinto, A.V.; Castro-Fonseca, E.; da Silva, C.G.; Guimarães, D.M.; Szczupak, D.; Parente-Bruno, D.R.; Carvalho, L.R.B.; Polichiso, L.; Gomes, B.V.; et al. Cell Number Changes in Alzheimer’s Disease Relate to Dementia, Not to Plaques and Tangles. Brain 2013, 136, 3738–3752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J. Association of Apolipoprotein E Allele Epsilon 4 with Late-Onset Familial and Sporadic Alzheimer’s Disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emrani, S.; Arain, H.A.; DeMarshall, C.; Nuriel, T. APOE4 Is Associated with Cognitive and Pathological Heterogeneity in Patients with Alzheimer’s Disease: A Systematic Review. Alzheimers Res. 2020, 12, 141. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zipfel, G.J.; Han, H.; Ford, A.L.; Lee, J.-M. Cerebral Amyloid Angiopathy: Progressive Disruption of the Neurovascular Unit. Stroke 2009, 40, S16–S19. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Shi, J.; Smallman, R.; Iwatsubo, T.; Mann, D.M.A. Relationships in Alzheimer’s Disease between the Extent of Abeta Deposition in Cerebral Blood Vessel Walls, as Cerebral Amyloid Angiopathy, and the Amount of Cerebrovascular Smooth Muscle Cells and Collagen. Neuropathol. Appl. Neurobiol. 2006, 32, 332–340. [Google Scholar] [CrossRef]
- Freeze, W.M.; Bacskai, B.J.; Frosch, M.P.; Jacobs, H.I.L.; Backes, W.H.; Greenberg, S.M.; van Veluw, S.J. Blood-Brain Barrier Leakage and Microvascular Lesions in Cerebral Amyloid Angiopathy. Stroke 2019, 50, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Zhao, Z.; Sagare, A.P.; Wu, Y.; Wang, M.; Owens, N.C.; Verghese, P.B.; Herz, J.; Holtzman, D.M.; Zlokovic, B.V. Blood-Brain Barrier-Associated Pericytes Internalize and Clear Aggregated Amyloid-Β42 by LRP1-Dependent Apolipoprotein E Isoform-Specific Mechanism. Mol. Neurodegener. 2018, 13, 57. [Google Scholar] [CrossRef] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte Loss Influences Alzheimer-like Neurodegeneration in Mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [Green Version]
- Schultz, N.; Brännström, K.; Byman, E.; Moussaud, S.; Nielsen, H.M.; Netherlands Brain Bank; Olofsson, A.; Wennström, M. Amyloid-Beta 1-40 Is Associated with Alterations in NG2+ Pericyte Population Ex Vivo and in Vitro. Aging Cell 2018, 17, e12728. [Google Scholar] [CrossRef] [Green Version]
- Kimbrough, I.F.; Robel, S.; Roberson, E.D.; Sontheimer, H. Vascular Amyloidosis Impairs the Gliovascular Unit in a Mouse Model of Alzheimer’s Disease. Brain 2015, 138, 3716–3733. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; del Alonso, A.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau Pathology in Alzheimer Disease and Other Tauopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Carranza, D.L.; Nilson, A.N.; Van Skike, C.E.; Jahrling, J.B.; Patel, K.; Garach, P.; Gerson, J.E.; Sengupta, U.; Abisambra, J.; Nelson, P.; et al. Cerebral Microvascular Accumulation of Tau Oligomers in Alzheimer’s Disease and Related Tauopathies. Aging Dis. 2017, 8, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canepa, E.; Fossati, S. Impact of Tau on Neurovascular Pathology in Alzheimer’s Disease. Front. Neurol. 2021, 11, 573324. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau Induces Blood Vessel Abnormalities and Angiogenesis-Related Gene Expression in P301L Transgenic Mice and Human Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalicova, A.; Majerova, P.; Kovac, A. Tau Protein and Its Role in Blood–Brain Barrier Dysfunction. Front. Mol. Neurosci. 2020, 13, 570045. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular Mechanisms of Alzheimer’s Neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef]
- Albrecht, D.; Isenberg, A.L.; Stradford, J.; Monreal, T.; Sagare, A.; Pachicano, M.; Sweeney, M.; Toga, A.; Zlokovic, B.; Chui, H.; et al. Associations between Vascular Function and Tau PET Are Associated with Global Cognition and Amyloid. J. Neurosci. 2020, 40, 8573–8586. [Google Scholar] [CrossRef]
- Liu, C.-C.; Yamazaki, Y.; Heckman, M.G.; Martens, Y.A.; Jia, L.; Yamazaki, A.; Diehl, N.N.; Zhao, J.; Zhao, N.; DeTure, M.; et al. Tau and Apolipoprotein E Modulate Cerebrovascular Tight Junction Integrity Independent of Cerebral Amyloid Angiopathy in Alzheimer’s Disease. Alzheimers Dement. 2020, 16, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Weekman, E.M.; Wilcock, D.M. Matrix Metalloproteinase in Blood-Brain Barrier Breakdown in Dementia. J. Alzheimers Dis. 2016, 49, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Brilha, S.; Ong, C.W.M.; Weksler, B.; Romero, N.; Couraud, P.-O.; Friedland, J.S. Matrix Metalloproteinase-9 Activity and a Downregulated Hedgehog Pathway Impair Blood-Brain Barrier Function in an in Vitro Model of CNS Tuberculosis. Sci. Rep. 2017, 7, 16031. [Google Scholar] [CrossRef] [PubMed]
- Brkic, M.; Balusu, S.; Libert, C.; Vandenbroucke, R.E. Friends or Foes: Matrix Metalloproteinases and Their Multifaceted Roles in Neurodegenerative Diseases. Mediat. Inflamm. 2015, 2015, 620581. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Shinohara, M.; Shinohara, M.; Yamazaki, A.; Murray, M.E.; Liesinger, A.M.; Heckman, M.G.; Lesser, E.R.; Parisi, J.E.; Petersen, R.C.; et al. Selective Loss of Cortical Endothelial Tight Junction Proteins during Alzheimer’s Disease Progression. Brain 2019, 142, 1077–1092. [Google Scholar] [CrossRef]
- Lee, D.; Cho, S.-J.; Lim, H.J.; Seok, J.; Jo, C.; Jo, S.A.; Park, M.H.; Han, C.; Kowall, N.; Ryu, H.; et al. Alteration of Vascular Endothelial Cadherin in Alzheimer’s Disease Patient and Mouse Model. BioRxiv 2018, 430140. [Google Scholar] [CrossRef]
- Vogelsang, P.; Giil, L.M.; Lund, A.; Vedeler, C.A.; Parkar, A.P.; Nordrehaug, J.E.; Kristoffersen, E.K. Reduced Glucose Transporter-1 in Brain Derived Circulating Endothelial Cells in Mild Alzheimer’s Disease Patients. Brain Res. 2018, 1678, 304–309. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 Reductions Exacerbate Alzheimer’s Disease Vasculo-Neuronal Dysfunction and Degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and Amyloid-Beta Peptide Neurotoxicity in Alzheimer’s Disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Jeynes, B.; Provias, J. Evidence for Altered LRP/RAGE Expression in Alzheimer Lesion Pathogenesis. Curr. Alzheimer Res. 2008, 5, 432–437. [Google Scholar] [CrossRef]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A.; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and Amyloid-Beta Protein in Alzheimer’s Disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-Glycoprotein Expression and Amyloid Accumulation in Human Aging and Alzheimer’s Disease: Preliminary Observations. NeuroBiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guo, H.; Chow, N.; Sallstrom, J.; Bell, R.D.; Deane, R.; Brooks, A.I.; Kanagala, S.; Rubio, A.; Sagare, A.; et al. Role of the MEOX2 Homeobox Gene in Neurovascular Dysfunction in Alzheimer Disease. Nat. Med. 2005, 11, 959–965. [Google Scholar] [CrossRef]
