
Signals and Mechanisms Regulating Monocyte and Macrophage Activation in the Pathogenesis of Juvenile Idiopathic Arthritis


Abstract
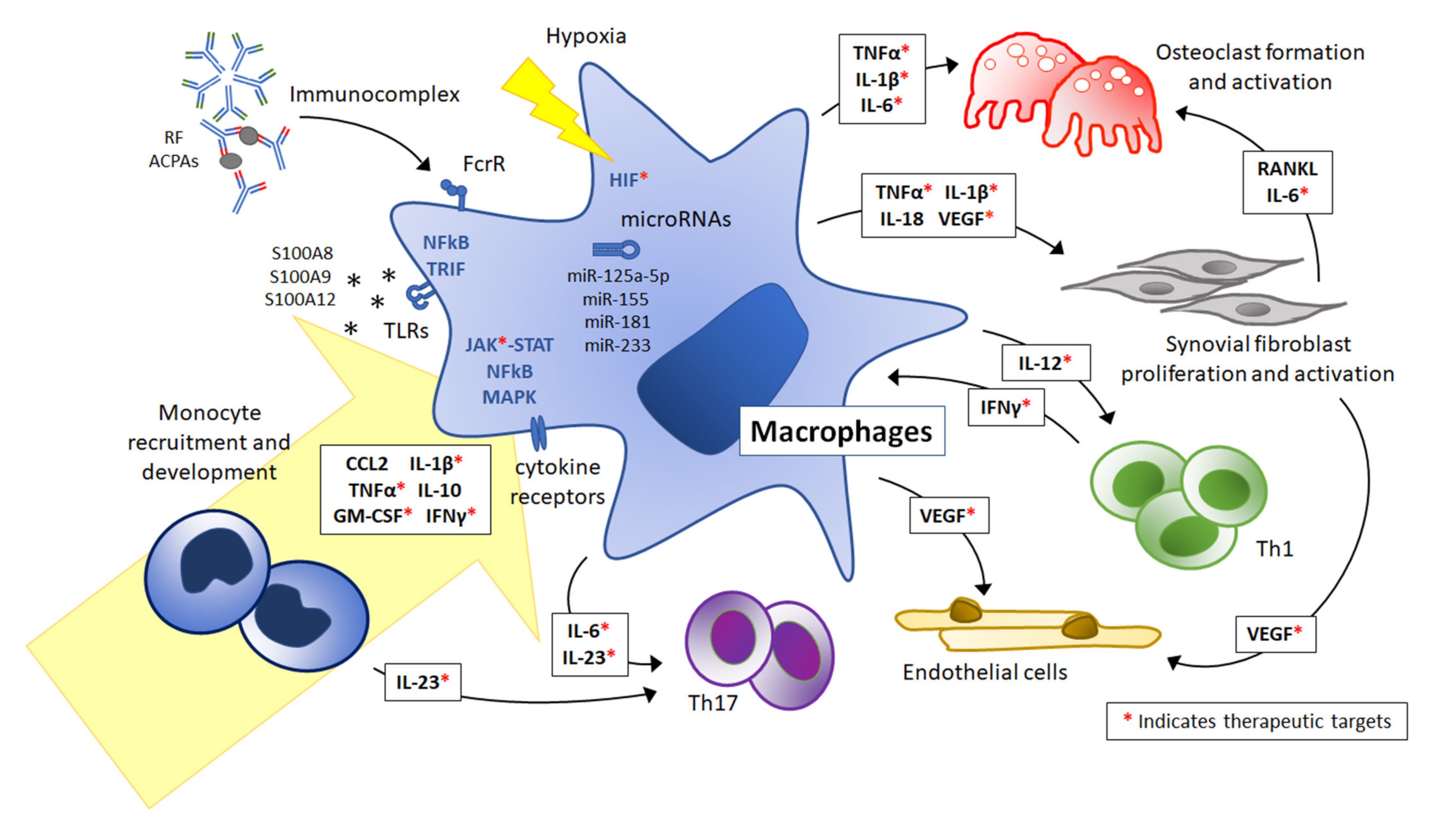
:1. Introduction: Overview of the Pathogenesis of Juvenile Idiopathic Arthritis
2. The Origin of Synovial Resident Macrophages
3. Characteristics of Monocytes and Macrophages in Juvenile Idiopathic Arthritis
3.1. Monocyte Subsets
3.2. Monocyte/Macrophage Polarization
4. Mediators Directing Monocyte/Macrophage Activation and Polarization
4.1. Cytokines
4.1.1. GM-CSF
4.1.2. TNFα
4.1.3. IFNγ
4.1.4. IL-10
4.2. Toll-Like Receptor Signaling
4.3. Autoantibodies and Immunocomplexes
4.4. Hypoxia
4.5. MicroRNAs
5. Effect of Monocyte/Macrophage-Produced Cytokines and Chemokines
5.1. TNFα
5.2. IL-1β
5.3. IL-6
5.4. IL-18
5.5. IL-12/IL-23
5.6. IL-10
5.7. Chemokines
5.8. Vascular Endothelial Growth Factor
6. Available Treatments and Emerging Therapeutic Opportunities
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ACPA | anti-citrullinated protein antibody |
| AS | ankylosing spondylitis |
| CCL | C-C motif chemokine ligand |
| CXCL | C-X-C motif chemokine ligand |
| DMARD | disease-modifying antirheumatic drug |
| ERA | enthesitis-related arthritis |
| FcγR | Fcγ receptor |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| HIF | hypoxia-inducible factor |
| IC | immunocomplex |
| ICAM-1 | intracellular adhesion molecule 1 |
| IL | interleukin |
| ILAR | International League of Associations for Rheumatology |
| IFN | Interferon |
| IP-10 | IFNγ inducible protein 10 |
| IRAK | IL-1R-activating kinase |
| IRF | interferon regulatory factor |
| JAK | Janus kinase |
| JIA | juvenile idiopathic arthritis |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MAS | macrophage activation syndrome |
| MCP-1 | monocyte chemoattractant protein-1 |
| MMP | matrix metalloproteinases |
| Mφ | macrophage |
| Mo | monocyte |
| MTX | methotrexate |
| MyD88 | myeloid differentiation primary response protein 88 |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | natural killer |
| NLRP3 | NOD-, LRR-, and pyrin domain-containing protein 3 |
| PBMCs | peripheral blood mononuclear cells |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| RA | rheumatoid arthritis |
| RANTES | regulated upon activation, normal T cell expressed and presumably secreted |
| RF | rheumatoid factor |
| RANK | receptor activator of NFκB |
| RANKL | RANK ligand |
| SF | synovial fluid |
| sJIA | systemic onset JIA |
| SOCS | suppressor of cytokine signaling |
| STAT | signal transducer and activator of transcription |
| TGFβ | transforming growth factor beta |
| Th1 | type 1 helper T cells |
| Th17 | type 17 helper T cells |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TREM | triggering receptor expressed on myeloid cells |
| TRAF | TNF receptor associated factor |
| VEGF | vascular endothelial growth factor |
References
- Prakken, B.; Albani, S.; Martini, A. Juvenile idiopathic arthritis. Lancet 2011, 377, 2138–2149. [Google Scholar] [CrossRef] [Green Version]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.M.; et al. International League of Associations for, R., International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar]
