
Human Oligodendrocytes and Myelin In Vitro to Evaluate Developmental Neurotoxicity


1
Center for Alternatives to Animal Testing (CAAT), Johns Hopkins Bloomberg School of Public Health, Baltimore, MD 21205, USA
2
Center for Alternatives to Animal Testing (CAAT-Europe), University of Konstanz, 78464 Konstanz, Germany
3
Department of Physiology, University of Lausanne, 1005 Lausanne, Switzerland
4
Swiss Centre for Applied Human Toxicology (SCAHT), 4055 Basel, Switzerland
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(15), 7929; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157929
Submission received: 9 June 2021
/
Revised: 14 July 2021
/
Accepted: 21 July 2021
/
Published: 25 July 2021
(This article belongs to the Special Issue Novel In Vitro and In Silico Approaches to Study Developmental Neurotoxicity)
{kind=link}
{kind=link}
Abstract
:Neurodevelopment is uniquely sensitive to toxic insults and there are concerns that environmental chemicals are contributing to widespread subclinical developmental neurotoxicity (DNT). Increased DNT evaluation is needed due to the lack of such information for most chemicals in common use, but in vivo studies recommended in regulatory guidelines are not practical for the large-scale screening of potential DNT chemicals. It is widely acknowledged that developmental neurotoxicity is a consequence of disruptions to basic processes in neurodevelopment and that testing strategies using human cell-based in vitro systems that mimic these processes could aid in prioritizing chemicals with DNT potential. Myelination is a fundamental process in neurodevelopment that should be included in a DNT testing strategy, but there are very few in vitro models of myelination. Thus, there is a need to establish an in vitro myelination assay for DNT. Here, we summarize the routes of myelin toxicity and the known models to study this particular endpoint.
1. Introduction: Role of Environmental Factors in an Epidemic of Adverse Neurodevelopmental Outcomes
One in every six children in the United States, or about ten million children, were diagnosed with a developmental disorder from 2006 to 2008, according to the latest report by the Centers for Disease Control and Prevention (CDC) National Center on Birth Defects and Developmental Disabilities (NCBDDD) [1]. This represents an increase in prevalence of about 17% in the 12 year period from 1997 to 2008, or nearly 1.8 million more children with at least one developmental disability from 2006 to 2008 compared to the previous decade [1]. It was reported that this trend was primarily due to an increase in the prevalence of neurodevelopmental disorders [1]. Children with neurodevelopmental disorders can exhibit a wide range of functional deficits, including: learning disabilities; limited control of executive functions; sensory impairments; language, social, or motor limitations; and an inability to achieve expected milestones in development [2].
In recent decades, the prevalence of certain neurodevelopmental disorders, such as autism spectrum disorder (ASD) and attention deficit/hyperactivity disorder (ADHD), has increased considerably [1,2,3,4,5,6,7]. The CDC Autism and Developmental Disabilities Monitoring (ADDM) Network estimates that one in 54 children has been identified with ASD [8], and the NCBDDD of the CDC reports that 9.4% (6.1 million) of children in the United States were diagnosed with ADHD in 2016 [9]. Although an increase in prevalence could be influenced by several other factors, including the increased awareness and recognition of symptoms or behavioral phenotypes, improved diagnostic capabilities, changes in the definitions of conditions, better reporting, older parental age, or medical advances that allow increased survival rates of preterm infants with borderline viability [1,10], increased exposure to environmental contaminants is thought to play a substantial role [11,12,13].
Despite intense research, the exact etiologies of most neurodevelopmental disorders remain undetermined [11,12,13,14]. Although many neurodevelopmental disorders are thought to have a genetic basis, there is increasing evidence of an environmental influence [15,16,17], as demonstrated in monozygotic twin studies [18,19,20]. Overall, it has been estimated that about 3% of developmental defects are directly attributable to maternal exposure to chemical or physical agents in the environment, and approximately 25% are due to gene–environment interactions in genetically predisposed individuals (NRC 2000). However, these rough approximations are reported to likely underestimate the magnitude of the environmental influence on neurodevelopment [21].
Furthermore, there are concerns that environmental chemicals are contributing to a subclinical pandemic of neurodevelopmental toxicity, with reductions in cognitive function characterized by a slight decrease in intelligence quotient (IQ). These diseases might even be more prevalent than neurodevelopmental disorders with clinically observable behavioral phenotypes (such as ASD and ADHD) [11]. Although it is difficult to determine that prior exposure to a toxic agent during neurodevelopment is the cause of lowered IQ [22], this has been documented as a result of lead exposure, in which an inverse relationship was identified between blood-lead level and IQ score [23]. Such outcomes have significant repercussions on society as a whole [22]. It has been estimated that a relatively small drop (five points) in the average IQ of a cohort of lead-exposed children greatly affects the IQ distribution in a population, resulting in a more than 50% decrease in the number of gifted individuals (IQ > 130) and more than 50% increase in the number of individuals with significant cognitive impairment (IQ < 70) [22,24]. Given the potential societal implications of reduced cognitive function resulting from childhood exposures, the environmental influence on neurodevelopment is a major public health concern, particularly because children are a uniquely susceptible population [11].
1.1. Pre- and Postnatal Susceptibility to Environmental Exposure
Compared to adults, infants and children have a higher air, food, and water intake relative to their body weight, and, consequently, they are exposed to higher doses of environmental contaminants [24,25,26]. In addition, their immature metabolism increases their susceptibility to adverse effects following exposure to a toxic agent [12,27]. Postnatal exposure to xenobiotics can also occur during breastfeeding; lipophilic compounds stored in maternal adipose tissue are mobilized with fat in breast milk production and can diffuse into the mammary glands and be excreted during lactation [28,29]. In addition, the unique activities of children, such as hand-to-mouth behavior and crawling or playing close to the ground, also make children particularly susceptible to environmental exposure [26,27].
Although the susceptibility of infants and children has been long recognized, prenatal vulnerability to environmental exposure was not acknowledged until the causal association was made between birth defects and thalidomide (a drug that had been prescribed to pregnant women for morning sickness) [21]. Prior to the recognition of birth defects attributed to thalidomide in the 1960s, chemicals were not routinely tested for their teratogenicity, due to the assumption that the placental barrier completely protected the fetus from toxicant exposure [21]. The placenta provides the exchange of nutrients, oxygen, and fetal waste products between the fetal and maternal circulatory systems, and the placental barrier describes the interface at which this exchange occurs [30]. The placenta is not completely protective, because the transplacental passage of xenobiotics and their metabolites across the placental barrier can occur [28]. The ability of a compound to cross the placental barrier depends on its physical and chemical properties, and the mechanisms of exchange across the placental barrier include the passive diffusion of lipophilic compounds, facilitated diffusion by transporters, endocytosis, and active transport [30,31]. Compounds can also be metabolized by the placenta, as it is capable of both phase I and phase II biotransformation, which can alter transplacental passage [30]. In addition, polar metabolites formed in the fetal compartment can accumulate, as their lipophobicity may prevent transport across the placental barrier [32].
The blood–brain barrier (BBB) permeability to xenobiotics or their metabolites is also an important factor in pre- and postnatal susceptibility. The BBB separates the central nervous system (CNS) from the bloodstream [28,33,34], and the capillaries that form the BBB are comprised of tight junctions of adjacent endothelial cells, effectively preventing the paracellular transport of hydrophilic compounds that can occur in other organs [28], thus protecting the brain from many neurotoxicants [33,35]. The endothelial cells of the BBB also contain efflux transporters, which actively transport certain nonpolar compounds across endothelial cell membranes back into the blood to prevent entry by diffusion into the brain [28]. Although the BBB is protective, lipophilic compounds may still bypass efflux transporters to cross endothelial cell plasma membranes, and certain polar compounds may be actively transported across the BBB [28,36]. In addition, the BBB is not established until about 23–32 weeks of gestation [37,38], and thus, the central nervous system is less protected from hydrophilic compounds in the very early stages of development, during which time the developing brain is particularly vulnerable.
1.2. Vulnerability of the Developing Brain to Environmental Exposures
Human brain development involves complex molecular, cellular, and environmental interactions that begin during the first month of gestation [39,40,41,42]. Due to neural plasticity—the adaptability of the brain—the developing human brain is uniquely vulnerable to toxic insult [42,43]. Moreover, nervous system development is progressive, meaning that emerging structures provide the basis for further development [42]. Considering this progression and adaptability, there exist critical windows in embryonic and fetal development in which the consequences of inhibited or altered neurodevelopment can be permanent, and there is often a delay between chemical exposure during development and an adverse neurologic outcome [12,34,41,43,44,45]. Furthermore, even minor changes in brain development can result in a dramatic loss of cognitive function [34]. Consequently, developmental neurotoxicity (DNT) is distinct from adult neurotoxicity [12]. DNT can manifest in many other forms, including differences in cell type proportions, altered cell migration and organization, functional changes, or a lack of network connectivity [39]. In addition, DNT often occurs at doses lower than those, which cause adult neurotoxicity and transient insults that result in reversible effects in adults (and can cause irreversible damage during neurodevelopment) [12].
2. Limited Current Knowledge of Chemicals That Disrupt Neurodevelopment
DNT is among the least studied forms of toxicology [46,47], and most substances in commercial use today, even of those classified as high-production-volume chemicals, have not been screened for DNT [15,48,49,50]. Traditionally, DNT has only been studied following a poisoning incident or after neurotoxicity was observed in adults [15], and regulatory action often lags far behind the first recognition of DNT [24]. Two classic examples of developmental neurotoxicants are lead and methylmercury, and it took about 25 years for lead to be recognized as a developmental neurotoxicant and removed from gasoline [51]. About 50 years after the first report of methylmercury-induced neurodevelopmental effects, it was finally considered in risk assessment [52].
A scope review conducted by Grandjean and Landrigan (2006) described the extent of the current knowledge of chemicals that disrupt neurodevelopment as “the tip of a very large iceberg” [15]. This review identified only five compounds that had been convincingly demonstrated to cause DNT in humans [15]. The five DNT compounds included lead, methylmercury, polychlorinated biphenyls (PCBs), arsenic, and toluene [15]. This list did not include ethanol, as maternal exposure to ethanol was considered voluntary [15]. The list of DNT compounds was later expanded to include tetrachloroethylene, manganese, chlorpyrifos, brominated diphenyl ethers (BDEs), dichlorodiphenyltrichloroethane (DDT), fluoride, and dichlorodiphenyldichloroethylene (DDE) [11], and recent clinical studies have suggested the addition of valproic acid to the list of known DNT compounds [44,46,53]. Thus, only 14 chemicals are known to be toxic to human neurodevelopment [11,15,44,46,53]. This number is likely not representative of the actual number of compounds that cause DNT, emphasizing the need for improved methods of DNT evaluation to expand current knowledge and inform regulatory decision making [15]. In addition, as few chemicals are classified as being developmentally neurotoxic, it is also difficult to establish reference compounds to evaluate the specific DNT assays. However, suggestions of criteria to select reference compounds for developmental neurotoxicity (DNT) have been published recently [44]. The authors of this review convened in a workshop to define criteria for the selection of positive and negative reference compounds [44].
2.1. Constraints of Traditional Developmental Neurotoxicity Testing
Regulation focused on protecting neurodevelopment is restricted by current methods in regulatory developmental neurotoxicity testing [11,12,15]. Current DNT guidelines from both the US (USEPA 712-C-98-155 239) and internationally (OECD TG 426) recommend the use of animal testing with rodents to classify developmental neurotoxicants [54,55]. To meet the requirements of both the Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA) and the Toxic Substances Control Act (TSCA), which was recently reauthorized as the Frank Lautenberg Chemical Safety for the 21st Century Act in 2016, the US EPA recommends exposure at three doses with a control and several groups of pregnant rats exposed during gestation and early lactation. At least 20 litters for each dose level are recommended, along with the evaluation of maternal toxicity and DNT evaluation based on a series of tests to examine motor activity, auditory startle, learning and memory, brain weight, and neuropathology in offspring through adulthood [54]. Similarly, the OECD advises the use of rats, 20 litters of per dose level, and the observation of gross neurologic and behavioral abnormalities with evaluations of physical development, behavioral ontogeny, motor activity, motor and sensory function, learning and memory, brain weight, and neuropathology during postnatal development and adulthood [55]. These testing methods are time-consuming, resource-intensive, and expensive [12]. It is estimated that a guideline DNT study for one chemical takes about 3 months, costs approximately USD 1.4 million, and requires a minimum of 140 mated female rats to produce 1000 rat pups [12]. It is therefore not feasible to screen the thousands of untested chemicals with potential to cause DNT using in vivo guideline studies [12].