- Lau, S.-F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-Nucleus Transcriptome Analysis Reveals Dysregulation of Angiogenic Endothelial Cells and Neuroprotective Glia in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavalu, K.; Zhang, C.; Zhang, X.; Tanzi, R.E.; Sisodia, S.S. Age-Dependent, Non-Cell-Autonomous Deposition of Amyloid from Synthesis of β-Amyloid by Cells Other than Excitatory Neurons. J. Neurosci. 2014, 34, 3668–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grolla, A.A.; Fakhfouri, G.; Balzaretti, G.; Marcello, E.; Gardoni, F.; Canonico, P.L.; DiLuca, M.; Genazzani, A.A.; Lim, D. Aβ Leads to Ca2+ Signaling Alterations and Transcriptional Changes in Glial Cells. NeuroBiol. Aging 2013, 34, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Gubert, O.M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant IPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem. Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Astrogliosis: An Integral Player in the Pathogenesis of Alzheimer’s Disease. Prog. NeuroBiol. 2016, 144, 121–141. [Google Scholar] [CrossRef]
- Li, X.; Li, M.; Tian, L.; Chen, J.; Liu, R.; Ning, B. Reactive Astrogliosis: Implications in Spinal Cord Injury Progression and Therapy. Oxid. Med. Cell. Longev. 2020, 2020, 9494352. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.-I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.-S.; Kwon, S.-H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 Astrocyte Conversion by Microglia Is Neuroprotective in Models of Parkinson’s Disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Taylor, X.; Cisternas, P.; You, Y.; You, Y.; Xiang, S.; Marambio, Y.; Zhang, J.; Vidal, R.; Lasagna-Reeves, C.A. A1 Reactive Astrocytes and a Loss of TREM2 Are Associated with an Early Stage of Pathology in a Mouse Model of Cerebral Amyloid Angiopathy. J. Neuroinflamm. 2020, 17, 223. [Google Scholar] [CrossRef]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Concomitant Astroglial Atrophy and Astrogliosis in a Triple Transgenic Animal Model of Alzheimer’s Disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Kulijewicz-Nawrot, M.; Verkhratsky, A.; Chvátal, A.; Syková, E.; Rodríguez, J.J. Astrocytic Cytoskeletal Atrophy in the Medial Prefrontal Cortex of a Triple Transgenic Mouse Model of Alzheimer’s Disease. J. Anat. 2012, 221, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.M.; Rozemuller, A.J.M.; van Haastert, E.S.; Eikelenboom, P.; van Gool, W.A. Neuroinflammation in Alzheimer’s Disease Wanes with Age. J. Neuroinflamm. 2011, 8, 171. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular Amyloid Alters Astrocytic Water and Potassium Channels in Mouse Models and Humans with Alzheimer’s Disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.J.; Duan, T.; Verkman, A.S. Aquaporin-4 Reduces Neuropathology in a Mouse Model of Alzheimer’s Disease by Remodeling Peri-Plaque Astrocyte Structure. Acta Neuropathol. Commun. 2019, 7, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, R.; Hase, Y.; Ameen-Ali, K.E.; Ndung’u, M.; Stevenson, W.; Barsby, J.; Gourlay, R.; Akinyemi, T.; Akinyemi, R.; Uemura, M.T.; et al. Loss of Capillary Pericytes and the Blood-Brain Barrier in White Matter in Poststroke and Vascular Dementias and Alzheimer’s Disease. Brain Pathol. 2020, 30, 1087–1101. [Google Scholar] [CrossRef]
- Halliday, M.R.; Rege, S.V.; Ma, Q.; Zhao, Z.; Miller, C.A.; Winkler, E.A.; Zlokovic, B.V. Accelerated Pericyte Degeneration and Blood-Brain Barrier Breakdown in Apolipoprotein E4 Carriers with Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2016, 36, 216–227. [Google Scholar] [CrossRef]
- Ishii, M.; Iadecola, C. Risk Factor for Alzheimer’s Disease Breaks the Blood-Brain Barrier. Nature 2020, 581, 31–32. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. The Low-Density Lipoprotein Receptor-Related Protein 1 and Amyloid-β Clearance in Alzheimer’s Disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Shinohara, M.; Yamazaki, A.; Ren, Y.; Asmann, Y.W.; Kanekiyo, T.; Bu, G. ApoE (Apolipoprotein E) in Brain Pericytes Regulates Endothelial Function in an Isoform-Dependent Manner by Modulating Basement Membrane Components. Arter. Thromb. Vasc. Biol. 2020, 40, 128–144. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Hosono, T.; Nakamura, T.; Bu, G.; Michikawa, M. Apolipoprotein E Regulates the Integrity of Tight Junctions in an Isoform-Dependent Manner in an in Vitro Blood-Brain Barrier Model. J. Biol. Chem. 2011, 286, 17536–17542. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, J.W.; Bula, M.; Davila-Velderrain, J.; Akay, L.A.; Zhu, L.; Frank, A.; Victor, M.B.; Bonner, J.M.; Mathys, H.; Lin, Y.-T.; et al. Reconstruction of the Human Blood-Brain Barrier in Vitro Reveals a Pathogenic Mechanism of APOE4 in Pericytes. Nat. Med. 2020, 26, 952–963. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral Blood Flow Regulation and Neurovascular Dysfunction in Alzheimer Disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β Oligomers Constrict Human Capillaries in Alzheimer’s Disease via Signaling to Pericytes. Science 2019, 365, 9518. [Google Scholar] [CrossRef]
- Montagne, A.; Nikolakopoulou, A.M.; Huuskonen, M.T.; Sagare, A.P.; Lawson, E.J.; Lazic, D.; Rege, S.V.; Grond, A.; Zuniga, E.; Barnes, S.R.; et al. Author Correction: APOE4 Accelerates Advanced-Stage Vascular and Neurodegenerative Disorder in Old Alzheimer’s Mice via Cyclophilin A Independently of Amyloid-β. Nat. Aging 2021. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E Controls Cerebrovascular Integrity via Cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Gate, D.; Town, T. CNS Infiltration of Peripheral Immune Cells: D-Day for Neurodegenerative Disease? J. Neuroimmune Pharm. 2009, 4, 462–475. [Google Scholar] [CrossRef] [Green Version]
- Hohsfield, L.A.; Humpel, C. Migration of Blood Cells to β-Amyloid Plaques in Alzheimer’s Disease. Exp. Gerontol. 2015, 65, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting Microglial and Peripheral Immune Cell Crosstalk to Treat Alzheimer’s Disease. J. Neuroinflamm. 2019, 16, 74. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.H.; Weninger, W.; Hunter, C.A. Trafficking of Immune Cells in the Central Nervous System. J. Clin. Investig. 2010, 120, 1368–1379. [Google Scholar] [CrossRef] [Green Version]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Chen, S.; Klebe, D.; Zhang, J.H.; Tang, J. Adhesion Molecules in CNS Disorders: Biomarker and Therapeutic Targets. CNS Neurol. Disord Drug Targets 2013, 12, 392–404. [Google Scholar] [CrossRef] [Green Version]
- Wennström, M.; Nielsen, H.M. Cell Adhesion Molecules in Alzheimer’s Disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Liesz, A.; Zhou, W.; Mracskó, É.; Karcher, S.; Bauer, H.; Schwarting, S.; Sun, L.; Bruder, D.; Stegemann, S.; Cerwenka, A.; et al. Inhibition of Lymphocyte Trafficking Shields the Brain against Deleterious Neuroinflammation after Stroke. Brain 2011, 134, 704–720. [Google Scholar] [CrossRef] [Green Version]
- Dexter, D.T.; Jenner, P. Parkinson Disease: From Pathology to Molecular Disease Mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef]