- Petty, R.E.; Laxer, R.M.; Lindsley, C.B.; Wedderburn, L.; Fuhlbriggezzz, R.C.; Mellins, E.D. Juvenile Idiopathic Arthritis: Classification and Basic Concepts. In Textbook of Pediatric Rheumatology, 8th ed.; Elsevier: Philadelphia, PA, USA, 2021; pp. 209–215. [Google Scholar]
- Lin, Y.T.; Wang, C.T.; Gershwin, M.E.; Chiang, B.L. The pathogenesis of oligoarticular/polyarticular vs systemic juvenile idiopathic arthritis. Autoimmun. Rev. 2011, 10, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.A.; Binstadt, B.A. Autoantibodies in the Pathogenesis, Diagnosis, and Prognosis of Juvenile Idiopathic Arthritis. Front. Immunol. 2018, 9, 3168. [Google Scholar] [CrossRef] [Green Version]
- Leong, J.Y.; Guan, Y.J.; Albani, S.; Arkachaisri, T. Recent advances in our understanding of the pathogenesis of juvenile idiopathic arthritis and their potential clinical implications. Expert. Rev. Clin. Immunol. 2018, 14, 933–944. [Google Scholar] [CrossRef]
- Laria, A.; Lurati, A.; Marrazza, M.; Mazzocchi, D.; Re, K.A.; Scarpellini, M. The macrophages in rheumatic diseases. J. Inflamm. Res. 2016, 9, 1–11. [Google Scholar] [PubMed] [Green Version]
- Ma, W.T.; Gao, F.; Gu, K.; Chen, D.K. The Role of Monocytes and Macrophages in Autoimmune Diseases: A Comprehensive Review. Front. Immunol. 2019, 10, 1140. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.T.; Chang, C.; Gershwin, M.E.; Lian, Z.X. Development of autoantibodies precedes clinical manifestations of autoimmune diseases: A comprehensive review. J. Autoimmun. 2017, 83, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Hunter, P.J.; Nistala, K.; Jina, N.; Eddaoudi, A.; Thomson, W.; Hubank, M.; Wedderburn, L.R. Biologic predictors of extension of oligoarticular juvenile idiopathic arthritis as determined from synovial fluid cellular composition and gene expression. Arthritis Rheum. 2010, 62, 896–907. [Google Scholar] [CrossRef] [Green Version]
- Barnes, M.G.; Grom, A.A.; Thompson, S.D.; Griffin, T.A.; Luyrink, L.K.; Colbert, R.A.; Glass, D.N. Biologic similarities based on age at onset in oligoarticular and polyarticular subtypes of juvenile idiopathic arthritis. Arthritis Rheum. 2010, 62, 3249–3258. [Google Scholar] [CrossRef] [Green Version]
- Griffin, T.A.; Barnes, M.G.; Ilowite, N.T.; Olson, J.C.; Sherry, D.D.; Gottlieb, B.S.; Aronow, B.J.; Pavlidis, P.; Hinze, C.H.; Thornton, S.; et al. Gene expression signatures in polyarticular juvenile idiopathic arthritis demonstrate disease heterogeneity and offer a molecular classification of disease subsets. Arthritis Rheum. 2009, 60, 2113–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Erny, D.; Hagemeyer, N. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 2017, 18, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Pluddemann, A. The Mononuclear Phagocytic System. Generation of Diversity. Front. Immunol. 2019, 10, 1893. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Yang, X.; Chang, Y.; Wei, W. Emerging role of targeting macrophages in rheumatoid arthritis: Focus on polarization, metabolism and apoptosis. Cell Prolif. 2020, 53, e12854. [Google Scholar] [CrossRef]