Moreover, there are concerns regarding the physiological relevance of extrapolating the results from animal studies to human health effects [12,56,57]. There is evidence of considerable disagreement between the animal and human response to toxic agents [56]. The repercussions of interspecies differences are most evident in clinical trials for drugs [58], in which about 92% of substances fail—often due to effects in humans that were not observed in preclinical animal studies [59]. The human and rodent brain also differ considerably [60,61]. Despite the evolutionary conservation of genes important to neurodevelopment across species, gene expression during development is not conserved between rodents and humans [62]. Furthermore, studies conducted to ascertain differential developmental neurotoxicity comparing the response of rodent and human neural progenitor cells in vitro have elucidated species-specific effects of compounds on key neurodevelopmental events, such as proliferation, migration, and differentiation [63,64].
2.2. Opportunities for Integrated Developmental Neurotoxicity Testing Strategies
The use of alternative methods in toxicology, either as replacements or supplements to animal testing, will allow more cost-effective and human-relevant chemical screening for DNT [49,65]. Toxicology is currently experiencing a paradigm shift from the reliance on observational animal experimentation to mechanism-based science, which supports this concept [56,66,67,68,69]. The paradigm shift is outlined in the 2007 U.S. National 188 Research Council (NRC) report Toxicity Testing in the 21st Century: A Vision and a Strategy, in which the NRC Committee on Toxicity Testing and Assessment of Environmental Agents recommended that toxicity testing should focus on the mechanistic evaluation of toxicity pathways through the combined use of in vitro and in silico technologies, with less reliance on conventional in vivo animal experiments [70]. This transition to the use of alternatives to animal testing is also supported by recent regulatory efforts, such as the EU Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) of June 2007, which recommended the replacement, reduction, and refinement of animal testing, as well as the Frank R. Lautenberg Chemical Safety for the 21st Century Act, which amended the Toxic Substances Control Act (TSCA) in June of 2016 in the US and recommended methods that reduce or replace the use of vertebrate animals1. Thus, the development of novel alternative methods is considered the “future of toxicology” [69]. An important consideration, however, is that a single assay does not provide sufficient information to replace an animal model or facilitate regulatory decision making, and, as a result, current efforts are focused on the development of integrated testing strategies that systematically combine information from multiple sources and test systems to quickly and reliably identify toxicity [71].
It is widely acknowledged that DNT is a consequence of disruption to the fundamental processes that define neurodevelopment at the cellular level [12,46,72,73]. A comprehensive in vitro developmental neurotoxicity testing strategy should, therefore, consider each of these processes, which include proliferation, migration, differentiation, synaptogenesis, myelination, apoptosis, and the formation of functional neuronal networks [28,39,40,41,42,65]. The ultimate goals of these new methods are their usage for regulatory purposes [74]. OECD/EFSA have recently started a program with the consensus of the DNT community after several workshops [75] and meetings to develop a testing battery for DNT, in which the aim is to present enough data and outline what could become an integrated approach to testing and assessment (IATA) for the purposes of screening and prioritization or hazard assessment [75]. Moreover, with regard to the federal insecticide, fungicide, and rodenticide act (FIFRA), the US EPA recently had a public virtual peer review meeting for the use of new approach methodologies (NAMs) to evaluate DNT for human risk assessment. Among the NAMs discussed were two assays performed by the EPA and the whole testing battery evaluated by EFSA [76] (see above). The overall response by the scientific advisory panel to NAMs to DNT was positive. However, one of the main criticisms was the lack of assays in the battery addressing glial toxicity, including myelination.
3. Myelination as a Developmental Neurotoxicity Endpoint
3.1. Oligodendrocyte Differentiation, Maturation, and Myelination
Myelination is a particularly important endpoint to consider in a DNT testing strategy due to the significant involvement of the process in the maturation and function of the human brain [77,78,79]. Myelination begins around the fifth month of fetal development but is prolonged through adolescence [80,81,82]. A peak in myelination rate occurs at 2–3 years of age [83], but ongoing myelination and increased white matter volume continue in children aged 12–18 years [84,85]. Around age 20 and throughout life, gray matter volume starts to decline [86,87]. The formation of myelin begins with the specification of oligodendrocyte precursor cells (OPCs) from neural progenitor cells (NPCs) [79,88,89]. OPCs then migrate to their target axons, differentiate into pre-oligodendrocytes (pre-OLs), then differentiate into pre-myelinating oligodendrocytes (pre-myelinating OLs), and finally differentiate into mature and/or myelinating oligodendrocytes (OLs) [79,88,89]. The differentiation of oligodendrocytes following axogenesis is believed to be mediated by complex trophic signals between the oligodendrocytes and neurons. During differentiation into mature oligodendrocytes, the morphology changes to a large network of branching processes [90].
3.2. Functions of Oligodendrocytes and Myelin
The myelination of axons in the central nervous system (CNS) is crucial for brain function [77] and is one of the key events in brain development. Myelin is formed from the processes of oligodendrocytes that provide electrical insulation and accelerate transmissions along the axon [91]. In the CNS, one oligodendrocyte can produce about 100 myelin sheaths for several different neurons [92]. The sheaths are formed by multiple alternating protein–lipid layers with a characteristic composition different from other cellular membranes [93]. It consists of 70–85% lipids (mainly phospholipids, glycolipids, and cholesterol) and 20–30% proteins, such as myelin basic protein (MBP), proteolipid protein (PLP), 2′3′-cyclic-nucleotide 3′-phospodiesterase (CNP), myelin-associated glycoprotein (MAG), and myelin oligodendrocyte glycoprotein (MOG) [93,94,95,96]. The myelin sheaths along the axons are separated by gaps exposed to the extracellular space, referred to as nodes of Ranvier, which are highly enriched in ion channels and mediate the exchange of ions during the action potential [77,79,97,98]. Propagation of the action potential between the nodes in myelinated axons (saltatory conduction) is much faster than in unmyelinated axons [95]. The increase in action potential speed reduces the need for the ATP-dependent sodium–potassium exchange required for the maintenance of the resting membrane potential along axons [89]. However, oligodendrocytes have other important roles during development and in the mature brain, such as the regulation of neurotransmitter release and metabolism [99,100], synaptic plasticity [101,102,103], axonal conductance and synaptic efficacy [99,100], neuronal signaling networks [104,105,106], neuronal excitability [107], and axonal growth, metabolism, integrity, and survival [97,108,109,110,111,112,113]. Therefore, the failure to form or maintain myelin can disrupt neuronal signal transmission or trigger the degradation of axons, and a reduction in the velocity of action potentials can lead to physical or mental disability and induce severe neurological symptoms [79]. Myelin development is also associated with improved language, memory [114], motor-skill learning [115,116], and reading performance in childhood [117]. Animal studies have implicated white matter deficits during critical periods of development in learning and memory impairments [118,119] and in an inability to learn motor skills [116]. Moreover, there is evidence for oligodendrocyte and myelin dysfunction in 258 neurodevelopmental disorders with cognitive symptoms (e.g., Williams syndrome, Pitt-Hopkins syndrome, and autism) [120,121].
4. Potential Mechanisms of Toxicity to Oligodendrocytes and Myelin
As oligodendrocytes are operating near their metabolic capacity, they are especially sensitive to chemical disturbances and alternations in oligodendrocyte proliferation [122]. Chemicals proposed to have an effect on the myelination process include ethanol [123], tellurium [124,125,126], Bisphenol A (BPA) [127,128,129], TDCPP [130], Cuprizone [131], sodium metavanadate [132], methylmercury [133], and lead [134,135,136,137,138]. However, there is limited information on the mechanism involved, and only few chemicals have been identified to include DNT. Possible mechanisms of toxicity to oligodendrocyte differentiation include the inflammatory cytokine release by microglia [139], thyroid hormone disruption [65], glutamate excitotoxicity [140,141,142], as well as the disruption of cholinergic signaling (e.g., acetylcholinesterase inhibition) [141,143] (Figure 1). Many of these mechanisms involve toxicity pathways that ultimately result in cell death by oxidative stress [139,144].
4.1. Inflammatory Cytokine Release
Following systemic infection, inflammation, or neural damage, microglia in the brain can become activated and release inflammatory cytokines [139], such as interleukins (IL-1 and IL-6), tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), and transforming growth factor beta (TGFβ). These cytokines have been shown to induce white matter lesions in the CNS [145]. Maternal immune activation has also been shown to alter myelination during neurodevelopment [146,147], and the proinflammatory cytokines IL-6, IL-1, and TNF-α are thought to play roles in prenatal white matter damage that occurs following maternal intrauterine infection [148]. In addition, the prenatal exposure to lipopolysaccharide (LPS), a bacterial endotoxin that elicits an immune response [28], has been shown to alter myelination in rats [149].
4.2. Thyroid Hormone Disruption
Alterations of thyroid hormone homeostasis during this time span might affect the myelination process and cause persistent adverse outcomes [150]. There is strong evidence demonstrating that thyroid hormone (TH) signaling is integral to myelin formation because TH is required for the regulation of OL differentiation and maturation [151,152,153,154,155,156,157]. Moreover, there is evidence that environmental chemicals that disrupt thyroid hormone function also disrupt myelin formation [158,159,160,161]. For example, the polybrominated diphenyl ether (PBDE) flame retardant BDE-99 was recently demonstrated to inhibit NPC differentiation into the OL lineage in vitro [162], and PBDE flame retardants are known thyroid hormone disruptors [163]. In addition, the leading commercial pentaBDE technical mixture, DE-71, which contains BDE-99 [164], has also been shown to downregulate the gene expression of myelin basic protein in exposed zebrafish embryos [158,160].
4.3. Glutamate Excitotoxicity
Glutamate excitotoxicity from the sustained activation of OL glutamate receptors can lead to oxidative stress in oligodendrocytes through an influx of calcium and subsequent production of reactive oxygen species (ROS) [140,142,165]. Because the calcium influx occurs through calcium-permeable kainate and α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors, which are expressed at higher levels in developing OLs [140,166], they are more vulnerable to excitotoxicity than mature OLs are [140,142]. Excitotoxicity can also be mediated by other cell types in the brain [139,167]. For example, astrocytes regulate glutamate homeostasis through glutamate uptake [167] and activated microglia can generate ROS and reactive nitrogen species (RNS), which reduce the expression of glutamate transporters and prevent glutamate uptake. OLs are more susceptible to ROS (see below), which can contribute to increased OL susceptibility to glutamate excitotoxicity [139,167].
4.4. Disruption of Cholinergic Signaling
Oligodendrocytes express muscarinic and nicotinic acetylcholine (ACh) receptors [103,168,169,170,171,172], and acetylcholine signaling is thought to play a role in the production and maintenance of myelin [103]. An environmental compound that acts as an agonist for the nicotinic ACh receptors is nicotine. Exposure to nicotine has been shown to affect oligodendrocyte maturation and myelin formation [103], and maternal nicotine exposure has been demonstrated to alter myelin-related gene expression in adolescent rats [173] and zebrafish offspring [174].
Inhibitors of acetylcholinesterase (AChE), the enzyme that hydrolyzes acetylcholine to acetate and choline, have been shown to inhibit myelin formation as well [103]. Environmental AChE inhibitors include organophosphates or carbamates nerve agents and insecticides, and any axoglial communication required for myelin formation that relies on cholinergic signaling could potentially be affected by AChE inhibitors [103]. Chlorpyrifos, an organophosphate insecticide known to inhibit AChE, is thought to be associated with autism risk [175] and has shown DNT effects on oligodendrocyte differentiation and myelin formation at concentrations below those that cause systemic toxicity due to AChE inhibition [176,177]. The US EPA has updated its Human Health Risk Assessment in 2016 to reflect the possibility of toxicity at exposures below levels that cause 10% red blood cell AChE inhibition—the level that was used previously to predict DNT [175]. Notably, organophosphate flame retardants (OPFRs), while structurally similar to organophosphate pesticides, do not as effectively inhibit acetylcholinesterase [178,179]. However, OPFRs have still been demonstrated to elicit DNT effects on oligodendrocytes, such as 1,3-dichloro 2-propyl phosphate (TDCPP), which has been studied in zebrafish embryos and is thought to act through thyroid hormone disruption [130]. Furthermore, several OPFRs affected the expression of genes involved in myelination in a 3D rat primary in vitro model [180].