- Kalia, L.V.; Kalia, S.K. α-Synuclein and Lewy Pathology in Parkinson’s Disease. Curr. Opin. Neurol. 2015, 28, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood-Brain Barrier Dysfunction in Parkinsonian Midbrain in Vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L.; Willemsen, A.T.M.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.H.; Portman, A.; Leenders, K.L. Decreased Blood-Brain Barrier P-Glycoprotein Function in the Progression of Parkinson’s Disease, PSP and MSA. J. Neural Transm. (Vienna) 2008, 115, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Shin, J.-Y.; Lee, Y.-S.; Yun, S.P.; Maeng, H.-J.; Lee, Y. Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway. Int. J. Mol. Sci. 2020, 21, 8538. [Google Scholar] [CrossRef]
- Al-Bachari, S.; Vidyasagar, R.; Emsley, H.; Parkes, L. PO071 Mri Assessment of Neurovascular Changes in Idiopathic Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, A3–A30. [Google Scholar] [CrossRef]
- Ham, J.H.; Yi, H.; Sunwoo, M.K.; Hong, J.Y.; Sohn, Y.H.; Lee, P.H. Cerebral Microbleeds in Patients with Parkinson’s Disease. J. Neurol. 2014, 261, 1628–1635. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal Blood-Brain Barrier Permeability in Parkinson’s Disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-Brain Stimulation Associates with Improved Microvascular Integrity in the Subthalamic Nucleus in Parkinson’s Disease. NeuroBiol. Dis. 2015, 74, 392–405. [Google Scholar] [CrossRef]
- Liguori, C.; Olivola, E.; Pierantozzi, M.; Cerroni, R.; Galati, S.; Saviozzi, V.; Mercuri, N.B.; Stefani, A. Cerebrospinal-Fluid Alzheimer’s Disease Biomarkers and Blood-Brain Barrier Integrity in a Natural Population of Cognitive Intact Parkinson’s Disease Patients. CNS Neurol. Disord. Drug Targets 2017, 16, 339–345. [Google Scholar] [CrossRef]
- Pisani, V.; Stefani, A.; Pierantozzi, M.; Natoli, S.; Stanzione, P.; Franciotta, D.; Pisani, A. Increased Blood-Cerebrospinal Fluid Transfer of Albumin in Advanced Parkinson’s Disease. J. Neuroinflamm. 2012, 9, 188. [Google Scholar] [CrossRef] [Green Version]
- Janelidze, S.; Lindqvist, D.; Francardo, V.; Hall, S.; Zetterberg, H.; Blennow, K.; Adler, C.H.; Beach, T.G.; Serrano, G.E.; van Westen, D.; et al. Increased CSF Biomarkers of Angiogenesis in Parkinson Disease. Neurology 2015, 85, 1834–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-Induced Alterations in Blood-Brain Barrier Permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Olmedo-Díaz, S.; Estévez-Silva, H.; Orädd, G.; af Bjerkén, S.; Marcellino, D.; Virel, A. An Altered Blood–Brain Barrier Contributes to Brain Iron Accumulation and Neuroinflammation in the 6-OHDA Rat Model of Parkinson’s Disease. Neuroscience 2017, 362, 141–151. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP Mouse Models of Parkinson’s Disease: An Update. J. Parkinson’s Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.C.; Kim, Y.-S.; Bok, E.; Yune, T.Y.; Maeng, S.; Jin, B.K. MMP-3 Contributes to Nigrostriatal Dopaminergic Neuronal Loss, BBB Damage, and Neuroinflammation in an MPTP Mouse Model of Parkinson’s Disease. Mediat. Inflamm. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Ling, Z.; Newman, M.B.; Bhatia, A.; Carvey, P.M. TNF-α Knockout and Minocycline Treatment Attenuates Blood–Brain Barrier Leakage in MPTP-Treated Mice. Neurobiol. Dis. 2007, 26, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yunfu, W.; Guangjian, L.; Ping, Z.; Yanpeng, S.; Xiaoxia, F.; Wei, H.; Jiang, Y.; Jingquan, H.; Songlin, W.; Hongyan, Z.; et al. PINK1 and Its Familial Parkinson’s Disease-Associated Mutation Regulate Brain Vascular Endothelial Inflammation. J. Mol. Neurosci. 2014, 53, 109–116. [Google Scholar] [CrossRef]
- Hongge, L.; Kexin, G.; Xiaojie, M.; Nian, X.; Jinsha, H. The Role of LRRK2 in the Regulation of Monocyte Adhesion to Endothelial Cells. J. Mol. Neurosci. 2015, 55, 233–239. [Google Scholar] [CrossRef]
- Sui, Y.-T.; Bullock, K.M.; Erickson, M.A.; Zhang, J.; Banks, W.A. Alpha Synuclein Is Transported into and out of the Brain by the Blood-Brain Barrier. Peptides 2014, 62, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein Strains Cause Distinct Synucleinopathies after Local and Systemic Administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma Exosomal α-Synuclein Is Likely CNS-Derived and Increased in Parkinson’s Disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef]
- Yang, P.; Min, X.-L.; Mohammadi, M.; Turner, C.; Faull, R.; Waldvogel, H.; Dragunow, M.; Guan, J. Endothelial Degeneration of Parkinson’s Disease Is Related to Alpha-Synuclein Aggregation. J. Alzheimers Dis. Parkinsonism 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Yamauchi, A.; Kataoka, Y. Monomeric α-Synuclein Induces Blood-Brain Barrier Dysfunction through Activated Brain Pericytes Releasing Inflammatory Mediators in Vitro. Microvasc. Res. 2019, 124, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Elabi, O.; Gaceb, A.; Carlsson, R.; Padel, T.; Soylu-Kucharz, R.; Cortijo, I.; Li, W.; Li, J.-Y.; Paul, G. Human α-Synuclein Overexpression in a Mouse Model of Parkinson’s Disease Leads to Vascular Pathology, Blood Brain Barrier Leakage and Pericyte Activation. Sci. Rep. 2021, 11, 1120. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The Epidemiology of ALS: A Conspiracy of Genes, Environment and Time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.O.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.V.; Sanberg, P.R. Impaired Blood-Brain/Spinal Cord Barrier in ALS Patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Bowser, R.; Appel, S.H. DECREASED MRNA EXPRESSION OF TIGHT JUNCTION PROTEINS IN LUMBAR SPINAL CORDS OF PATIENTS WITH ALS. Neurology 2009, 72, 1614–1616. [Google Scholar] [CrossRef]
- Kwan, K.Y.; Sestan, N.; Anton, E.S. Transcriptional Co-Regulation of Neuronal Migration and Laminar Identity in the Neocortex. Development 2012, 139, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of Neurovascular Unit Prior to Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood-Spinal Cord Barrier Breakdown and Pericyte Reductions in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2013, 125, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamadera, M.; Fujimura, H.; Inoue, K.; Toyooka, K.; Mori, C.; Hirano, H.; Sakoda, S. Microvascular Disturbance with Decreased Pericyte Coverage Is Prominent in the Ventral Horn of Patients with Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 393–401. [Google Scholar] [CrossRef]
- Zhong, Z.; Deane, R.; Ali, Z.; Parisi, M.; Shapovalov, Y.; O’Banion, M.K.; Stojanovic, K.; Sagare, A.; Boillee, S.; Cleveland, D.W.; et al. ALS-Causing SOD1 Mutants Generate Vascular Changes Prior to Motor Neuron Degeneration. Nat. Neurosci. 2008, 11, 420–422. [Google Scholar] [CrossRef] [Green Version]
- Garbuzova-Davis, S.; Haller, E.; Saporta, S.; Kolomey, I.; Nicosia, S.V.; Sanberg, P.R. Ultrastructure of Blood–Brain Barrier and Blood–Spinal Cord Barrier in SOD1 Mice Modeling ALS. Brain Res. 2007, 1157, 126–137. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Saporta, S.; Haller, E.; Kolomey, I.; Bennett, S.P.; Potter, H.; Sanberg, P.R. Evidence of Compromised Blood-Spinal Cord Barrier in Early and Late Symptomatic SOD1 Mice Modeling ALS. PLoS ONE 2007, 2, e1205. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.C.; Serres, S.; Khrapitchev, A.A.; Stolp, H.B.; Anthony, D.C.; Talbot, K.; Turner, M.R.; Sibson, N.R. T2 -Weighted MRI Detects Presymptomatic Pathology in the SOD1 Mouse Model of ALS. J. Cereb. Blood Flow Metab. 2014, 34, 785–793. [Google Scholar] [CrossRef]