- Yao, T.C.; Kuo, M.L.; See, L.C.; Ou, L.S.; Lee, W.I.; Chan, C.K.; Huang, J.L. RANTES and monocyte chemoattractant protein 1 as sensitive markers of disease activity in patients with juvenile rheumatoid arthritis: A six-year longitudinal study. Arthritis Rheum. 2006, 54, 2585–2593. [Google Scholar] [CrossRef]
- De Jager, W.; Hoppenreijs, E.P.; Wulffraat, N.M.; Wedderburn, L.R.; Kuis, W.; Prakken, B.J. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: A cross-sectional study. Ann. Rheum. Dis. 2007, 66, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A. Colony-stimulating factors in inflammation and autoimmunity. Nat. Rev. Immunol. 2008, 8, 533–544. [Google Scholar] [CrossRef]
- Hashimoto, D.; Miller, J.; Merad, M. Dendritic cell and macrophage heterogeneity in vivo. Immunity 2011, 35, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. Blood Monocytes and Their Subsets: Established Features and Open Questions. Front. Immunol. 2015, 6, 423. [Google Scholar] [CrossRef]
- Passlick, B.; Flieger, D.; Ziegler-Heitbrock, H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989, 74, 2527–2534. [Google Scholar] [CrossRef] [Green Version]
- Gaur, P.; Myles, A.; Misra, R.; Aggarwal, A. Intermediate monocytes are increased in enthesitis-related arthritis, a category of juvenile idiopathic arthritis. Clin. Exp. Immunol. 2017, 187, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Throm, A.A.; Moncrieffe, H.; Orandi, A.B.; Pingel, J.T.; Geurs, T.L.; Miller, H.L.; Daugherty, A.L.; Malkova, O.N.; Lovell, D.J.; Thompson, S.D.; et al. Identification of enhanced IFN-gamma signaling in polyarticular juvenile idiopathic arthritis with mass cytometry. JCI Insight 2018, 3, e121544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, B.R.; Yoo, S.J.; Choi, Y.; Chung, Y.H.; Kim, J.; Yoo, I.S.; Kang, S.W.; Lee, W.W. Functional phenotype of synovial monocytes modulating inflammatory T-cell responses in rheumatoid arthritis (RA). PLoS ONE 2014, 9, e109775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanaka, N.; Yamamura, M.; Aita, T.; Morita, Y.; Okamoto, A.; Kawashima, M.; Iwahashi, M.; Ueno, A.; Ohmoto, Y.; Makino, H. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002, 46, 2578–2586. [Google Scholar] [CrossRef] [PubMed]
- Macaubas, C.; Nguyen, K.D.; Peck, A.; Buckingham, J.; Deshpande, C.; Wong, E.; Alexander, H.C.; Chang, S.Y.; Begovich, A.; Sun, Y.; et al. Alternative activation in systemic juvenile idiopathic arthritis monocytes. Clin. Immunol. 2012, 142, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.; Berthold, E.; Arve-Butler, S.; Gullstrand, B.; Mossberg, A.; Kahn, F.; Bengtsson, A.A.; Mansson, B.; Kahn, R. Children with oligoarticular juvenile idiopathic arthritis have skewed synovial monocyte polarization pattern with functional impairment-a distinct inflammatory pattern for oligoarticular juvenile arthritis. Arthritis Res. Ther. 2020, 22, 186. [Google Scholar] [CrossRef]