4.5. Oxidative Stress
Oligodendrocytes are known to be sensitive to oxidative stress caused by reactive oxygen and nitrogen species (ROS and RNS, respectively) [90,140,181]. ROS are produced by oxidative phosphorylation, and a main source is the mitochondrial electron transport chain [182,183]. At very low levels, ROS and RNS act as second messenger signaling molecules [184]; however, when levels exceed antioxidant levels, this can result in mitochondrial dysfunction and oxidative stress [182]. The level of sensitivity to oxidative stress depends on the OL differentiation stage [144,185]. Oligodendrocyte precursor cells (OPCs) are particularly susceptible to oxidative stress because they have lower levels of antioxidants, such as glutathione, and higher levels of iron compared to mature OLs [140,185]. The maturation-dependent susceptibility to oxidative stress has been demonstrated by depleting intracellular glutathione, a regulator of intrinsic antioxidant protection, in OLs at various differentiation stages in vitro, demonstrating that mature OLs were less vulnerable to cell death caused by oxidative stress than their precursors were [144].
4.6. Disruption of Ion Channel Signaling
Myelin isolates the axons of neurons to favor a more efficient action potential propagation by saltatory conduction. This process requires a high density of voltage-gated channels in the areas without myelin (called the nodes of Ranvier) [186]. These specialized axonal domains differ dramatically from internodal axonal regions, with high density in voltage-dependent Na1 (Nav) channels, to juxtaparanodal domains, where Kv channels are clustered beneath the myelin sheath [187]. The lack of oligodendroglial signals observed in neuroinflammatory disorders, such as multiple sclerosis (MS) or in experimental animal models of demyelination, has shown the reorganization of voltage-gated ion channels to compensate the saltatory loss. Animal studies have suggested that changes in the subunit expression and redistribution of these channels can follow traumatic nerve injury [188]. In addition, brain disorders associated with neuronal excitability, such as epilepsy, are linked to Nav channel alterations, suggesting the importance of this structure [189].
Ca2+ activity is also very important during myelin sheath formation and growth [190]. In vivo studies have indicated that local Ca2+ signaling regulates distinct stages of myelination [191]. In vitro studies have shown how compounds such as BDE-47 and 6-OH-BDE-47 interfere with Ca2+ signaling during development [192] and can alter oligodendrocytes differentiation [193].
5. Existing NAMs Used to Study Toxicity to Oligodendrocytes and Myelination
There are several existing methods for evaluating oligodendrocyte toxicity in vitro, with pure cultures of OPCs or OLs and corresponding assays for proliferation, migration, and differentiation, and this has been reviewed elsewhere [194]. In addition, new protocols to generate oligodendrocytes from human iPSCs have been developed [195,196]. However, there are challenges to culturing these cells for a longer period of time, and they also lack the maturation diversity and cytoarchitecture that oligodendrocytes present in vivo. The central limitation of pure OPC/OL culture systems is that cell–matrix or cell–cell interactions between oligodendrocytes and neurons or other glial cells cannot be studied [194]. To observe these interactions, methods of co-culturing neurons and oligodendrocytes have been developed [197,198,199]. Still, these systems do not incorporate other types of glial cells and most are cultured under 2D conditions [194]. The majority of in vitro models of oligodendrocytes and myelination that comprise more than neurons and OLs are derived from rodent primary or stem cells [65], including ex vivo rodent brain slice cultures [200,201,202], 3D organotypic cultures [203,204,205,206,207], and microfluidic cell culture systems [208]. However, the study of myelin formation during neurodevelopment warrants human cell-based DNT testing methods due to species differences. Differential gene expression analysis has revealed 244 genes expressed in human OPCs that are not expressed in their mouse counterparts [209]. In addition, the protein composition of myelin is not fully conserved across species [210]. A 3D model derived from primary fetal human neural progenitor cells (hNPCs) has been successfully used to determine the effect of DNT compounds in oligodendrocyte differentiation [193], showing the advantages of 3D cultures for studying oligodendrocyte maturation. Unfortunately, very few in vitro human cell-based organotypic models of myelination exist. This is partly because there are limited protocols available to efficiently myelinate axons in vitro, and sufficient myelination by oligodendrocytes typically requires in vivo transplantation in animal models [211].
Organotypic Human Models to Study Myelin
Only a few human 3D models have been reported to include myelination by oligodendrocytes. Most of these models are generated from iPSCs; human embryonic stem cells, however, have also been used. Sandstrom and collaborators have developed a human embryonic stem cell-derived 3D neural tissue model of neurons, astrocytes, and oligodendrocytes in which immature myelin sheaths are formed around axons after two months in culture [212]. In the case of iPSC-derived organoids, different approaches have been applied to achieve myelination. For example, Madhavan and collaborators used iPSC-derived spheroids (oligocortical spheroids; OCS) to achieve myelination. The OCS generated robust populations of oligodendrocytes across different cell lines, showing expressions of proteolipid protein 1 (PLP1) (oligodendrocyte membrane protein) and MYRF25, a transcription factor specifically expressed in oligodendrocytes in the CNS. After 20 weeks in culture, they were able to observe myelin compaction [213]. Furthermore, Kim and collaborators generated fused forebrain organoids (FFOs) by combining ventral forebrain organoids (VFOs) with dorsal forebrain organoids (DFOs) that were able to promote oligodendroglia maturation in 12 weeks [214]. This model was extensively characterized, with compact myelination produced by the present oligodendrocytes. The oligodendroglia in this study seems to be dorsally derived, and the authors believed that the observed processes recapitulate the third wave of oligodendrogenesis [214]. Furthermore, Pamies and collaborators were able to generate a large number of myelinated axons (between 40 and 50% of total axons) using a simpler and faster protocol. In only 8 weeks, these iPSC-derived organotypic models (BrainSpheres) were differentiated into approximately 350 μm spheroids consisting of different cell types that facilitated cell–cell interactions and neuron–glial function (such as myelination) [215]. An increase in myelination as the system matured was confirmed with confocal imaging and electron microscopy that showed multiple layers of myelin wrapping the axonal structures [215]. Similarly, Pasca and collaborators used their human iPSC-derived model (human oligodendrocyte spheroids; hOLS) to generate oligodendrocytes. They claimed that in 37 days, they could generate transcriptionally similar stages to human primary oligodendrocytes [216]. Another approach has been to generate differentiated oligodendrocytes in a monolayer (70% of O4+ oligodendrocytes) within 28 days and then introduce them to 3D nanofiber scaffolds with neurons [196]. In addition, the quantification of this endpoint is crucial for DNT and NT assessment. To this point, only two models have shown the ability to quantify myelin [217,218]. Furthermore, Chesnut and collaborators were able to determine four reference compounds for chemical-induced myelin disruption, and they evaluated them with several myelin quantification endpoints [218].
The advantages of these models are that iPSCs can be used to explore sensitivities associated with different genetic backgrounds [14,34], and their differentiation into neural cell lineages is comparable to brain development in utero [14,219]. However, it should be acknowledged that more complex models bring some limitations and disadvantages compared to in vivo studies and simpler in vitro systems. Some of these limitations and disadvantages have been summarized elsewhere [220,221] and include, e.g., lower reproducibility, in many cases, the lack of immune cells and hormone systems, and the lack of vasculature and organ–organ interactions. Nevertheless, they have the potential to surpass animal models as they more closely mimic human physiology.
6. Endpoints to Study Toxicity of Oligodendrocytes and Myelination
Cytotoxicity assays (such as resazurin, LDH, and MTT) are used extensively in toxicology to evaluate cytotoxicity. However, they cannot be utilized to determine specific toxicity, such as neuro or oligodendrocyte toxicity, in heterogeneous cell models. Toxicity to different cell phenotypes can instead give an idea of the compound’s specificity and provide information about the molecular mechanism of action. For this reason, methods that discriminate from general toxicity effects to a more functional or structural method are preferable, especially in developmental neurotoxicity, where effects are generally below cytotoxic levels (Figure 1).
Several markers associated with oligodendrocytes and myelin can be studied by different techniques to assess glia toxicity. Commonly used oligodendroglia lineage developmental markers have been summarized by Deng and Poretz in a previously published review [90]. These markers cover immature to mature oligodendroglia stages and include maturation and differentiation. These markers can be used to quantify oligodendrocytes and myelin with techniques such as image analysis combined with immunohistochemistry and flow cytometry. In image analysis, pictures can be obtained manually [99] or by using new high-throughput imaging instruments (such as Aperio T2 ScanScope, MicroMatrices microTMA, or PerkinElmer Opera Phoenix) [222,223]. Many of these high-throughput imaging technologies have been summarized elsewhere [224]. Oligodendrocytes can then be counted manually [225] or by image analysis tools (such as simple total fluorescent quantification), more complex methods (such as semi-automated computer platforms) [208], or very sophisticated single-cell resolution quantification [226]. Flow cytometry has also been applied to quantify oligodendrocytes [196]; this technique, however, may be difficult for 3D multicellular models, as disaggregation to single cells can damage the cells.
The same markers can also be studied by gene expression analysis [180]. For example, the gene expressions of different myelin and oligodendrocyte makers were assessed in BrainSpheres after the exposure to different demyelinating compounds [218]. However, gene expression changes could be temporary and do not necessarily lead to adverse outcomes. For this reason, it is important to combine gene expression with more functional assays, such as protein analysis or morphological evaluations.
Other, more specific endpoints can be measured to understand the molecular mechanism of certain compounds (e.g., specific growth factors and cytokines that regulate oligodendrocytes, neurotransmitters, glutamate excitotoxicity, and oxidative stress). However, these endpoints can be challenging and labor-intensive to study in multicellular models, either in vivo or in vitro, where cell–cell and organ–organ interactions can contribute or protect the cells from the compound’s target mechanism. The use of 3D models in which the cellular density is higher than in traditional monolayer cultures complicates single-cell evaluation even more. To determine cell-specific effects, the use of a homogenous cell population would be advantageous. However, this would reduce the complexity and physiological relevance, as the cell–cell interactions are of high importance for the function of an organ, especially the brain. New technologies are emerging with higher resolution (single-cell resolution), allowing the combining of more complex models (in vivo or 3D organotypic cultures) with very specific cellular measurements. For example, the use of single-cell sequencing can distinguish between genetic changes in the oligodendrocyte population versus changes in other CNS cell types [227,228]. Many other methods, such as imaging (single-cell-resolution microscopy) [229] and single-cell metabolomics [230], can allow for single-cell resolution as well.
7. Conclusions
The evaluation of oligodendrocytes and myelin is of great importance, both in developmental neurotoxicity and in studies of demyelinating disorders. Still, there are very few suitable, human-relevant in vitro and in vivo models. It is crucial to further develop and improve upon existing models and assays to study this important cell type. This will allow the improvement of current testing strategies and permit the development of treatments for myelin-related diseases. The development of new models that better mimic human brain physiology has been a priority in recent years, giving rise to next-generation 3D human cell cultures that simulate myelination in vitro. New technologies in stem cells and cell culturing, such as human 3D organotypic models (where it is possible to obtain myelinated axons) [212,213,214,215], are bringing us closer to the in vivo situation of the human brain and have the potential to replace or reduce the use of in vivo and in vitro rodent models. However, these cell cultures also have limitations and associated challenges [220,221,222,223,224,225,226,227,228,229,230,231,232]. It is therefore important to ensure that the application of these models is fit-for-purpose. Drug development for demyelination diseases could clearly be enhanced by these models, to help study the effects of protective compounds, but also the possible mechanisms and interactions with other cell types. In addition, advances in instrumentation, such as microscopy and omics technologies for single-cell resolution and high-throughput screening adaptation, will enable the study of valuable endpoints such as myelin formation.