- Nicaise, C.; Mitrecic, D.; Demetter, P.; De Decker, R.; Authelet, M.; Boom, A.; Pochet, R. Impaired Blood–Brain and Blood–Spinal Cord Barriers in Mutant SOD1-Linked ALS Rat. Brain Res. 2009, 1301, 152–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, E.A.; Sengillo, J.D.; Sagare, A.P.; Zhao, Z.; Ma, Q.; Zuniga, E.; Wang, Y.; Zhong, Z.; Sullivan, J.S.; Griffin, J.H.; et al. Blood-Spinal Cord Barrier Disruption Contributes to Early Motor-Neuron Degeneration in ALS-Model Mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1035–E1042. [Google Scholar] [CrossRef] [Green Version]
- Andjus, P.R.; BataveljicÌ, D.; Vanhoutte, G.; Mitrecic, D.; Pizzolante, F.; Djogo, N.; Nicaise, C.; Kengne, F.G.; Gangitano, C.; Michetti, F.; et al. In Vivo Morphological Changes in Animal Models of Amyotrophic Lateral Sclerosis and Alzheimer’s-Like Disease: MRI Approach. Anat. Rec. 2009, 292, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, C.; Soyfoo, M.S.; Authelet, M.; De Decker, R.; Bataveljic, D.; Delporte, C.; Pochet, R. Aquaporin-4 Overexpression in Rat ALS Model. Anat. Rec. 2009, 292, 207–213. [Google Scholar] [CrossRef]
- Bataveljić, D.; Nikolić, L.; Milosević, M.; Todorović, N.; Andjus, P.R. Changes in the Astrocytic Aquaporin-4 and Inwardly Rectifying Potassium Channel Expression in the Brain of the Amyotrophic Lateral Sclerosis SOD1 G93A Rat Model. Glia 2012, 60, 1991–2003. [Google Scholar] [CrossRef]
- Bataveljić, D.; Stamenković, S.; Bačić, G.; Andjus, P. Imaging Cellular Markers of Neuroinflammation in the Brain of the Rat Model of Amyotrophic Lateral Sclerosis. Acta Physiol. Hung. 2011, 98, 27–31. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein Aggregation in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.A.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 Distinguishes Sporadic Amyotrophic Lateral Sclerosis from Amyotrophic Lateral Sclerosis with SOD1 Mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- Dayalu, P.; Albin, R.L. Huntington Disease: Pathogenesis and Treatment. Neurol. Clin. 2015, 33, 101–114. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s Disease Protein Interacts with P53 and CREB-Binding Protein and Represses Transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, A.; Goldstein, D.R.; Hodges, A.; Strand, A.D.; Sengstag, T.; Kooperberg, C.; Becanovic, K.; Pouladi, M.A.; Sathasivam, K.; Cha, J.-H.J.; et al. Mutant Huntingtin’s Effects on Striatal Gene Expression in Mice Recapitulate Changes Observed in Human Huntington’s Disease Brain and Do Not Differ with Mutant Huntingtin Length or Wild-Type Huntingtin Dosage. Hum. Mol. Genet. 2007, 16, 1845–1861. [Google Scholar] [CrossRef] [Green Version]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L.F. Huntingtin Facilitates Dynein/Dynactin-Mediated Vesicle Transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, E.J.; Shaler, T.A.; Woodman, B.; Ryu, K.-Y.; Zaitseva, T.S.; Becker, C.H.; Bates, G.P.; Schulman, H.; Kopito, R.R. Global Changes to the Ubiquitin System in Huntington’s Disease. Nature 2007, 448, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. The Regulation of Autophagosome Dynamics by Huntingtin and HAP1 Is Disrupted by Expression of Mutant Huntingtin, Leading to Defective Cargo Degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1alpha by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Reddy, P.H. Abnormal Mitochondrial Dynamics, Mitochondrial Loss and Mutant Huntingtin Oligomers in Huntington’s Disease: Implications for Selective Neuronal Damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, T.-S.; Slow, E.; Lupu, V.; Stavrovskaya, I.G.; Sugimori, M.; Llinás, R.; Kristal, B.S.; Hayden, M.R.; Bezprozvanny, I. Disturbed Ca2+ Signaling and Apoptosis of Medium Spiny Neurons in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2005, 102, 2602–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aylward, E.H.; Li, Q.; Stine, O.C.; Ranen, N.; Sherr, M.; Barta, P.E.; Bylsma, F.W.; Pearlson, G.D.; Ross, C.A. Longitudinal Change in Basal Ganglia Volume in Patients with Huntington’s Disease. Neurology 1997, 48, 394–399. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Scahill, R.I.; Owen, G.; Durr, A.; Leavitt, B.R.; Roos, R.A.; Borowsky, B.; Landwehrmeyer, B.; Frost, C.; Johnson, H.; et al. Predictors of Phenotypic Progression and Disease Onset in Premanifest and Early-Stage Huntington’s Disease in the TRACK-HD Study: Analysis of 36-Month Observational Data. Lancet Neurol. 2013, 12, 637–649. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; DiFiglia, M. Huntington Disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [Green Version]
- Drouin-Ouellet, J.; Sawiak, S.J.; Cisbani, G.; Lagacé, M.; Kuan, W.-L.; Saint-Pierre, M.; Dury, R.J.; Alata, W.; St-Amour, I.; Mason, S.L.; et al. Cerebrovascular and Blood-Brain Barrier Impairments in Huntington’s Disease: Potential Implications for Its Pathophysiology. Ann. Neurol. 2015, 78, 160–177. [Google Scholar] [CrossRef]
- Hsiao, H.-Y.; Chen, Y.-C.; Huang, C.-H.; Chen, C.-C.; Hsu, Y.-H.; Chen, H.-M.; Chiu, F.-L.; Kuo, H.-C.; Chang, C.; Chern, Y. Aberrant Astrocytes Impair Vascular Reactivity in Huntington Disease. Ann. Neurol. 2015, 78, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Hsu, Y.-H.; Lin, M.-H.; Yang, T.-H.; Chen, H.-M.; Chen, Y.-C.; Hsiao, H.-Y.; Chen, C.-C.; Chern, Y.; Chang, C. Neurovascular Abnormalities in Humans and Mice with Huntington’s Disease. Exp. Neurol. 2013, 250, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Di Pardo, A.; Amico, E.; Scalabrì, F.; Pepe, G.; Castaldo, S.; Elifani, F.; Capocci, L.; De Sanctis, C.; Comerci, L.; Pompeo, F.; et al. Impairment of Blood-Brain Barrier Is an Early Event in R6/2 Mouse Model of Huntington Disease. Sci. Rep. 2017, 7, 41316. [Google Scholar] [CrossRef] [PubMed]
- Di Pardo, A.; Carrizzo, A.; Damato, A.; Castaldo, S.; Amico, E.; Capocci, L.; Ambrosio, M.; Pompeo, F.; De Sanctis, C.; Spinelli, C.C.; et al. Motor Phenotype Is Not Associated with Vascular Dysfunction in Symptomatic Huntington’s Disease Transgenic R6/2 (160 CAG) Mice. Sci. Rep. 2017, 7, 42797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Teshigawara, R.; Cho, J.; Kameda, M.; Tada, T. Mechanism of Human Somatic Reprogramming to IPS Cell. Lab. Investig. 2017, 97, 1152–1157. [Google Scholar] [CrossRef]
- Malik, N.; Rao, M.S. A Review of the Methods for Human IPSC Derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negoro, T.; Okura, H.; Matsuyama, A. Induced Pluripotent Stem Cells: Global Research Trends. BioResearch Open Access 2017, 6, 63–73. [Google Scholar] [CrossRef] [Green Version]
- de Rus Jacquet, A.; Denis, H.L.; Cicchetti, F.; Alpaugh, M. Current and Future Applications of Induced Pluripotent Stem Cell-Based Models to Study Pathological Proteins in Neurodegenerative Disorders. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Ting, H.-C.; Liu, C.-A.; Su, H.-L.; Chiou, T.-W.; Lin, S.-Z.; Harn, H.-J.; Ho, T.-J. Induced Pluripotent Stem Cell (IPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 2020, 25, 2000. [Google Scholar] [CrossRef]