- Macaubas, C.; Nguyen, K.; Deshpande, C.; Phillips, C.; Peck, A.; Lee, T.; Park, J.L.; Sandborg, C.; Mellins, E.D. Distribution of circulating cells in systemic juvenile idiopathic arthritis across disease activity states. Clin. Immunol. 2010, 134, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Macaubas, C.; Deshpande, C.; Alexander, H.C.; Chang, S.Y.; Sun, Y.; Park, J.L.; Lee, T.; Begovich, A.; Mellins, E.D. Monocytes are resistant to apoptosis in systemic juvenile idiopathic arthritis. Clin. Immunol. 2010, 136, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Ziegler-Heitbrock, L.; Hofer, T.P. Toward a refined definition of monocyte subsets. Front. Immunol. 2013, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.L.; Yeap, W.H.; Tai, J.J.; Ong, S.M.; Dang, T.M.; Wong, S.C. The three human monocyte subsets: Implications for health and disease. Immunol. Res. 2012, 53, 41–57. [Google Scholar] [CrossRef]
- Rossol, M.; Kraus, S.; Pierer, M.; Baerwald, C.; Wagner, U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum. 2012, 64, 671–677. [Google Scholar] [CrossRef]
- Fabriek, B.O.; van Bruggen, R.; Deng, D.M.; Ligtenberg, A.J.; Nazmi, K.; Schornagel, K.; Vloet, R.P.; Dijkstra, C.D.; van den Berg, T.K. The macrophage scavenger receptor CD163 functions as an innate immune sensor for bacteria. Blood 2009, 113, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.M.; Rogacev, K.S.; Rotter, B.; Winter, P.; Marell, R.R.; Fliser, D.; Heine, G.H. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 2011, 118, e50–e61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Ambarus, C.A.; Noordenbos, T.; de Hair, M.J.; Tak, P.P.; Baeten, D.L. Intimal lining layer macrophages but not synovial sublining macrophages display an IL-10 polarized-like phenotype in chronic synovitis. Arthritis Res. Ther. 2012, 14, R74. [Google Scholar] [CrossRef] [Green Version]
- Soler Palacios, B.; Estrada-Capetillo, L.; Izquierdo, E.; Criado, G.; Nieto, C.; Municio, C.; Gonzalez-Alvaro, I.; Sanchez-Mateos, P.; Pablos, J.L.; Corbi, A.L.; et al. Macrophages from the synovium of active rheumatoid arthritis exhibit an activin A-dependent pro-inflammatory profile. J. Pathol. 2015, 235, 515–526. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Vandooren, B.; Noordenbos, T.; Ambarus, C.; Krausz, S.; Cantaert, T.; Yeremenko, N.; Boumans, M.; Lutter, R.; Tak, P.P.; Baeten, D. Absence of a classically activated macrophage cytokine signature in peripheral spondylarthritis, including psoriatic arthritis. Arthritis Rheum. 2009, 60, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Schulert, G.S.; Fall, N.; Harley, J.B.; Shen, N.; Lovell, D.J.; Thornton, S.; Grom, A.A. Monocyte MicroRNA Expression in Active Systemic Juvenile Idiopathic Arthritis Implicates MicroRNA-125a-5p in Polarized Monocyte Phenotypes. Arthritis Rheumatol. 2016, 68, 2300–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellins, E.D.; Macaubas, C.; Grom, A.A. Pathogenesis of systemic juvenile idiopathic arthritis: Some answers, more questions. Nat. Rev. Rheumatol. 2011, 7, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Yadav, A.; Aggarwal, A. Evidence for M2 macrophage activation in patients with enthesitis-related arthritis category of juvenile idiopathic arthritis. Clin. Rheumatol. 2019, 38, 1715–1719. [Google Scholar] [CrossRef]