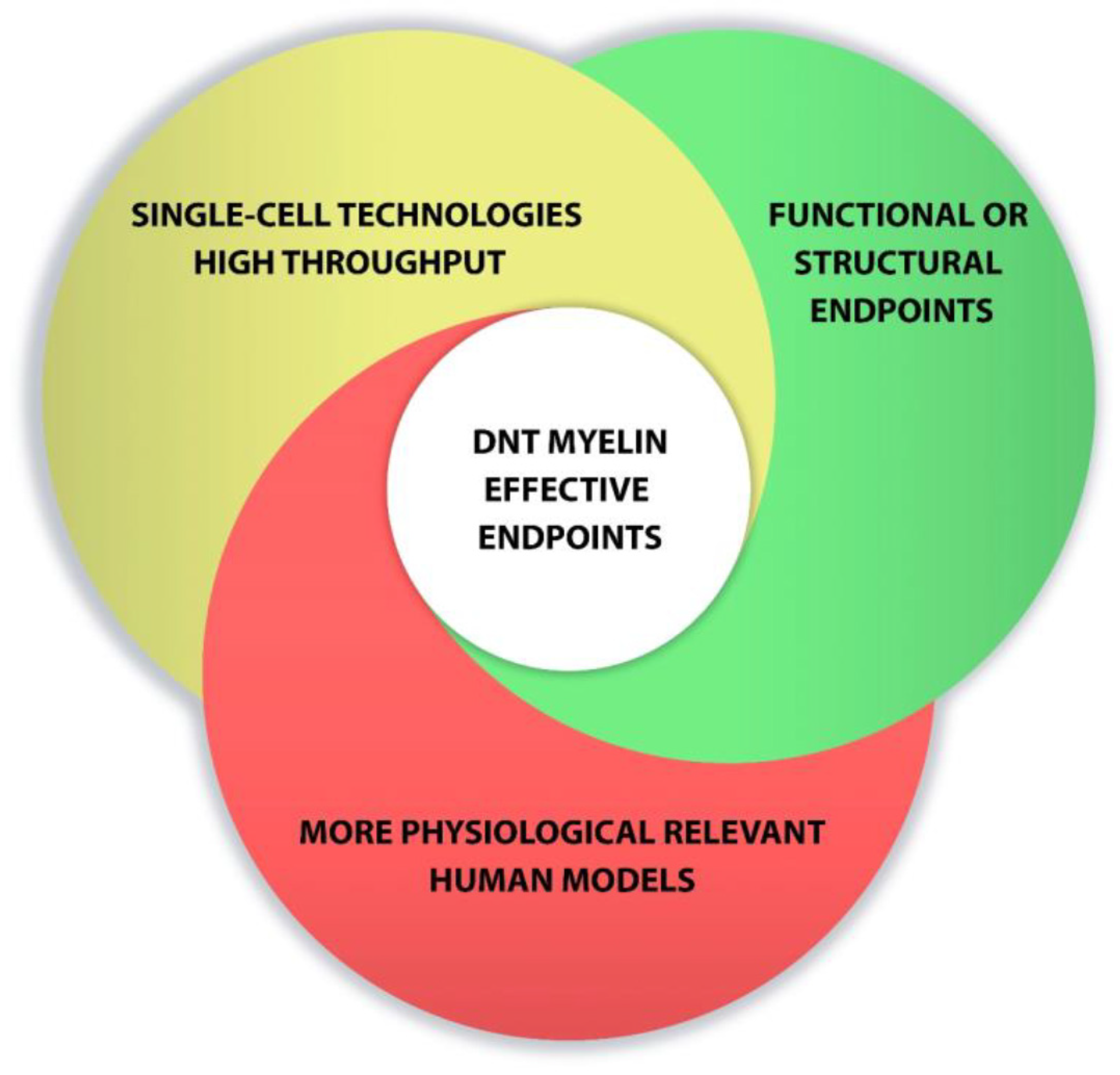
Overall, new advances in cell culture and the development of new methods to detect 469 developmental neurotoxicity disorders are enhancing progress toward relevant tests to predict human toxicity. Combination of single-cell technologies, high-throughput screening, functional endpoints and the use of more physiological relevant models could favor the development of DNT myelin effective endpoints (Figure 2)
Author Contributions
All the authors contributed to the manuscript equally. All authors have read and agreed to the published version of the manuscript.
Funding
This research was in part funded by a Colgate-Palmolive Grant for Alternative Research (PI: HH); David Pamies is financially supported by the Swiss Centre for Applied Human Toxicology (SCAHT).
Acknowledgments
We are grateful to Michael Hughes for editorial work.
Conflicts of Interest
T.H., H.H. and D.P. are named inventors on a patent by Johns Hopkins University on the production of BrainSpheres mentioned in this review, which is licensed to AxoSim, New Orleans, LA, USA. They consult AxoSim.
References
- Boyle, C.A.; Boulet, S.; Schieve, L.A.; Cohen, R.A.; Blumberg, S.J.; Yeargin-Allsopp, M.; Visser, S.; Kogan, M.D. Trends in the Prevalence of Developmental Disabilities in US Children, 1997–2008. Pediatrics 2011, 127, 1034–1042. [Google Scholar] [CrossRef] [Green Version]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2014. [Google Scholar]
- ADDM. Prevalence of autism spectrum disorders--autism and developmental disabilities monitoring network, 14 sites, united states. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2012, 61, 1–19. [Google Scholar]
- Atladottir, H.O.; Parner, E.T.; Schendel, D.; Dalsgaard, S.; Thomsen, P.H.; Thorsen, P. Time trends in reported diagnoses of childhood neuropsychiatric disorders—A Danish cohort study. Arch. Pediat. Adol. Med. 2007, 161, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, J.G.; Fritz, M.S.; Ness, K.K.; Sievers, P.; Newschaffer, C.J.; Shapiro, E.G. Analysis of Prevalence Trends of Autism Spectrum Disorder in Minnesota. Arch. Pediatr. Adolesc. Med. 2003, 157, 622–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robison, L.M.; Skaer, T.L.; Sclar, D.A.; Galin, R.S. Is attention deficit hyperactivity disorder increasing among girls in the US? Trends in diagnosis and the prescribing of stimulants. CNS Drugs 2002, 16, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Schechter, R.; Grether, J.K. Continuing increases in autism reported to California’s developmental services system. Arch. Gen. Psychiat. 2008, 65, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pet-tygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States. Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef]
- Danielson, M.; Bitsko, R.H.; Ghandour, R.M.; Holbrook, J.R.; Kogan, M.D.; Blumberg, S.J. Prevalence of Parent-Reported ADHD Diagnosis and Associated Treatment Among U.S. Children and Adolescents, 2016. J. Clin. Child. Adolesc. Psychol. 2018, 47, 199–212. [Google Scholar] [CrossRef]
- Hansen, S.N.; Schendel, D.E.; Parner, E.T. Explaining the increase in the prevalence of autism spectrum disorders: The proportion attributable to changes in reporting practices. JAMA Pediatr. 2015, 169, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Grandjean, P.; Landrigan, P. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014, 13, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, L.; Hogberg, H.T.; Leist, M.; Hartung, T. Developmental neurotoxicity—Challenges in the 21st Century and In Vitro Opportunities. ALTEX 2014, 31, 129–156. [Google Scholar] [CrossRef] [Green Version]
- Visser, S.N.; Danielson, M.; Bitsko, R.H.; Holbrook, J.R.; Kogan, M.D.; Ghandour, R.M.; Perou, R.; Blumberg, S.J. Trends in the Parent-Report of Health Care Provider-Diagnosed and Medicated Attention-Deficit/Hyperactivity Disorder: United States, 2003–2011. J. Am. Acad. Child. Adolesc. Psychiatry 2014, 53, 34–46.e2. [Google Scholar] [CrossRef] [Green Version]
- Ardhanareeswaran, K.; Mariani, J.; Coppola, G.; Abyzov, A.; Vaccarino, K.A.J.M.G.C.F.M. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat. Rev. Neurol. 2017, 13, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Landrigan, P. Developmental neurotoxicity of industrial chemicals. Lancet 2006, 368, 2167–2178. [Google Scholar] [CrossRef]
- Heyer, D.B.; Meredith, R.M. Environmental toxicology: Sensitive periods of development and neurodevelopmental disorders. NeuroToxicology 2017, 58, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Schettler, T. Toxic threats to neurologic development of children. Environ. Health Persp. 2001, 109, 813–816. [Google Scholar]
- Hur, Y.-M. Increasing Phenotypic and Genetic variations in Hyperactivity/Inattention Problems from Age 3 to 13 Years: A Cross-Sectional Twin Study. Twin Res. Hum. Genet. 2014, 17, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingstone, L.T.; Coventry, W.L.; Corley, R.P.; Willcutt, E.G.; Samuelsson, S.; Olson, R.K.; Byrne, B. Does the Environment Have an Enduring Effect on ADHD? A Longitudinal Study of Monozygotic Twin Differences in Children. J. Abnorm. Child. Psychol. 2016, 44, 1487–1501. [Google Scholar] [CrossRef] [Green Version]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.L.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Academies Press. Scientific Frontiers in Developmental Toxicology and Risk Assessment; National Academies Press (US): Washington, DC, USA, 2000.
- Bondy, S.C.; Campbell, A. Developmental neurotoxicology. J. Neurosci. Res. 2005, 81, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Lanphear, B.P.; Hornung, R.; Khoury, J.; Yolton, K.; Baghurstl, P.; Bellinger, D.C.; Canfield, R.L.; Dietrich, K.N.; Bornschein, R.; Greene, T.; et al. Low-level envi-ronmental lead exposure and children’s intellectual function: An international pooled analysis. Environ. Health Persp. 2005, 113, 894–899. [Google Scholar] [CrossRef]
- Landrigan, P.J.; Goldman, L.R. Children’s Vulnerability to Toxic Chemicals: A Challenge and Opportunity to Strengthen Health and Environmental Policy. Health Aff. 2011, 30, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrigan, P.J. Children as a vulnerable population. Hum. Ecol. Risk. Assess. 2005, 11, 235–247. [Google Scholar] [CrossRef]
- USEPA. America’s Children and The Environment (ace), 3rd ed.; National Service Center for Environmental Publications (NSCEP): Cincinnati, OH, USA, 2013. [Google Scholar]
- Ferguson, A.; Penney, R.; Solo-Gabriele, H. A Review of the Field on Children’s Exposure to Environmental Contaminants: A Risk Assessment Approach. Int. J. Environ. Res. Public Health 2017, 14, 265. [Google Scholar] [CrossRef]
- Klaassen, C.D. Casarett and Doull’s Toxicology: The Basic Science of Poisons; McGraw-Hill: New York, NY, USA, 2008. [Google Scholar]
- Landrigan, P.J.; Sonawane, B.; Mattison, D.; McCally, M.; Garg, A. Chemical contaminants in breast milk and their impacts on children’s health: An overview. Environ. Health Persp. 2002, 110, A313–A315. [Google Scholar] [CrossRef] [Green Version]
- Al-Enazy, S.; Ali, S.; Albekairi, N.; El-Tawil, M.; Rytting, E. Placental control of drug delivery. Adv. Drug Deliv. Rev. 2017, 116, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Staud, F.; Červený, L.; Ceckova, M. Pharmacotherapy in pregnancy; effect of ABC and SLC transporters on drug transport across the placenta and fetal drug exposure. J. Drug Target. 2012, 20, 736–763. [Google Scholar] [CrossRef] [PubMed]
- Syme, M.R.; Paxton, J.W.; Keelan, J.A. Drug Transfer and Metabolism by the Human Placenta. Clin. Pharmacokinet. 2004, 43, 487–514. [Google Scholar] [CrossRef]
- Alépée, N.; Bahinski, A.; Daneshian, M.; De Wever, B.; Fritsche, E.; Goldberg, A.; Hansmann, J.; Hartung, T.; Haycock, J.; Hogberg, H.T.; et al. State-of-the-art of 3D cultures (organs-on-a-chip) in safety testing and pathophysiology. ALTEX 2014, 31, 441–477. [Google Scholar] [CrossRef]
- Schmidt, B.Z.; Lehmann, M.; Gutbier, S.; Nembo, E.; Noel, S.; Smirnova, L.; Forsby, A.; Hescheler, J.; Avci, H.X.; Hartung, T.; et al. In vitro acute and developmental neurotoxicity screening: An overview of cellular platforms and high-throughput technical possibilities. Arch. Toxicol. 2017, 91, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Cecchelli, R.; Berezowski, V.; Lundquist, S.; Culot, M.; Renftel, M.; Dehouck, M.-P.; Fenart, L. Modelling of the blood–brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 2007, 6, 650–661. [Google Scholar] [CrossRef]
- Hawkins, B.; Davis, T. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryo-genesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B. Development of the blood-brain barrier. Cell Tissue Res. 2003, 314, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L. Trajectories of brain development: Point of vulnerability or window of opportunity? Neurosci. Biobehav. Rev. 2003, 27, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Bayer, S.A.; Altman, J.; Russo, R.J.; Zhang, X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. NeuroToxicology 1993, 14, 83–144. [Google Scholar]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108, 511–533. [Google Scholar] [PubMed]
- Stiles, J. The Fundamentals of Brain Development. Integrating Nature and Nurture; Harvard University Press: Cambridge, MA, USA, 2008. [Google Scholar]
- Meredith, R.M. Sensitive and critical periods during neurotypical and aberrant neurodevelopment: A framework for neu-rodevelopmental disorders. Neurosci. Biobehav. Rev. 2015, 50, 180–188. [Google Scholar] [CrossRef]
- Aschner, M.; Ceccatelli, S.; Daneshian, M.; Fritsche, E.; Hasiwa, N.; Hartung, T.; Hogberg, H.T.; Leist, M.; Li, A.; Mundi, W.R.; et al. Reference compounds for alternative test methods to indicate developmental neurotoxicity (DNT) potential of chemicals: Example lists and criteria for their selection and use. ALTEX 2016, 34, 49–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miodovnik, A. Environmental Neurotoxicants and Developing Brain. Mt. Sinai. J. Med. J. Transl. Pers. Med. 2011, 78, 58–77. [Google Scholar] [CrossRef]
- Kadereit, S.; Zimmer, B.; van Thriel, C.; Hengstler, J.G.; Leist, M. Compound selection for in vitro modeling of developmental neurotoxicity. Front. Biosci. 2012, 17, 2442–2460. [Google Scholar] [CrossRef] [Green Version]