- Pasteuning-Vuhman, S.; de Jongh, R.; Timmers, A.; Pasterkamp, R.J. Towards Advanced IPSC-Based Drug Development for Neurodegenerative Disease. Trends Mol. Med. 2021, 27, 263–279. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Al-Ahmad, A.; Azarin, S.M.; Palecek, S.P.; Shusta, E.V. A Retinoic Acid-Enhanced, Multicellular Human Blood-Brain Barrier Model Derived from Stem Cell Sources. Sci. Rep. 2014, 4, 4160. [Google Scholar] [CrossRef] [Green Version]
- Qian, T.; Maguire, S.E.; Canfield, S.G.; Bao, X.; Olson, W.R.; Shusta, E.V.; Palecek, S.P. Directed Differentiation of Human Pluripotent Stem Cells to Blood-Brain Barrier Endothelial Cells. Sci. Adv. 2017, 3, e1701679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollmann, E.K.; Bailey, A.K.; Potharazu, A.V.; Neely, M.D.; Bowman, A.B.; Lippmann, E.S. Accelerated Differentiation of Human Induced Pluripotent Stem Cells to Blood-Brain Barrier Endothelial Cells. Fluids Barriers CNS 2017, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.H.; Marinelli, N.A.; Shi, Y.; McClatchey, P.M.; Balotin, K.M.; Gullett, D.R.; Hagerla, K.A.; Bowman, A.B.; Ess, K.C.; Wikswo, J.P.; et al. A Simplified, Fully Defined Differentiation Scheme for Producing Blood-Brain Barrier Endothelial Cells from Human IPSCs. Stem Cell Rep. 2019, 12, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praça, C.; Rosa, S.C.; Sevin, E.; Cecchelli, R.; Dehouck, M.-P.; Ferreira, L.S. Derivation of Brain Capillary-like Endothelial Cells from Human Pluripotent Stem Cell-Derived Endothelial Progenitor Cells. Stem Cell Rep. 2019, 13, 599–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.M.; Barcia Durán, J.G.; Houghton, S.; Rafii, S.; Redmond, D.; Lis, R. Human Induced Pluripotent Stem Cell-Derived Brain Endothelial Cells: Current Controversies. Front. Physiol. 2021, 12, 642812. [Google Scholar] [CrossRef]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory Cell Trafficking across the Blood-Brain Barrier: Chemokine Regulation and in Vitro Models: Leukocyte Migration and in Vitro BBB Model. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, H.; Gastfriend, B.D.; Soldati, S.; Perriot, S.; Mathias, A.; Sano, Y.; Shimizu, F.; Gosselet, F.; Kanda, T.; Palecek, S.P.; et al. Advancing Human Induced Pluripotent Stem Cell-Derived Blood-Brain Barrier Models for Studying Immune Cell Interactions. FASEB J. 2020, 34, 16693–16715. [Google Scholar] [CrossRef]
- Delsing, L.; Dönnes, P.; Sánchez, J.; Clausen, M.; Voulgaris, D.; Falk, A.; Herland, A.; Brolén, G.; Zetterberg, H.; Hicks, R.; et al. Barrier Properties and Transcriptome Expression in Human IPSC-Derived Models of the Blood-Brain Barrier. Stem Cells 2018, 36, 1816–1827. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.M.; Houghton, S.; Magdeldin, T.; Durán, J.G.B.; Minotti, A.P.; Snead, A.; Sproul, A.; Nguyen, D.-H.T.; Xiang, J.; Fine, H.A.; et al. Pluripotent Stem Cell-Derived Epithelium Misidentified as Brain Microvascular Endothelium Requires ETS Factors to Acquire Vascular Fate. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Krencik, R.; Zhang, S.-C. Directed Differentiation of Functional Astroglial Subtypes from Human Pluripotent Stem Cells. Nat. Protoc. 2011, 6, 1710–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte Pathology and the Absence of Non-Cell Autonomy in an Induced Pluripotent Stem Cell Model of TDP-43 Proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaltouki, A.; Peng, J.; Liu, Q.; Rao, M.S.; Zeng, X. Efficient Generation of Astrocytes from Human Pluripotent Stem Cells in Defined Conditions. Stem Cells 2013, 31, 941–952. [Google Scholar] [CrossRef]
- Santos, R.; Vadodaria, K.C.; Jaeger, B.N.; Mei, A.; Lefcochilos-Fogelquist, S.; Mendes, A.P.D.; Erikson, G.; Shokhirev, M.; Randolph-Moore, L.; Fredlender, C.; et al. Differentiation of Inflammation-Responsive Astrocytes from Glial Progenitors Generated from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 1757–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tcw, J.; Wang, M.; Pimenova, A.A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef] [Green Version]
- Perriot, S.; Mathias, A.; Perriard, G.; Canales, M.; Jonkmans, N.; Merienne, N.; Meunier, C.; El Kassar, L.; Perrier, A.L.; Laplaud, D.-A.; et al. Human Induced Pluripotent Stem Cell-Derived Astrocytes Are Differentially Activated by Multiple Sclerosis-Associated Cytokines. Stem Cell Rep. 2018, 11, 1199–1210. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, T.; Mukouyama, Y.-S. Tissue Specific Origin, Development, and Pathological Perspectives of Pericytes. Front. Cardiovasc. Med. 2018, 5, 78. [Google Scholar] [CrossRef] [Green Version]
- Etchevers, H.C.; Vincent, C.; Le Douarin, N.M.; Couly, G.F. The Cephalic Neural Crest Provides Pericytes and Smooth Muscle Cells to All Blood Vessels of the Face and Forebrain. Development 2001, 128, 1059–1068. [Google Scholar] [CrossRef]
- Faal, T.; Phan, D.T.T.; Davtyan, H.; Scarfone, V.M.; Varady, E.; Blurton-Jones, M.; Hughes, C.C.W.; Inlay, M.A. Induction of Mesoderm and Neural Crest-Derived Pericytes from Human Pluripotent Stem Cells to Study Blood-Brain Barrier Interactions. Stem Cell Rep. 2019, 12, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Stebbins, M.J.; Gastfriend, B.D.; Canfield, S.G.; Lee, M.-S.; Richards, D.; Faubion, M.G.; Li, W.-J.; Daneman, R.; Palecek, S.P.; Shusta, E.V. Human Pluripotent Stem Cell-Derived Brain Pericyte-like Cells Induce Blood-Brain Barrier Properties. Sci. Adv. 2019, 5, 7375. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Huang, Y.; Gong, J.; Wang, J.; Fan, Y.; Cai, J.; Wang, Y.; Qiu, Y.; Wei, Y.; Xiong, C.; et al. Transplantation of HPSC-Derived Pericyte-like Cells Promotes Functional Recovery in Ischemic Stroke Mice. Nat. Commun. 2020, 11, 5196. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; D’Souza, S.S.; Moskvin, O.V.; Toh, H.; Wang, B.; Zhang, J.; Swanson, S.; Guo, L.-W.; Thomson, J.A.; Slukvin, I.I. Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Rep. 2017, 19, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelleher, J.; Dickinson, A.; Cain, S.; Hu, Y.; Bates, N.; Harvey, A.; Ren, J.; Zhang, W.; Moreton, F.C.; Muir, K.W.; et al. Patient-Specific IPSC Model of a Genetic Vascular Dementia Syndrome Reveals Failure of Mural Cells to Stabilize Capillary Structures. Stem Cell Rep. 2019, 13, 817–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Kojima, K.; Taura, D.; Sone, M.; Washida, K.; Egawa, N.; Kondo, T.; Minakawa, E.N.; Tsukita, K.; Enami, T.; et al. Human IPS Cell-Derived Mural Cells as an in Vitro Model of Hereditary Cerebral Small Vessel Disease. Mol. Brain 2020, 13, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, J.J.; Linville, R.M.; Ding, Y.Y.; Gerecht, S.; Searson, P.C. Role of IPSC-Derived Pericytes on Barrier Function of IPSC-Derived Brain Microvascular Endothelial Cells in 2D and 3D. Fluids Barriers CNS 2019, 16, 15. [Google Scholar] [CrossRef]