- Makris, S.L.; Raffaele, K.; Allen, S.; Bowers, W.J.; Hass, U.; Alleva, E.; Calamandrei, G.; Sheets, L.; Amcoff, P.; Delrue, N.; et al. A Retrospective Performance Assessment of the Developmental Neurotoxicity Study in Support of OECD Test Guideline. Environ. Health Perspect. 2009, 117, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bal-Price, A.; Crofton, K.; Leist, M.; Allen, S.; Arand, M.; Buetler, T.; Delrue, N.; Fitzgerald, R.E.; Hartung, T.; Heinonen, T.; et al. International STakeholder NETwork (ISTNET): Creating a developmental neurotoxicity (DNT) testing road map for regulatory purposes. Arch. Toxicol. 2015, 89, 269–287. [Google Scholar] [CrossRef] [Green Version]
- Crofton, K.M.; Mundy, W.R.; Shafer, T. Developmental neurotoxicity testing: A path forward. Congenit. Anomalies 2012, 52, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hogberg, H.T.; Bressler, J.; Christian, K.M.; Harris, G.; Makri, G.; O’Driscoll, C.; Pamies, D.; Smirnova, L.; Wen, Z.; Hartung, T. Toward a 3D model of human brain development for studying gene/environment interactions. Stem Cell Res. Ther. 2013, 4, S4. [Google Scholar] [CrossRef] [Green Version]
- Needleman, H.L. The Removal of Lead from Gasoline: Historical and Personal Reflections. Environ. Res. 2000, 84, 20–35. [Google Scholar] [CrossRef] [Green Version]
- Grandjean, P.; Satoh, H.; Murata, K.; Eto, K. Adverse effects of methylmercury: Environmental health research implications. Environ. Health Perspect. 2010, 118, 1137–1145. [Google Scholar] [CrossRef] [Green Version]
- Balmer, N.V.; Weng, M.K.; Zimmer, B.; Ivanova, V.N.; Chambers, S.M.; Nikolaeva, E.; Jagtap, S.; Sachinidis, A.; Hescheler, J.; Waldmann, T.; et al. Epigenetic changes and disturbed neural development in a human embryonic stem cell-based model relating to the fetal valproate syndrome. Hum. Mol. Genet. 2012, 21, 4104–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- USEPA. Health Effects Test Guidelines Oppts 870.6300 Developmental Neurotoxicity Study; National Service Center for Environ-mental Publications (NSCEP): Cincinnati, OH, USA, 1998. [Google Scholar]
- OECD iLibrary. OECD Guidelines for the Testing of Chemicals, Section 5; OECD iLibrary: Paris, Frence, 2007. [Google Scholar]
- Hartung, T.; Leist, M. Food for thought … on the evolution of toxicology and the phasing out of animal testing. ALTEX 2008, 25, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Hartung, T. Food for thought … on animal tests. ALTEX 2008, 25, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Leist, M.; Hartung, T. Inflammatory findings on species extrapolations: Humans are definitely no 70-kg mice. Arch. Toxicol. 2013, 87, 563–567. [Google Scholar] [CrossRef] [Green Version]
- USFDA. Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products; National Service Center for Environmental Publications (NSCEP): Cincinnati, OH, USA, 2004. [Google Scholar]
- Florio, M.; Huttner, W.B. Neural progenitors, neurogenesis and the evolution of the neocortex. Development 2014, 141, 2182–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herculano-Houzel, S. Not All Brains Are Made the Same: New Views on Brain Scaling in Evolution. Brain, Behav. Evol. 2011, 78, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Fougerousse, F.; Bullen, P.; Herasse, M.; Lindsay, S.; Richard, I.; Wilson, D.; Suel, L.; Durand, M.; Robson, S.; Abitbol, M.; et al. Human-mouse differences in the embryonic expression patterns of developmental control genes and disease genes. Hum. Mol. Genet. 2000, 9, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Baumann, J.; Barenys, M.; Gassmann, K.; Fritsche, E. Comparative Human and Rat “Neurosphere Assay” for Developmental Neurotoxicity Testing. Curr. Protoc. Toxicol. 2014, 59, 12.21.1–12.21.24. [Google Scholar] [CrossRef]
- Gassmann, K.; Abel, J.; Bothe, H.; Haarmann-Stemmann, T.; Merk, H.F.; Quasthoff, K.N.; Rockel, T.D.; Schreiber, T.; Fritsche, E. Species-Specific Differential AhR Expression Protects Human Neural Progenitor Cells against Developmental Neurotoxicity of PAHs. Environ. Heal. Perspect. 2010, 118, 1571–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, E.A.; Baumann, J.H.; Geerts, L.; Håkansson, H.; Masjosthusmann, S.; Witters, H. Literature review on in vitro and alternative Developmental Neurotoxicity (DNT) testing methods. EFSA Supporting Publ. 2015, 12, 778. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Zhang, Q.; Carmichael, P.L.; Boekelheide, K.; Andersen, M.E. Toxicity Testing in the 21(st) Century: De-fining New Risk Assessment Approaches Based on Perturbation of Intracellular Toxicity Pathways. PLoS ONE 2011, 6, e20887. [Google Scholar] [CrossRef] [PubMed]
- Hartung, T.; McBride, M. Food for Thought … on mapping the human toxome. ALTEX 2011, 28, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Leist, M.; Hasiwa, N.; Rovida, C.; Daneshian, M.; Basketter, D.; Kimber, I.; Clewell, H.; Gocht, T.; Goldberg, A.; Busquet, F.; et al. Consensus Report on the Future of Animal-Free Systemic Toxicity Testing. Altex-Altern. Anim. Ex. 2014, 31, 341–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leist, M.; Hartung, T.; Nicotera, P. The dawning of a new age of toxicology. ALTEX 2008, 25, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NRC. Toxicity Testing in the 21st Century: A Vision and A Strategy; National Academies Press: Washington, DC, USA, 2007. [Google Scholar]
- Rovida, C.; Alépée, N.; Api, A.M.; Basketter, D.A.; Bois, F.Y.; Caloni, F.; Corsini, E.; Daneshian, M.; Eskes, C.; Ezendam, J.; et al. Integrated testing strategies (ITS) for safety assessment. ALTEX 2015, 32, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bal-Price, A.K.; Coecke, S.; Costa, L.; Crofton, K.M.; Fritsche, E.; Goldberg, A.; Grandjean, P.; Lein, P.J.; Li, A.; Lucchini, R.; et al. Advancing the Science of Developmental Neurotoxicity (DNT): Testing for Better Safety Evaluation. Altex-Altern. Anim. Ex. 2012, 29, 202–215. [Google Scholar]
- Kuegler, P.B.; Zimmer, B.; Waldmann, T.; Baudis, B.; Ilmjärv, S.; Hescheler, J.; Gaughwin, P.; Brundin, P.; Mundy, W.; Bal-Price, A.K.; et al. Markers of murine embryonic and neural stem cells, neurons and astrocytes: Reference points for developmental neurotoxicity testing. ALTEX 2010, 27, 17–42. [Google Scholar] [CrossRef]
- Fritsche, E.; Grandjean, P.; Crofton, K.M.; Aschner, M.; Goldberg, A.; Heinonen, T.; Hessel, V.; Hogberg, H.T.; Bennekou, S.H.; Lein, P.J.; et al. Consensus statement on the need for innovation, transition and implementation of developmental neurotoxicity (DNT) testing for regulatory purposes. Toxicol. Appl. Pharmacol. 2018, 354, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, E.; Crofton, K.M.; Hernandez, A.F.; Hougaard Bennekou, S.; Leist, M.; Bal-Price, A.; Reaves, E.; Wilks, M.F.; Terron, A.; Solecki, R.; et al. OECD/EFSA workshop on developmental neurotoxicity (DNT): The use of non-animal test methods for regulatory purposes. ALTEX 2017, 34, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Masjosthusmann, S.B.J.; Bartmann, K.; Dolde, X.; Holzer, A.K.; Stürzl, L.C.; Waldmann, T.; Kisitu, J.; Suciu, I.; Leist, M. Establishment of an a priori protocol for the implementation and interpretation of an in-vitro testing battery for the assessment of developmental neurotoxicity. EFSA Supporting Publ. 2020, 17, 10. [Google Scholar] [CrossRef]
- Bunge, R.P. Glial cells and the central myelin sheath. Physiol. Rev. 1968, 48, 197–251. [Google Scholar] [CrossRef]
- Jernigan, T.L.; Stiles, J. Construction of the human forebrain. Wiley Interdiscip. Rev. Cogn. Sci. 2016, 8, e1409. [Google Scholar] [CrossRef] [Green Version]
- Mathews, E.; Appel, B. Oligodendrocyte differentiation. Method Cell Biol. 2016, 134, 69–96. [Google Scholar] [CrossRef]
- Barkovich, A.J. Magnetic resonance techniques in the assessment of myelin and myelination. J. Inherit. Metab. Dis. 2005, 28, 311–343. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Turtle, M.; Khan, Y.; Farol, P. Myelination of a Key Relay Zone in the Hippocampal Formation Occurs in the Human Brain During Childhood, Adolescence, and Adulthood. Arch. Gen. Psychiatry 1994, 51, 477. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, P.I.L.A. The Myelogenetic Cycles of Regional Maturation of the Brain. Regional Development of the Brain in Early Life; Blackwell: Oxford, UK, 1967; pp. 3–70. [Google Scholar]
- Keshavan, M.S.; Diwadkar, V.A.; Debellis, M.; Dick, E.; Kotwal, R.; Rosenberg, D.R.; Sweeney, J.A.; Minshew, N.; Pettegrew, J.W. Development of the corpus callosum in childhood, adolescence and early adulthood. Life Sci. 2002, 70, 1909–1922. [Google Scholar] [CrossRef]
- Brouwer, R.M.; Mandl, R.C.W.; Schnack, H.G.; Van Soelen, I.L.C.; Van Baal, G.C.; Peper, J.S.; Kahn, R.S.; Boomsma, D.I.; Pol, H.H. White Matter Development in Early Puberty: A Longitudinal Volumetric and Diffusion Tensor Imaging Twin Study. PLoS ONE 2012, 7, e32316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giedd, J.N.; Blumenthal, J.; Jeffries, N.O.; Castellanos, F.; Liu, H.; Zijdenbos, A.; Paus, T.; Evans, A.C.; Rapoport, J.L. Brain development during childhood and adolescence: A longitudinal MRI study. Nat. Neurosci. 1999, 2, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.; Gee, M.; Camicioli, R.; Wieler, M.; Martin, W.W.; Beaulieu, C. Diffusion tensor imaging of white matter tract evolution over the lifespan. NeuroImage 2012, 60, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.; Beaulieu, C. Longitudinal Development of Human Brain Wiring Continues from Childhood into Adulthood. J. Neurosci. 2011, 31, 10937–10947. [Google Scholar] [CrossRef] [PubMed]
- Fancy, S.P.; Chan, J.R.; Baranzini, S.E.; Franklin, R.J.; Rowitch, D.H. Myelin Regeneration: A Recapitulation of Development? Annu. Rev. Neurosci. 2011, 34, 21–43. [Google Scholar] [CrossRef]
- Nave, K.A.; Werner, H.B. Myelination of the Nervous System: Mechanisms and Functions. Annu. Rev. Cell Dev. Bi. 2014, 30, 503–533. [Google Scholar] [CrossRef]
- Deng, W.; Poretz, R.D. Oligodendroglia in Developmental Neurotoxicity. NeuroToxicology 2003, 24, 161–178. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef]
- Hughes, E.G.; Orthmann-Murphy, J.L.; Langseth, A.J.; Bergles, D.E. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 2018, 21, 696–706. [Google Scholar] [CrossRef]
- Podbielska, M.; Hogan, E.L. Molecular and immunogenic features of myelin lipids: Incitants or modulators of multiple scle-rosis? Mult. Scler. J. 2009, 15, 1011–1029. [Google Scholar] [CrossRef]
- Barkovich, A.J. Concepts of myelin and myelination in neuroradiology. Am. J. Neuroradiol. 2000, 21, 1099–1109. [Google Scholar]