- Tigges, U.; Welser-Alves, J.V.; Boroujerdi, A.; Milner, R. A Novel and Simple Method for Culturing Pericytes from Mouse Brain. Microvasc. Res. 2012, 84, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Smyth, L.C.D.; Rustenhoven, J.; Scotter, E.L.; Schweder, P.; Faull, R.L.M.; Park, T.I.H.; Dragunow, M. Markers for Human Brain Pericytes and Smooth Muscle Cells. J. Chem. Neuroanat. 2018, 92, 48–60. [Google Scholar] [CrossRef]
- Stone, N.L.; England, T.J.; O’Sullivan, S.E. A Novel Transwell Blood Brain Barrier Model Using Primary Human Cells. Front. Cell Neurosci. 2019, 13, 230. [Google Scholar] [CrossRef] [Green Version]
- Ruck, T.; Bittner, S.; Meuth, S.G. Blood-Brain Barrier Modeling: Challenges and Perspectives. Neural Regen. Res. 2015, 10, 889–891. [Google Scholar] [CrossRef]
- Stebbins, M.J.; Wilson, H.K.; Canfield, S.G.; Qian, T.; Palecek, S.P.; Shusta, E.V. Differentiation and Characterization of Human Pluripotent Stem Cell-Derived Brain Microvascular Endothelial Cells. Methods 2016, 101, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Oikari, L.E.; Pandit, R.; Stewart, R.; Cuní-López, C.; Quek, H.; Sutharsan, R.; Rantanen, L.M.; Oksanen, M.; Lehtonen, S.; de Boer, C.M.; et al. Altered Brain Endothelial Cell Phenotype from a Familial Alzheimer Mutation and Its Potential Implications for Amyloid Clearance and Drug Delivery. Stem Cell Rep. 2020, 14, 924–939. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of Shear Stress on Endothelial Cells: Go with the Flow. Acta Physiol. (Oxf.) 2017, 219, 382–408. [Google Scholar] [CrossRef] [PubMed]
- DeStefano, J.G.; Xu, Z.S.; Williams, A.J.; Yimam, N.; Searson, P.C. Effect of Shear Stress on IPSC-Derived Human Brain Microvascular Endothelial Cells (DhBMECs). Fluids Barriers CNS 2017, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Canfield, S.G.; Stebbins, M.J.; Morales, B.S.; Asai, S.W.; Vatine, G.D.; Svendsen, C.N.; Palecek, S.P.; Shusta, E.V. An Isogenic Blood-Brain Barrier Model Comprising Brain Endothelial Cells, Astrocytes, and Neurons Derived from Human Induced Pluripotent Stem Cells. J. Neurochem. 2017, 140, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Appelt-Menzel, A.; Cubukova, A.; Günther, K.; Edenhofer, F.; Piontek, J.; Krause, G.; Stüber, T.; Walles, H.; Neuhaus, W.; Metzger, M. Establishment of a Human Blood-Brain Barrier Co-Culture Model Mimicking the Neurovascular Unit Using Induced Pluri- and Multipotent Stem Cells. Stem Cell Rep. 2017, 8, 894–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [Google Scholar] [CrossRef] [Green Version]
- Urich, E.; Patsch, C.; Aigner, S.; Graf, M.; Iacone, R.; Freskgård, P.-O. Multicellular Self-Assembled Spheroidal Model of the Blood Brain Barrier. Sci. Rep. 2013, 3, 1500. [Google Scholar] [CrossRef] [Green Version]
- Nzou, G.; Wicks, R.T.; Wicks, E.E.; Seale, S.A.; Sane, C.H.; Chen, A.; Murphy, S.V.; Jackson, J.D.; Atala, A.J. Human Cortex Spheroid with a Functional Blood Brain Barrier for High-Throughput Neurotoxicity Screening and Disease Modeling. Sci. Rep. 2018, 8, 7413. [Google Scholar] [CrossRef] [Green Version]
- Ungrin, M.D.; Joshi, C.; Nica, A.; Bauwens, C.; Zandstra, P.W. Reproducible, Ultra High-Throughput Formation of Multicellular Organization from Single Cell Suspension-Derived Human Embryonic Stem Cell Aggregates. PLoS ONE 2008, 3, e1565. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.-F.; Wolfe, J.M.; Fadzen, C.M.; Calligaris, D.; Hornburg, K.; Chiocca, E.A.; Agar, N.Y.R.; Pentelute, B.L.; Lawler, S.E. Blood-Brain-Barrier Spheroids as an in Vitro Screening Platform for Brain-Penetrating Agents. Nat. Commun. 2017, 8, 15623. [Google Scholar] [CrossRef]
- Bhalerao, A.; Sivandzade, F.; Archie, S.R.; Chowdhury, E.A.; Noorani, B.; Cucullo, L. In Vitro Modeling of the Neurovascular Unit: Advances in the Field. Fluids Barriers CNS 2020, 17, 22. [Google Scholar] [CrossRef]
- Song, L.; Yuan, X.; Jones, Z.; Griffin, K.; Zhou, Y.; Ma, T.; Li, Y. Assembly of Human Stem Cell-Derived Cortical Spheroids and Vascular Spheroids to Model 3-D Brain-like Tissues. Sci. Rep. 2019, 9, 5977. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.S.Y.; Guo, F.; Zhang, B. Modeling Organ-Specific Vasculature with Organ-on-a-Chip Devices. Nanotechnology 2019, 30, 024002. [Google Scholar] [CrossRef]
- Sivandzade, F.; Cucullo, L. In-Vitro Blood-Brain Barrier Modeling: A Review of Modern and Fast-Advancing Technologies. J. Cereb. Blood Flow Metab. 2018, 38, 1667–1681. [Google Scholar] [CrossRef]
- van der Helm, M.W.; van der Meer, A.D.; Eijkel, J.C.T.; van den Berg, A.; Segerink, L.I. Microfluidic Organ-on-Chip Technology for Blood-Brain Barrier Research. Tissue Barriers 2016, 4, e1142493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.I.; Abaci, H.E.; Shuler, M.L. Microfluidic Blood–Brain Barrier Model Provides in Vivo-like Barrier Properties for Drug Permeability Screening. Biotechnol. Bioeng. 2017, 114, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Vatine, G.D.; Barrile, R.; Workman, M.J.; Sances, S.; Barriga, B.K.; Rahnama, M.; Barthakur, S.; Kasendra, M.; Lucchesi, C.; Kerns, J.; et al. Human IPSC-Derived Blood-Brain Barrier Chips Enable Disease Modeling and Personalized Medicine Applications. Cell Stem Cell 2019, 24, 995–1005.e6. [Google Scholar] [CrossRef] [PubMed]
- Park, T.-E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-Enhanced Blood-Brain Barrier Chip Recapitulates Human Barrier Function and Shuttling of Drugs and Antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef]
- Motallebnejad, P.; Thomas, A.; Swisher, S.L.; Azarin, S.M. An Isogenic HiPSC-Derived BBB-on-a-Chip. Biomicrofluidics 2019, 13, 064119. [Google Scholar] [CrossRef] [PubMed]
- Pediaditakis, I.; Kodella, K.R.; Manatakis, D.V.; Hinojosa, C.D.; Manolakos, E.S.; Rubin, L.L.; Hamilton, G.A.; Karalis, K. Modeling Alpha-Synuclein Pathology in a Human Brain-Chip to Assess. Blood-Brain Barrier Disruption in Parkinson’s Disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Halldorsson, S.; Lucumi, E.; Gómez-Sjöberg, R.; Fleming, R.M.T. Advantages and Challenges of Microfluidic Cell Culture in Polydimethylsiloxane Devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Li, S.; Zheng, J.; Li, Y.; Huang, H. Recent Progress in Microfluidic Models of the Blood-Brain Barrier. Micromachines 2019, 10, 375. [Google Scholar] [CrossRef] [Green Version]
- Campisi, M.; Shin, Y.; Osaki, T.; Hajal, C.; Chiono, V.; Kamm, R.D. 3D Self-Organized Microvascular Model of the Human Blood-Brain Barrier with Endothelial Cells, Pericytes and Astrocytes. Biomaterials 2018, 180, 117–129. [Google Scholar] [CrossRef]
- Faley, S.L.; Neal, E.H.; Wang, J.X.; Bosworth, A.M.; Weber, C.M.; Balotin, K.M.; Lippmann, E.S.; Bellan, L.M. IPSC-Derived Brain Endothelium Exhibits Stable, Long-Term Barrier Function in Perfused Hydrogel Scaffolds. Stem Cell Rep. 2019, 12, 474–487. [Google Scholar] [CrossRef] [Green Version]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of Human Vascularized Brain Organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.-J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.-S.; et al. Engineering of Human Brain Organoids with a Functional Vascular-like System. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Zhao, Z.; Vizetto-Duarte, C.; Moay, Z.K.; Setyawati, M.I.; Rakshit, M.; Kathawala, M.H.; Ng, K.W. Composite Hydrogels in Three-Dimensional in Vitro Models. Front. Bioeng. Biotechnol. 2020, 8, 611. [Google Scholar] [CrossRef] [PubMed]
- Logan, S.; Arzua, T.; Canfield, S.G.; Seminary, E.R.; Sison, S.L.; Ebert, A.D.; Bai, X. Studying Human Neurological Disorders Using Induced Pluripotent Stem Cells: From 2D Monolayer to 3D Organoid and Blood Brain Barrier Models. Compr. Physiol. 2019, 9, 565–611. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.; Shieh, J.C. Cell Culture Techniques. In Guide to Research Techniques in Neuroscience; Elsevier: Amsterdam, The Netherlands, 2010; pp. 281–296. ISBN 978-0-12-374849-2. [Google Scholar]
- de Fernandez, T.S.; de Souza, F.C.; Mencalha, A.L. Human Induced Pluripotent Stem Cells from Basic Research to Potential Clinical Applications in Cancer. BioMed. Res. Int. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Medvedev, S.P.; Shevchenko, A.I.; Zakian, S.M. Induced Pluripotent Stem Cells: Problems and Advantages When Applying Them in Regenerative Medicine. Acta Nat. 2010, 2, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.; Escott-Price, V. Polygenic Risk Scores in Alzheimer’s Disease: Current Applications and Future Directions. Front. Digit. Health 2020, 2, 14. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Noyce, A.J.; Hemani, G.; Nicolas, A.; Calvo, A.; Mora, G.; ITALSGEN Consortium; International ALS Genomics Consortium; Tienari, P.J.; Stone, D.J.; et al. Shared Polygenic Risk and Causal Inferences in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2019, 85, 470–481. [Google Scholar] [CrossRef]
- Escott-Price, V.; International Parkinson’s Disease Genomics Consortium; Nalls, M.A.; Morris, H.R.; Lubbe, S.; Brice, A.; Gasser, T.; Heutink, P.; Wood, N.W.; Hardy, J.; et al. Polygenic Risk of Parkinson Disease Is Correlated with Disease Age at Onset. Ann. Neurol. 2015, 77, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Raut, S.; Patel, R.; Al-Ahmad, A.J. Presence of a Mutation in PSEN1 or PSEN2 Gene Is Associated with an Impaired Brain Endothelial Cell Phenotype in Vitro. Fluids Barriers CNS 2021, 18, 3. [Google Scholar] [CrossRef]
- Katt, M.E.; Mayo, L.N.; Ellis, S.E.; Mahairaki, V.; Rothstein, J.D.; Cheng, L.; Searson, P.C. The Role of Mutations Associated with Familial Neurodegenerative Disorders on Blood-Brain Barrier Function in an IPSC Model. Fluids Barriers CNS 2019, 16, 20. [Google Scholar] [CrossRef]
- Patel, R.; Raut, S.; Al-Ahmad, A. Induced Pluripotent Stem Cells Derived Brain Endothelial Cells from Patients Suffering from Familial Form. of Alzheimer’s Disease Display Impaired Barrier Function and Cell Metabolism. Research 2020. [Google Scholar] [CrossRef]
- Rieker, C.; Migliavacca, E.; Vaucher, A.; Baud, G.; Marquis, J.; Charpagne, A.; Hegde, N.; Guignard, L.; McLachlan, M.; Pooler, A.M. Apolipoprotein E4 Expression Causes Gain of Toxic Function in Isogenic Human Induced Pluripotent Stem Cell-Derived Endothelial Cells. Arter. Thromb. Vasc. Biol. 2019, 39, e195–e207. [Google Scholar] [CrossRef]
- Lee, S.-I.; Jeong, W.; Cho, S.; Lee, H.; Jang, Y.; Lin, Y.-T.; Tsai, L.-H.; Seo, J. ApoE4-Carrying Human Astrocytes Oversupply Cholesterol into Neurons and Promote Aβ Generation. Res. Artic. 2020. [Google Scholar] [CrossRef]
- Zhao, J.; Davis, M.D.; Martens, Y.A.; Shinohara, M.; Graff-Radford, N.R.; Younkin, S.G.; Wszolek, Z.K.; Kanekiyo, T.; Bu, G. APOE Ε4/Ε4 Diminishes Neurotrophic Function of Human IPSC-Derived Astrocytes. Hum. Mol. Genet. 2017, 26, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific IPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, H.D.E.; Wessely, F.; Connor-Robson, N.; Rinaldi, F.; Vowles, J.; Browne, C.; Evetts, S.G.; Hu, M.T.; Cowley, S.A.; Webber, C.; et al. RNA Sequencing Reveals MMP2 and TGFB1 Downregulation in LRRK2 G2019S Parkinson’s IPSC-Derived Astrocytes. NeuroBiol. Dis. 2019, 129, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic Alterations in Parkinson’s Disease Astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef]
- Hall, C.E.; Yao, Z.; Choi, M.; Tyzack, G.E.; Serio, A.; Luisier, R.; Harley, J.; Preza, E.; Arber, C.; Crisp, S.J.; et al. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. 2017, 19, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Smethurst, P.; Risse, E.; Tyzack, G.E.; Mitchell, J.S.; Taha, D.M.; Chen, Y.-R.; Newcombe, J.; Collinge, J.; Sidle, K.; Patani, R. Distinct Responses of Neurons and Astrocytes to TDP-43 Proteinopathy in Amyotrophic Lateral Sclerosis. Brain 2020, 143, 430–440. [Google Scholar] [CrossRef]
- Qian, K.; Huang, H.; Peterson, A.; Hu, B.; Maragakis, N.J.; Ming, G.-L.; Chen, H.; Zhang, S.-C. Sporadic ALS Astrocytes Induce Neuronal Degeneration In Vivo. Stem Cell Rep. 2017, 8, 843–855. [Google Scholar] [CrossRef]
- Madill, M.; McDonagh, K.; Ma, J.; Vajda, A.; McLoughlin, P.; O’Brien, T.; Hardiman, O.; Shen, S. Amyotrophic Lateral Sclerosis Patient IPSC-Derived Astrocytes Impair Autophagy via Non-Cell Autonomous Mechanisms. Mol. Brain 2017, 10, 22. [Google Scholar] [CrossRef]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human IPSC-Derived Astrocytes from ALS Patients with Mutated C9ORF72 Show Increased Oxidative Stress and Neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Devlin, A.-C.; Chouhan, A.K.; Selvaraj, B.T.; Stavrou, M.; Burr, K.; Brivio, V.; He, X.; Mehta, A.R.; Story, D.; et al. Mutant C9orf72 Human IPSC-Derived Astrocytes Cause Non-Cell Autonomous Motor Neuron Pathophysiology. Glia 2020, 68, 1046–1064. [Google Scholar] [CrossRef] [Green Version]
- Qosa, H.; Lichter, J.; Sarlo, M.; Markandaiah, S.S.; McAvoy, K.; Richard, J.-P.; Jablonski, M.R.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Astrocytes Drive Upregulation of the Multidrug Resistance Transporter ABCB1 (P-Glycoprotein) in Endothelial Cells of the Blood-Brain Barrier in Mutant Superoxide Dismutase 1-Linked Amyotrophic Lateral Sclerosis. Glia 2016, 64, 1298–1313. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, L.A.; Markandaiah, S.S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Excess Glutamate Secreted from Astrocytes Drives Upregulation of P-Glycoprotein in Endothelial Cells in Amyotrophic Lateral Sclerosis. Exp. Neurol. 2019, 316, 27–38. [Google Scholar] [CrossRef]
- Lim, R.G.; Quan, C.; Reyes-Ortiz, A.M.; Lutz, S.E.; Kedaigle, A.J.; Gipson, T.A.; Wu, J.; Vatine, G.D.; Stocksdale, J.; Casale, M.S.; et al. Huntington’s Disease IPSC-Derived Brain Microvascular Endothelial Cells Reveal WNT-Mediated Angiogenic and Blood-Brain Barrier Deficits. Cell Rep. 2017, 19, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Juopperi, T.A.; Kim, W.R.; Chiang, C.-H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.; Song, H. Astrocytes Generated from Patient Induced Pluripotent Stem Cells Recapitulate Features of Huntington’s Disease Patient Cells. Mol. Brain 2012, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Garcia, V.J.; Rushton, D.J.; Tom, C.M.; Allen, N.D.; Kemp, P.J.; Svendsen, C.N.; Mattis, V.B. Huntington’s Disease Patient-Derived Astrocytes Display Electrophysiological Impairments and Reduced Neuronal Support. Front. Neurosci. 2019, 13, 669. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Citation | Experiment Conditions * | Main Readouts ** |