- Laule, C.; Vavasour, I.M.; Kolind, S.H.; Li, D.K.B.; Traboulsee, T.L.; Moore, G.R.W.; Mackay, A.L. Magnetic resonance imaging of myelin. Neurotherapeutics 2007, 4, 460–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Knaap, M.; Valk, J. Magnetic resonance of myelination and myelin disorders. J. Neuroradiol. 2006, 33, 132. [Google Scholar] [CrossRef]
- Branson, H.M. Normal Myelination: A Practical Pictorial Review. Neuroimag. Clin. N. Am. 2013, 23, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Guleria, S.; Kelly, T.G. Myelin, Myelination, and Corresponding Magnetic Resonance Imaging Changes. Radiol. Clin. N. Am. 2014, 52, 227–239. [Google Scholar] [CrossRef]
- Xin, W.; Mironova, Y.A.; Shen, H.; Marino, R.A.; Waisman, A.; Lamers, W.H.; Bergles, D.E.; Bonci, A. Oligodendrocytes Support Neuronal Glutamatergic Transmission via Expression of Glutamine Synthetase. Cell Rep. 2019, 27, 2262–2271.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.; Gould, E.; Xu, J.; Kim, E.J.; Kim, J.H. Oligodendrocytes regulate presynaptic properties and neurotransmission through BDNF signaling in the mouse brainstem. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Zemmar, A.; Chen, C.-C.; Weinmann, O.; Kast, B.; Vajda, F.; Bozeman, J.; Isaad, N.; Zuo, Y.; Schwab, M.E. Oligodendrocyte- and Neuron-Specific Nogo-A Restrict Dendritic Branching and Spine Density in the Adult Mouse Motor Cortex. Cereb. Cortex 2018, 28, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Ren, S.-Y.; Chen, J.-F.; Liu, K.; Li, R.-X.; Li, Z.-F.; Hu, B.; Niu, J.-Q.; Xiao, L.; Chan, J.R.; et al. Myelin degeneration and diminished myelin renewal contribute to age-related deficits in memory. Nat. Neurosci. 2020, 23, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D.; Dutta, D.J.; Belgrad, J.; Robnett, M. Cholinergic signaling in myelination. Glia 2017, 65, 687–698. [Google Scholar] [CrossRef]
- Bergles, D.E.; Roberts, J.D.; Somogyi, P.; Jahr, C.E. Glutamatergic synapses on oligodendrocyte precursor cells in the hip-pocampus. Nature 2000, 405, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Káradóttir, R.; Hamilton, N.B.; Bakiri, Y.; Attwell, D. Spiking and nonspiking classes of oligodendrocyte precursor glia in CNS white matter. Nat. Neurosci. 2008, 11, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-C.; Bergles, D. Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat. Neurosci. 2003, 7, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Battefeld, A.; Klooster, J.; Kole, M.H.P. Myelinating satellite oligodendrocytes are integrated in a glial syncytium constraining neuronal high-frequency activity. Nat. Commun. 2016, 7, 11298. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.T.; Witt, A.S.; Kirkpatrick, L.L.; De Waegh, S.M.; Readhead, C.; Tu, P.-H.; Lee, V.M.-Y. Formation of Compact Myelin Is Required for Maturation of the Axonal Cytoskeleton. J. Neurosci. 1999, 19, 7278–7288. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.T.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegenera-tion. Nature 2012, 487, 443–U1502. [Google Scholar] [CrossRef]
- Miller, R.H. Regulation of oligodendrocyte development in the vertebrate CNS. Prog. Neurobiol. 2002, 67, 451–467. [Google Scholar] [CrossRef]
- Navel, K.A.; Salzer, J.L. Axonal regulation of myelination by neuregulin. Curr. Opin. Neurobiol. 2006, 16, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.-A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef]
- Nagy, Z.; Westerberg, H.; Klingberg, T. Maturation of White Matter is Associated with the Development of Cognitive Functions during Childhood. J. Cogn. Neurosci. 2004, 16, 1227–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Ohayon, D.; McKenzie, I.A.; Sinclair-Wilson, A.; Wright, J.; Fudge, A.D.; Emery, B.; Li, H.; Richardson, W.D. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat. Neurosci. 2016, 19, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, I.A.; Ohayon, D.; Li, H.; de Faria, J.P.; Emery, B.; Tohyama, K.; Richardson, W.D. Motor skill learning requires active central myelination. Science 2014, 346, 318–322. [Google Scholar] [CrossRef]
- Deutsch, G.K.; Dougherty, R.F.; Bammer, R.; Siok, W.T.; Gabrieli, J.D.E.; Wandell, B. Children’s reading performance is correlated with white matter structure measured by diffusion tensor imaging. Cortex 2005, 41, 354–363. [Google Scholar] [CrossRef]
- Liu, J.; Dietz, K.C.; DeLoyht, J.M.; Pedreiturria, X.; Kelkar, D.; Kaur, J.; Vialou, V.; Lobo, M.K.; Dietz, D.; Nestler, E.J.; et al. Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nat. Neurosci. 2012, 15, 1621–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makinodan, M.; Rosen, K.M.; Ito, S.; Corfas, G. A Critical Period for Social Experience-Dependent Oligodendrocyte Matura-tion and Myelination. Science 2012, 337, 1357–1360. [Google Scholar] [CrossRef] [Green Version]
- Barak, B.; Zhang, Z.C.; Liu, Y.Y.; Nir, A.; Trangle, S.S.; Ennis, M.; Levandowski, K.M.; Wang, D.Q.; Quast, K.; Boulting, G.L.; et al. Neuronal deletion of Gtf2i, associated with Williams syndrome, causes behavioral and myelin alterations rescuable by a remyelinating drug (vol 22, pg 700, 2019). Nat. Neurosci. 2019, 22, 1197. [Google Scholar] [CrossRef]
- Phan, B.N.; Bohlen, J.F.; Davis, B.A.; Ye, Z.; Chen, H.-Y.; Mayfield, B.; Sripathy, S.R.; Page, S.C.; Campbell, M.N.; Smith, H.L.; et al. A myelin-related transcriptomic profile is shared by Pitt–Hopkins syndrome models and human autism spectrum disorder. Nat. Neurosci. 2020, 23, 375–385. [Google Scholar] [CrossRef]
- Wiggins, R.C. Myelination: A critical stage in development. NeuroToxicology 1986, 7, 103–120. [Google Scholar]
- Okamoto, H.; Miki, T.; Lee, K.-Y.; Yokoyama, T.; Kuma, H.; Wang, Z.-Y.; Gu, H.; Li, H.-P.; Matsumoto, Y.; Irawan, S.; et al. Oligodendrocyte myelin glycoprotein (OMgp) in rat hippocampus is depleted by chronic ethanol consumption. Neurosci. Lett. 2006, 406, 76–80. [Google Scholar] [CrossRef]
- Bouldin, T.W.; Samsa, G.; Earnhardt, T.S.; Krigman, M.R. Selective Vulnerability Among Schwann Cells to Tellurium-Induced Demyelination. J. Neuropathol. Exp. Neurol. 1986, 45, 353. [Google Scholar] [CrossRef]
- Toews, A.D.; Wagner-Recio, M.; Harry, J.; Popko, B.; Morell, P. Metabolic Alterations during Tellurium-Induced Demyelination and Subsequent Remyelination. Ann. N. Y. Acad. Sci. 1990, 605, 363–364. [Google Scholar] [CrossRef]
- Morell, P.; Toews, A.D.; Wagner, M.; Goodrum, J.F. Gene expression during tellurium-induced primary demyelination. NeuroToxicology 1994, 15, 171–180. [Google Scholar]
- Xu, X.B.; Fan, S.J.; He, Y.; Ke, X.; Song, C.; Xiao, Y.; Zhang, W.H.; Zhang, J.Y.; Yin, X.P.; Kato, N.; et al. Loss of Hippocampal Oligodendrocytes Contributes to the Deficit of Contextual Fear Learning in Adult Rats Experiencing Early Bi-sphenol A Exposure. Mol. Neurobiol. 2017, 54, 4524–4536. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.K.; Agarwal, S.; Chauhan, L.K.S.; Mishra, V.N.; Chaturvedi, R.K. Correction to: Bisphenol-A Impairs Myelination Potential During Development in the Hippocampus of the Rat Brain. Mol. Neurobiol. 2019, 56, 5270–5271. [Google Scholar] [CrossRef] [Green Version]
- Seiwa, C.; Nakahara, J.; Komiyama, T.; Katsu, Y.; Iguchi, T.; Asou, H. Bisphenol A Exerts Thyroid-Hormone-Like Effects on Mouse Oligodendrocyte Precursor Cells. Neuroendocrinology 2004, 80, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lai, N.L.; Wang, X.; Guo, Y.; Lam, P.K.; Lam, J.C.; Zhou, B. Bioconcentration and transfer of the organophorous flame retardant 1,3-dichloro-2-propyl phosphate causes thyroid endocrine disruption and developmental neurotoxicity in zebrafish larvae. Environ. Sci. Technol. 2015, 49, 5123–5132. [Google Scholar] [CrossRef]
- Cammer, W. The neurotoxicant, cuprizone, retards the differentiation of oligodendrocytes in vitro. J. Neurol. Sci. 1999, 168, 116–120. [Google Scholar] [CrossRef]
- Mustapha, O.; Oke, B.; Offen, N.; Siren, A.L.; Olopade, J. Neurobehavioral and cytotoxic effects of vanadium during oli-godendrocyte maturation: A protective role for erythropoietin. Environ. Toxicol. Phar. 2014, 38, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Padhi, B.K.; Pelletier, G. Perturbation of Myelin basic protein (Mbp) splice variant expression in developing rat cerebellum following perinatal exposure to methylmercury. Toxicol. Lett. 2012, 213, 374–380. [Google Scholar] [CrossRef] [PubMed]
- David, P.; Subramaniam, K. Prenatal alcohol exposure and early postnatal changes in the developing nerve-muscle system. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 897–903. [Google Scholar] [CrossRef]
- Harry, G.; Toews, A.D.; Krigman, M.R.; Morell, P. The effect of lead toxicity and milk deprivation on myelination in the rat. Toxicol. Appl. Pharmacol. 1985, 77, 458–464. [Google Scholar] [CrossRef]
- Rothenberg, S.J.; Poblano, A.; Garza-Morales, S. Prenatal and perinatal low level lead exposure alters brainstem auditory evoked responses in infants. NeuroToxicology 1994, 15, 695–699. [Google Scholar]
- Tiffany-Castiglioni, E. Cell culture models for lead toxicity in neuronal and glial cells. NeuroToxicology 1993, 14, 513–536. [Google Scholar]
- Tiffany-Castiglioni, E.; Zmudzki, J.; Bratton, G.R. Cellular targets of lead neurotoxicity: In vitro models. Toxicology 1986, 42, 303–315. [Google Scholar] [CrossRef]
- Volpe, J.J.; Kinney, H.C.; Jensen, F.E.; Rosenberg, P. Reprint of “The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. ” Int. J. Dev. Neurosci. 2011, 29, 565–582. [Google Scholar] [CrossRef]
- Butts, B.D.; Houde, C.; Mehmet, H. Maturation-dependent sensitivity of oligodendrocyte lineage cells to apoptosis: Implica-tions for normal development and disease. Cell Death Differ. 2008, 15, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, J.S.; Kjellsen, B.R.; Tigert, J.; Miskimins, R. In utero PCP exposure alters oligodendrocyte differentiation and mye-lination in developing rat frontal cortex. Brain Res. 2008, 1234, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Oka, A.; Belliveau, M.J.; Rosenberg, P.A.; Volpe, J.J. Vulnerability of Oligodendroglia to Glutamate—Pharmacology, Mech-anisms, and Prevention. J. Neurosci. 1993, 13, 1441–1453. [Google Scholar] [CrossRef] [Green Version]
- Toran-Allerand, C. Acetylcholinesterase inhibition and myelinogenesis in vitro. Exp. Neurol. 1974, 43, 216–226. [Google Scholar] [CrossRef]
- Back, S.A.; Gan, X.D.; Li, Y.; Rosenberg, P.A.; Volpe, J.J. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J. Neurosci. 1998, 18, 6241–6253. [Google Scholar] [CrossRef]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Farrelly, L.; Foecking, M.; Piontkewitz, Y.; Dicker, P.; English, J.; Wynne, K.; Cannon, M.; Cagney, G.; Cotter, D. Maternal Immune Activation Induces Changes in Myelin and Metabolic Proteins, Some of Which Can Be Prevented with Risperidone in Adolescence. Dev. Neurosci. 2015, 37, 43–55. [Google Scholar] [CrossRef]
- Makinodan, M.; Tatsumi, K.; Manabe, T.; Yamauchi, T.; Makinodan, E.; Matsuyoshi, H.; Shimoda, S.; Noriyama, Y.; Kishimoto, T.; Wanaka, A. Maternal immune activation in mice delays myelination and axonal development in the hippocampus of the offspring. J. Neurosci. Res. 2008, 86, 2190–2200. [Google Scholar] [CrossRef] [PubMed]
- Dammann, O.; Leviton, A. Maternal Intrauterine Infection, Cytokines, and Brain Damage in the Preterm Newborn. Pediatr. Res. 1997, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wischhof, L.; Irrsack, E.; Osorio, C.; Koch, M. Prenatal LPS-exposure—A neurodevelopmental rat model of schizophrenia—Differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2015, 57, 17–30. [Google Scholar] [CrossRef]
- Rogister, B.; Ben-Hur, T.; Dubois-Dalcq, M. From Neural Stem Cells to Myelinating Oligodendrocytes. Mol. Cell. Neurosci. 1999, 14, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.M.; Aloise, P.A.; Soto, E.F.; Pasquini, J.M. Neonatal hyperthyroidism in the rat produces an increase in the activity of microperoxisomal marker enzymes coincident with biochemical signs of accelerated myelination. J. Neurosci. Res. 1990, 25, 353–359. [Google Scholar] [CrossRef]
- Billon, N.; Tokumoto, Y.; Forrest, D.; Raff, M. Role of Thyroid Hormone Receptors in Timing Oligodendrocyte Differentiation. Dev. Biol. 2001, 235, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarrola, N.; Rodríguez-Peña, A. Hypothyroidism coordinately and transiently affects myelin protein gene expression in most rat brain regions during postnatal development. Brain Res. 1997, 752, 285–293. [Google Scholar] [CrossRef]
- Lee, J.Y.; Petratos, S. Thyroid Hormone Signaling in Oligodendrocytes: From Extracellular Transport to Intracellular Signal. Mol. Neurobiol. 2016, 53, 6568–6583. [Google Scholar] [CrossRef]
- Marta, C.B.; Adamo, A.M.; Soto, E.F.; Pasquini, J.M. Sustained neonatal hyperthyroidism in the rat affects myelination in the central nervous system. J. Neurosci. Res. 1998, 53, 251–259. [Google Scholar] [CrossRef]
- Rodriguez-Pena, A.; Ibarrola, N.; Iñiguez, M. Ángel; Munoz, A.; Bernal, J. Neonatal hypothyroidism affects the timely expression of myelin-associated glycoprotein in the rat brain. J. Clin. Investig. 1993, 91, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Walters, S.N.; Morell, P. Effects of Altered Thyroid States on Myelinogenesis. J. Neurochem. 1981, 36, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, C.; Hu, C.; Yu, K.; Yang, L.; Zhou, B. Acute exposure to DE-71: Effects on locomotor behavior and devel-opmental neurotoxicity in zebrafish larvae. Environ. Toxicol. Chem. 2012, 31, 2338–2344. [Google Scholar] [CrossRef] [PubMed]
- Kodavanti, P.R.S.; Coburn, C.G.; Moser, V.C.; MacPhail, R.C.; Fenton, S.E.; Stoker, T.; Rayner, J.L.; Kannan, K.; Birnbaum, L. Developmental Exposure to a Commercial PBDE Mixture, DE-71: Neurobehavioral, Hormonal, and Reproductive Effects. Toxicol. Sci. 2010, 116, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, L.; Wang, Q.; Guo, Y.; Li, N.; Ma, M.; Zhou, B. The neurotoxicity of DE-71: Effects on neural development and impairment of serotonergic signaling in zebrafish larvae. J. Appl. Toxicol. 2016, 36, 1605–1613. [Google Scholar] [CrossRef]
- Zhou, T.; Taylor, M.M.; DeVito, M.J.; Crofton, K.M. Developmental Exposure to Brominated Diphenyl Ethers Results in Thyroid Hormone Disruption. Toxicol. Sci. 2002, 66, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dach, K.; Bendt, F.; Huebenthal, U.; Giersiefer, S.; Lein, P.J.; Heuer, H.; Fritsche, E. BDE-99 impairs differentiation of human and mouse NPCs into the oligodendroglial lineage by speciesspecific modes of action. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, H.; Li, J.; Shan, Z.; Teng, W.; Teng, X. The Correlation between Polybrominated Diphenyl Ethers (PBDEs) and Thyroid Hormones in the General Population: A Meta-Analysis. PLoS ONE 2015, 10, e0126989. [Google Scholar] [CrossRef] [Green Version]
- USEPA. Chlorpyrifos: Revised Human Health Risk Assessment; National Service Center for Environmental Publications (NSCEP): Cincinnati, OH, USA, 2006. [Google Scholar]
- Itoh, T.; Beesley, J.; Itoh, A.; Cohen, A.S.; Kavanaugh, B.; Coulter, D.A.; Grinspan, J.B.; Pleasure, D. AMPA glutamate re-ceptor-mediated calcium signaling is transiently enhanced during development of oligodendrocytes. J. Neurochem. 2002, 81, 390–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follett, P.L.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. NBQX attenuates excitotoxic injury in developing white matter (vol 20, pg 9235, 2000). J. Neurosci. 2001, 21, 1a. [Google Scholar]
- Matute, C.; Alberdi, E.; Domercq, M.; Gomez, M.V.S.; Samartin, A.L.P.; Rodríguez-Antigüedad, A.; Cerda, F.P. Excitotoxic damage to white matter. J. Anat. 2007, 210, 693–702. [Google Scholar] [CrossRef]
- Cohen, R.I.; Almazan, G. Rat Oligodendrocytes Express Muscarinic Receptors Coupled to Phosphoinositide Hydrolysis and Adenylyl Cyclase. Eur. J. Neurosci. 1994, 6, 1213–1224. [Google Scholar] [CrossRef]
- De Angelis, F.; Bernardo, A.; Magnaghi, V.; Minghetti, L.; Tata, A.M. Muscarinic receptor subtypes as potential targets to modulate oligodendrocyte progenitor survival, proliferation, and differentiation. Dev. Neurobiol. 2012, 72, 713–728. [Google Scholar] [CrossRef]
- Larocca, J.N.; Almazan, G. Acetylcholine agonists stimulate mitogen-activated protein kinase in oligodendrocyte progenitors by muscarinic receptors. J. Neurosci. Res. 1997, 50, 743–754. [Google Scholar] [CrossRef]
- Ragheb, F.; Molina-Holgado, E.; Cui, Q.L.; Khorchid, A.; Liu, H.N.; Larocca, J.N.; Almazan, G. Pharmacological and func-tional characterization of muscarinic receptor subtypes in developing oligodendrocytes. J. Neurochem. 2001, 77, 1396–1406. [Google Scholar] [CrossRef] [Green Version]
- Rogers, S.W.; Gregori, N.Z.; Carlson, N.; Gahring, L.C.; Noble, M. Neuronal nicotinic acetylcholine receptor expression by O2A/oligodendrocyte progenitor cells. Glia 2001, 33, 306–313. [Google Scholar] [CrossRef]
- Cao, J.; Wang, J.; Dwyer, J.B.; Gautier, N.M.; Wang, S.; Leslie, F.M.; Li, M.D. Gestational nicotine exposure modifies myelin gene expression in the brains of adolescent rats with sex differences. Transl. Psychiatry 2013, 3, e247. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Cui, W.-Y.; Cao, J.; Luo, C.; Fan, L.; Li, M.D. Impact of Maternal Nicotine Exposure on Expression of Myelin-Related Genes in Zebrafish Larvae. Zebrafish 2014, 11, 10–16. [Google Scholar] [CrossRef] [PubMed]
- US Enviromental Protection Agency. Chlorpyrifos: Revised Human Health Risk Assessment; US Enviromental Protection Agency: Washington, DC, USA, 2016. [Google Scholar]
- Betancourt, A.M.; Burgess, S.C.; Carr, R.L. Effect of Developmental Exposure to Chlorpyrifos on the Expression of Neurotrophin Growth Factors and Cell-Specific Markers in Neonatal Rat Brain. Toxicol. Sci. 2006, 92, 500–506. [Google Scholar] [CrossRef] [Green Version]
- Slotkin, T.A.; Seidler, F.J. Comparative developmental neurotoxicity of organophosphates in vivo: Transcriptional responses of pathways for brain cell development, cell signaling, cytotoxicity and neurotransmitter systems. Brain Res. Bull. 2007, 72, 232–274. [Google Scholar] [CrossRef] [Green Version]
- Dishaw, L.; Hunter, D.L.; Padnos, B.; Padilla, S.; Stapleton, H.M. Developmental Exposure to Organophosphate Flame Retardants Elicits Overt Toxicity and Alters Behavior in Early Life Stage Zebrafish (Danio rerio). Toxicol. Sci. 2014, 142, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Eldefrawi, A.T.; Mansour, N.A.; Brattsten, L.B.; Ahrens, V.D.; Lisk, D.J. Further toxicologic studies with commercial and candidate flame retardant chemicals. Part II. Bull. Environ. Contam. Toxicol. 1977, 17, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Hogberg, H.T.; Sá, R.D.C.D.S.E.; Kleensang, A.; Bouhifd, M.; Ulker, O.C.; Smirnova, L.; Behl, M.; Maertens, A.; Zhao, L.; Hartung, T. Organophosphorus flame retardants are developmental neurotoxicants in a rat primary brainsphere in vitro model. Arch. Toxicol. 2021, 95, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Riddle, A.; McClure, M.M. Maturation-Dependent Vulnerability of Perinatal White Matter in Premature Birth. Stroke 2007, 38, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free. Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Thorburne, S.K.; Juurlink, B.H. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J. Neurochem. 1996, 67, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.A.; Desmazières, A.; Fricker, D.; Lubetzki, C.; Sol-Foulon, N. Mechanisms of sodium channel clustering and its influence on axonal impulse conduction. Cell. Mol. Life Sci. 2016, 73, 723–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasband, M.N.; Trimmer, J.S. Developmental Clustering of Ion Channels at and near the Node of Ranvier. Dev. Biol. 2001, 236, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Calvo, M.; Richards, N.; Schmid, A.B.; Barroso, A.; Zhu, L.; Ivulic, D.; Zhu, N.; Anwandter, P.; Bhat, M.A.; Court, F.A.; et al. Author response: Altered potassium channel distribution and composition in myelinated axons suppresses hyperexcitability following injury. eLife 2016, 5, e12661. [Google Scholar] [CrossRef] [Green Version]
- Eijkelkamp, N.; Linley, J.; Baker, M.D.; Minett, M.S.; Cregg, R.; Werdehausen, R.; Rugiero, F.; Wood, J.N. Neurological perspectives on voltage-gated sodium channels. Brain 2012, 135, 2585–2612. [Google Scholar] [CrossRef]
- Krasnow, A.M.; Ford, M.C.; Valdivia, L.E.; Wilson, S.W.; Attwell, D. Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat. Neurosci. 2018, 21, 24–28. [Google Scholar] [CrossRef]
- Baraban, M.; Koudelka, S.; Lyons, D.A. Ca2+ activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci. 2018, 21, 19–23. [Google Scholar] [CrossRef]
- Gassmann, K.; Schreiber, T.; Dingemans, M.M.L.; Krause, G.; Roderigo, C.; Giersiefer, S.; Schuwald, J.; Moors, M.; Unfried, K.; Bergman, Å.; et al. BDE-47 and 6-OH-BDE-47 modulate calcium homeostasis in primary fetal human neural progenitor cells via ryanodine receptor-independent mechanisms. Arch. Toxicol. 2014, 88, 1537–1548. [Google Scholar] [CrossRef]