|---|---|---|
| Lippmann et al. 2012 [38] |  | TEER (Ω × cm2) Monoculture: 222 Co-culture: 1450, (IMR90-4), 777 (DF19-9-11T) |
| Lippmann et al. 2014 [191] |  | TEER (Ω × cm2) Monocultures: 3670 (IMR90-4), 1970 (DF19-9-11T), 1030 (hESC) Co-cultures: IMR90-4-ECs: 4450 (+pericytes), 5350 (+pericytes and NPCs) DF19-9-11T-ECs: 4740 (+pericytes and NPCs) hESC-ECs: 1680 (+pericytes and NPCs) |
| Qian et al. 2017 [192] |  | TEER (Ω × cm2) Monocultures: 3315 (IMR90-4), 1980 (H9), 3571 (DF19-9-11) Co-culture: ~30% increase compared to monoculture |
| Hollmann et al. 2017 [193] |  | TEER (Ω × cm2) Monoculture: 4227 Co-cultures: 5378 (+astrocytes), 5937 (+pericytes), 6635 (+astocytes, pericytes) Permeability, NaF (Pe × 10−7 cm/s) Monoculture: less than 1.97 in all lines |
| Appelt-Menzel et al. 2017 [225] |  | Max TEER (Ω × cm2) Monoculture: 2073 Co-cultures: 2024 (+primary astrocytes), 2485 (+iPSC-astrocytes), 2107 (+pericytes), 2171 (+hiPS-NSCs) Triple cultures: 2105 (+primary astrocytes, pericytes), 2293 (+primary astrocytes, hiPS-NSCs), 2556 (+pericytes, hiPS-NSCs) Quadruple cultures: 2489 (+primary astrocytes, pericytes and hiPS-NSCs) Permeability coefficient (µm/min) Monoculture: 1.52 (LY), 1.53 (fluorescein), 0.0166 (Dextran, 4 kDa), 0.0054 (Dextran, 40 kDa) Quadruple culture: 1.58 (LY), 1.33 (fluorescein), 0.0106 (Dextran, 4 kDa), 0.0030 (Dextran, 40 kDa) Other ↑ SLC2A1 in quadruple culture |
| Canfield et al. 2017 [224] |  | TEER (Ω × cm2) Monoculture: 153 Co-cultures: 491 (+neurons), 558 (+astrocytes), 611 (+NCPs), 784 (+primary astrocytes) Triple cultures: 661 (+neuron − astrocytes, 1:1), 886 (+neuron − astrocytes, 1:3) Permeability, NaF (Pe × 10−7 cm/s) Monoculture: 4.8 Co-cultures: 2.0 (+NCPs), 1.9 (+primary astrocytes) Triple cultures: 1.20 (+ neuron − astrocyte 1:3) Other Triple culture (neuron − astrocyte 1:3) increased TJ localization in iPSC-ECs, no change in transporter expression |
| Delsing et al. 2018 [199] |  | TEER (Ω × cm2) Monoculture: 773 (EC1), 52 (EC2) Co-culture: 1267 (EC1), 150 (EC2) Permeability Decreased NaF permeability in co-culture (EC1), exact values not told Other Changed gene expression in iPSC-ECs after co-culture: ↑ BCRP (EC1, EC2), ↑ P-gp (EC1), ↑ Glut1 (EC2), ↓ VE-cadherin (EC1), ↓ Caveolin 1 (EC1, EC2), ↑ occludin (EC2) |
| Neal et al. 2019 [194] |  | Max TEER (Ω × cm2) Monoculture: 8734 Co-culture: above 9000 Permeability, NaF (Pe × 10−7 cm/s) Monoculture: less than 2.5 |
| Stebbins et al. 2019 [210] |  | TEER (Ω × cm2) Monoculture: ≈300 (exact values not shown) Co-culture: 720 (+neurons and astrocytes), 503 (+pericyte), 1156 (+pericytes, astrocytes, and neurons) Permeability (Pe × 10−6 cm/s), NaF Monoculture: ≈6 Co-cultures: Five-fold decrease (exact values not shown) |
| Jamieson et al. 2019 [215] |  | TEER (Ω × cm2) Monoculture: 3510 Co-culture (non-contact): 3690 (1:50), 3600 (1:13), 3690 (1:5) Co-culture (contact, in gel): 2410 (1:13) |
| Faal et al. 2019 [209] |  | TEER Exact values not told, pericytes increased TEER values in co-culture. |
| Praça et al. 2019 [195] |  | TEER (Ω × cm2) Mono- and co-culture: ≈60, exact values not told Permeability, LY (Pe × 10−3 cm/min) ≈0.5, Slight decrease in co-culture compared to optimized monoculture |
| Blanchard et al. 2020 [107] |  | Average TEER (Ω × cm2) Monoculture: ≈5900 iBBB: ≈8300 Permeability, Dextran Values not told, decreased permeability in co-culture Other ↑ P-gp in co-culture |
| Nishihara et al. 2020 [198] |  | TEER (Ω × cm2) Monoculture: above 60 Co-culture: similar to monoculture Permeability, NaF (Pe × 10−3 cm/min) Monoculture: below 0.32 Co-culture: similar to monoculture Other more elongated morphology of ECs in co-culture compared to monoculture |
 | ||
| Citation | Experiment Conditions * | Main Readouts ** |
|---|---|---|
| Organ on chip | ||
| Wang et al. 2017 [236] |  | Max TEER (Ω× cm2) Monoculture: 368 Co-culture: 4399 Permeability, Dextrans (Papp× 10−8 cm/s) Co-culture: 8.43 (4 kDa), 2.18 (20 kDa) and 0.982 (70 kDa) |
| Vatine et al. 2019 [237] |  | Max TEER (Ω × cm2) Co-culture (+neural): 1500 Permeability, Dextran (Papp × 10−7 cm/s) Monoculture: ~3 (exact values not told) Co-cultures (+astrocyte, pericytes/ + neural): ~1 (exact values not told) Other ↓ ZO1 expression and ↓ permeability to Dextran after cytokine exposure (+astrocytes, pericytes) Spontaneous neuronal activity (+neural) |
| Park et al. 2019 [238] |  | Impedance (Ω) ~25 000 Permeability, Dextran (Papp × 10−8 cm/s) 8.9 (3 kDa), 1.1 (10 kDa) and 0.24 (70 kDa) Other Expression and functionality of efflux transporters |
| Motallebnejad et al. 2019 [239] |  | Max TEER (Ω × cm2) 1590 (0.4 µm membrane), 1369 (8.0 µm membrane) Co-culture increased TEER values Permeability, NaF (× 10−6) Below 1 Other Efflux transporter activity, decreased TEER after TGF-β1 exposure |
| Pediaditakis et al. 2020 preprint [240] |  | Permeability (Papp × 10−6 cm/s) range 1–3 (3 kDa), 4–6 (LY) Other RNAseq (brain side): more mature phenotype in the chip compared to conventional cell culture, chip recapitulated gene expression profile of primary tissue |
| Hydrogels/3D vessels | ||
| Campisi et al. 2018 [243] |  | Permeability coefficient, Dextran (× 10−7 cm/s) Monoculture: 6.6 (40 kDa), 12 (10 kDa) Co-culture (+pericytes): 2.5 (40 kDa), 4.8 (10 kDa) Triple co-culture (+pericytes, astrocytes): 0.89 (40 kDa), 2.2 (10 kDa) Other Complex and branched vascular network, ↑ ZO-1, claudin-5 and occludin expression in triple co-culture, ↑ gene expression of several transporters in triple co-culture |
| Faley et al. 2019 [244] |  | Permeability, Dextran 3 kDa (× 10−7 cm/s) Day 1: 1,2 (static), 1,9 (perfused) Day 7: 4.6 (static), 1.4 (perfused) Day 14: 11.7 (static), 0.23 (perfused) |
| Blanchard et al. 2020 [107] |  | Other Capillary structure ↑ CLDN5, JAMA, PGP, LRP1, RAGE and GLUT1 expression in co-culture |
| Vascularized organoids | ||
| Pham et al. 2018 [245] |  | Other Tubular structures in organoids, positive for CD31 |
| Cakir et al. 2019 [246] |  | TEER (Ω × cm2) Day 30: 186 (vhCO), 135 (hCO) Day 70: 351 (vhCO), 71 (hCO) Other Presence of vascular tubes positive for CD31, expression of BBB markers (ZO-1, occludin) Presence of astrocytes and pericytes |
 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-C.; Sonninen, T.-M.; Peltonen, S.; Koistinaho, J.; Lehtonen, Š. Blood–Brain Barrier and Neurodegenerative Diseases—Modeling with iPSC-Derived Brain Cells. Int. J. Mol. Sci. 2021, 22, 7710. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147710
Wu Y-C, Sonninen T-M, Peltonen S, Koistinaho J, Lehtonen Š. Blood–Brain Barrier and Neurodegenerative Diseases—Modeling with iPSC-Derived Brain Cells. International Journal of Molecular Sciences. 2021; 22(14):7710. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147710
Chicago/Turabian StyleWu, Ying-Chieh, Tuuli-Maria Sonninen, Sanni Peltonen, Jari Koistinaho, and Šárka Lehtonen. 2021. "Blood–Brain Barrier and Neurodegenerative Diseases—Modeling with iPSC-Derived Brain Cells" International Journal of Molecular Sciences 22, no. 14: 7710. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147710