- Schreiber, T.; Gassmann, K.; Götz, C.; Hübenthal, U.; Moors, M.; Krause, G.; Merk, H.F.; Nguyen, N.-H.; Scanlan, T.S.; Abel, J.; et al. Polybrominated Diphenyl Ethers Induce Developmental Neurotoxicity in a Human in Vitro Model: Evidence for Endocrine Disruption. Environ. Health Perspect. 2010, 118, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1843, 1917–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Bates, J.; Li, X.J.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-Derived Oligodendrocyte Progenitor Cells Can Myelinate and Rescue a Mouse Model of Congenital Hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M.; Mozafari, S.; Glatza, M.; Starost, L.; Velychko, S.; Hallmann, A.L.; Cui, Q.L.; Schambach, A.; Kim, K.P.; Bachelin, C.; et al. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using tran-scription factors. Proc. Natl. Acad. Sci. USA 2017, 114, E2243–E2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.R.; Watkins, T.A.; Cosgaya, J.M.; Zhang, C.; Chen, L.; Reichardt, L.F.; Shooter, E.M.; Barres, B.A. NGF Controls Axonal Receptivity to Myelination by Schwann Cells or Oligodendrocytes. Neuron 2004, 43, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lariosa-Willingham, K.D.; Rosler, E.S.; Tung, J.S.; Dugas, J.C.; Collins, T.L.; Leonoudakis, D. Development of a central nervous system axonal myelination assay for high throughput screening. BMC Neurosci. 2016, 17, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Colognato, H.; Ffrench-Constant, C. Contrasting effects of mitogenic growth factors on myelination in neu-ron-oligodendrocyte co-cultures. Glia 2007, 55, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Barateiro, A.; Domingues, H.S.; Fernandes, A.; Relvas, J.B.; Brites, R. Rat Cerebellar Slice Cultures Exposed to Bilirubin Evidence Reactive Gliosis, Excitotoxicity and Impaired Myelinogenesis that Is Prevented by AMPA and TNF-α Inhibitors. Mol. Neurobiol. 2013, 49, 424–439. [Google Scholar] [CrossRef]
- Hill, R.A.; Medved, J.; Patel, K.D.; Nishiyama, A. Organotypic Slice Cultures to Study Oligodendrocyte Dynamics and My-elination. Jove-J. Vis. Exp. 2014, 90. [Google Scholar]
- Miron, V.; Ludwin, S.K.; Darlington, P.; Jarjour, A.; Soliven, B.; Kennedy, T.E.; Antel, J. Fingolimod (FTY720) Enhances Remyelination Following Demyelination of Organotypic Cerebellar Slices. Am. J. Pathol. 2010, 176, 2682–2694. [Google Scholar] [CrossRef]
- Guentert-Lauber, B.; Monnet-Tschudi, F.; Omlin, F.; Favrod, P.; Honegger, P.; Guentert-Lauber, B.; Monnet-Tschudi, F.; Omlin, F.; Favrod, P.; Honegger, P. Serum-Free Aggregate Cultures of Rat CNS Glial Cells: Biochemical, Immunocytochemical and Morphological Characterization. Dev. Neurosci. 1985, 7, 33–44. [Google Scholar] [CrossRef]
- Honegger, P.; Zurich, M.-G. Preparation and Use of Serum-Free Aggregating Brain Cell Cultures for Routine Neurotoxicity Screening. Neuromethods 2011, 56, 105–128. [Google Scholar] [CrossRef]
- Honegger, P.; Defaux, A.; Monnet-Tschudi, F.; Zurich, M.-G. Preparation, Maintenance, and Use of Serum-Free Aggregating Brain Cell Cultures. Methods Mol. Biol. 2011, 758, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Zurich, M.G.; Honegger, P.; Schilter, B.; Costa, L.G.; Monnet-Tschudi, F. Use of aggregating brain cell cultures to study de-velopmental effects of organophosphorus insecticides. Neurotoxicology 2000, 21, 599–605. [Google Scholar] [PubMed]
- Zurich, M.-G.; Stanzel, S.; Kopp-Schneider, A.; Prieto, P.; Honegger, P. Evaluation of aggregating brain cell cultures for the detection of acute organ-specific toxicity. Toxicol. In Vitro 2013, 27, 1416–1424. [Google Scholar] [CrossRef]
- Kerman, B.; Kim, H.J.; Padmanabhan, K.; Mei, A.; Georges, S.; Joens, M.S.; Fitzpatrick, J.A.J.; Jappelli, R.; Chandross, K.J.; August, P.; et al. In vitro myelin formation using embryonic stem cells. Development 2015, 142, 2213–2225. [Google Scholar] [CrossRef] [Green Version]
- Sim, F.J.; Windrem, M.S.; Goldman, S.A. Fate determination of adult human glial progenitor cells. Neuron Glia Biol. 2009, 5, 45–55. [Google Scholar] [CrossRef] [Green Version]
- De Monasterio-Schrader, P.; Jahn, O.; Tenzer, S.; Wichert, S.P.; Patzig, J.; Werner, H.B. Systematic approaches to central nervous system myelin. Cell. Mol. Life Sci. 2012, 69, 2879–2894. [Google Scholar] [CrossRef]
- Mertens, J.; Marchetto, M.C.; Bardy, C.; Gage, F.H. Evaluating cell reprogramming, differentiation and conversion tech-nologies in neuroscience. Nat. Rev. Neurosci. 2016, 17, 424–437. [Google Scholar] [CrossRef]
- Sandstrom, J.; Eggermann, E.; Charvet, I.; Roux, A.; Toni, N.; Greggio, C.; Broyer, A.; Monnet-Tschudi, F.; Stoppini, L. De-velopment and characterization of a human embryonic stem cell-derived 3D neural tissue model for neurotoxicity testing. Toxicol In Vitro 2017, 38, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, M.; Nevin, Z.S.; Shick, H.E.; Garrison, E.; Clarkson-Paredes, C.; Karl, M.; Clayton, B.L.L.; Factor, D.C.; Allan, K.C.; Barbar, L.; et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 2018, 15, 700–706. [Google Scholar] [CrossRef]
- Kim, H.; Xu, R.; Padmashri, R.; Dunaevsky, A.; Liu, Y.; Dreyfus, C.F.; Jiang, P. Pluripotent Stem Cell-Derived Cerebral Or-ganoids Reveal Human Oligodendrogenesis with Dorsal and Ventral Origins. Stem Cell Rep. 2019, 12, 890–905. [Google Scholar] [CrossRef] [Green Version]
- Pamies, D.; Barreras, P.; Block, K.; Makri, G.; Kumar, A.; Wiersma, D.; Smirnova, L.; Zang, C.; Bressler, J.; Christian, K.M.; et al. A human brain mi-crophysiological system derived from induced pluripotent stem cells to study neurological diseases and toxicity. ALTEX 2017, 34, 362–376. [Google Scholar]
- Marton, R.M.; Miura, Y.; Sloan, S.A.; Li, Q.; Revah, O.; Levy, R.J.; Huguenard, J.R.; Pașca, S.P. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 2019, 22, 484–491. [Google Scholar] [CrossRef]
- James, O.G.; Selvaraj, B.T.; Magnani, D.; Burr, K.; Connick, P.; Barton, S.K.; Vasistha, N.A.; Hampton, D.W.; Story, D.; Smigiel, R.; et al. iPSC-derived myelinoids to study myelin biology of humans. Dev. Cell 2021, 56, 1346–1358.e6. [Google Scholar] [CrossRef]
- Chesnut, M.; Paschoud, H.; Repond, C.; Smirnova, L.; Hartung, T.; Zurich, M.; Hogberg, H.T.; Pamies, D. Human ipsc-derived model to study myelin disruption. Dev. Cell 2021, 56, 1346–1358. [Google Scholar]
- Nat, R.; DeChant, G. Milestones of Directed Differentiation of Mouse and Human Embryonic Stem Cells into Telencephalic Neurons Based on Neural Development In Vivo. Stem Cells Dev. 2011, 20, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Pamies, D.; Hartung, T. 21st Century Cell Culture for 21st Century Toxicology. Chem. Res. Toxicol. 2017, 30, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Pamies, D.; Bal-Price, A.; Chesne, C.; Coecke, S.; Dinnyes, A.; Eskes, C.; Grillari, R.; Gstraunthaler, G.; Hartung, T.; Jennings, P.; et al. Advanced Good Cell Culture Practice for human primary, stem cell-derived and organoid models as well as microphysiological systems. ALTEX 2018, 35, 353–378. [Google Scholar] [CrossRef] [PubMed]
- Jennings, A.; Carroll, W. Quantification of oligodendrocyte progenitor cells in human and cat optic nerve: Implications for endogenous repair in multiple sclerosis. Glia 2010, 58, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Plummer, S.; Wallace, S.; Ball, G.; Lloyd, R.; Schiapparelli, P.; Quinones-Hinojosa, A.; Hartung, T.; Pamies, D.A. Human iPSC-derived 3D platform using primary brain cancer cells to study drug development and personalized medicine. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Li, L.; Zhou, Q.; Voss, T.C.; Quick, K.L.; LaBarbera, D.V. High-throughput imaging: Focusing in on drug discovery in 3D. Methods 2016, 96, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Harris, G.; Smirnova, L.; Zufferey, V.; Sa, R.; Baldino Russo, F.; Baleeiro Beltrao Braga, P.C.; Chesnut, M.; Zurich, M.G.; Hogberg, H.T.; et al. Antidepressant Paroxetine Exerts Developmental Neurotoxicity in an iPSC-Derived 3D Human Brain Model. Front. Cell Neurosci. 2020, 14, 25. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.K.; Chitsaz, D.; Brown, R.A.; Cui, Q.L.; Dabarno, M.A.; Antel, J.; Kennedy, T.E. Deep learning for high-throughput quantification of oligodendrocyte ensheathment at single-cell resolution. Commun. Biol. 2019, 2, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, S.; van Bruggen, D.; Castelo-Branco, G. Single-Cell RNA Sequencing of Oligodendrocyte Lineage Cells from the Mouse Central Nervous System. Breast Cancer 2019, 1936, 1–21. [Google Scholar] [CrossRef]
- Avey, D.; Sankararaman, S.; Yim, A.K.; Barve, R.; Milbrandt, J.; Mitra, R.D. Single-Cell RNA-Seq Uncovers a Robust Transcriptional Response to Morphine by Glia. Cell Rep. 2018, 24, 3619–3629. [Google Scholar] [CrossRef] [Green Version]
- Van Ineveld, R.L.; Ariese, H.C.; Wehrens, E.J.; Dekkers, J.F.; Rios, A.C. Single-Cell Resolution Three-Dimensional Imaging of Intact Organoids. J. Vis. Exp. 2020, e60709. [Google Scholar] [CrossRef]
- Duncan, K.D.; Fyrestam, J.; Lanekoff, I. Advances in mass spectrometry based single-cell metabolomics. Analyst 2019, 144, 782–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamies, D.; Leist, M.; Coecke, S.; Bowe, G.; Allen, D.; Gstraunthaler, G.; Bal-Price, A.; Pistollato, F.; DeVries, R.; Hartung, T.; et al. Good Cell and Tissue Culture Practice 2.0 (GCCP 2.0)—Draft for stakeholder discussion and call for action. ALTEX 2020, 37, 490–492. [Google Scholar] [CrossRef]
- Pamies, D.; Zurich, M.-G.; Hartung, T. Organotypic Models to Study Human Glioblastoma: Studying the Beast in Its Ecosystem. iScience 2020, 23. [Google Scholar] [CrossRef]
Figure 1.
Graphical summary.

Figure 2.
Points to consider for developing DNT myelin effective endpoints.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chesnut, M.; Hartung, T.; Hogberg, H.; Pamies, D. Human Oligodendrocytes and Myelin In Vitro to Evaluate Developmental Neurotoxicity. Int. J. Mol. Sci. 2021, 22, 7929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157929
AMA Style
Chesnut M, Hartung T, Hogberg H, Pamies D. Human Oligodendrocytes and Myelin In Vitro to Evaluate Developmental Neurotoxicity. International Journal of Molecular Sciences. 2021; 22(15):7929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157929
Chicago/Turabian StyleChesnut, Megan, Thomas Hartung, Helena Hogberg, and David Pamies. 2021. "Human Oligodendrocytes and Myelin In Vitro to Evaluate Developmental Neurotoxicity" International Journal of Molecular Sciences 22, no. 15: 7929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157929
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.