- Sulahian, T.H.; Hogger, P.; Wahner, A.E.; Wardwell, K.; Goulding, N.J.; Sorg, C.; Droste, A.; Stehling, M.; Wallace, P.K.; Morganelli, P.M.; et al. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine 2000, 12, 1312–1321. [Google Scholar] [CrossRef]
- Baeten, D.; Demetter, P.; Cuvelier, C.A.; Kruithof, E.; Van Damme, N.; De Vos, M.; Veys, E.M.; De Keyser, F. Macrophages expressing the scavenger receptor CD163: A link between immune alterations of the gut and synovial inflammation in spondyloarthropathy. J. Pathol. 2002, 196, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Frings, W.; Dreier, J.; Sorg, C. Only the soluble form of the scavenger receptor CD163 acts inhibitory on phorbol ester-activated T-lymphocytes, whereas membrane-bound protein has no effect. FEBS Lett. 2002, 526, 93–96. [Google Scholar] [CrossRef]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Moestrup, S.K.; Moller, H.J. CD163: A regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann. Med. 2004, 36, 347–354. [Google Scholar] [CrossRef]
- Avci, A.B.; Feist, E.; Burmester, G.R. Targeting GM-CSF in rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 39–44. [Google Scholar]
- Piper, C.; Pesenacker, A.M.; Bending, D.; Thirugnanabalan, B.; Varsani, H.; Wedderburn, L.R.; Nistala, K. T cell expression of granulocyte-macrophage colony-stimulating factor in juvenile arthritis is contingent upon Th17 plasticity. Arthritis Rheumatol. 2014, 66, 1955–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swidrowska-Jaros, J.; Orczyk, K.; Smolewska, E. Macrophages—Silent enemies in juvenile idiopathic arthritis. Postepy. Hig. Med. Dosw. 2016, 70, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.E.; Winterrowd, G.E.; Krzesicki, R.F.; Sanders, M.E. Role of cytokines in inflammatory synovitis. The coordinate regulation of intercellular adhesion molecule 1 and HLA class I and class II antigens in rheumatoid synovial fibroblasts. Arthritis Rheum. 1990, 33, 1776–1786. [Google Scholar] [CrossRef]
- Haworth, C.; Brennan, F.M.; Chantry, D.; Turner, M.; Maini, R.N.; Feldmann, M. Expression of granulocyte-macrophage colony-stimulating factor in rheumatoid arthritis: Regulation by tumor necrosis factor-alpha. Eur. J. Immunol. 1991, 21, 2575–2579. [Google Scholar] [CrossRef]
- Butler, D.M.; Maini, R.N.; Feldmann, M.; Brennan, F.M. Modulation of proinflammatory cytokine release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF-alpha antibody with the interleukin-1 receptor antagonist. Eur. Cytokine Netw. 1995, 6, 225–230. [Google Scholar] [PubMed]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell. Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- De Benedetti, F.; Pignatti, P.; Massa, M.; Sartirana, P.; Ravelli, A.; Cassani, G.; Corti, A.; Martini, A. Soluble tumour necrosis factor receptor levels reflect coagulation abnormalities in systemic juvenile chronic arthritis. Br. J. Rheumatol. 1997, 36, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Degboe, Y.; Rauwel, B.; Baron, M.; Boyer, J.F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J.L. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism. Front. Immunol. 2019, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Catrina, A.I.; Trollmo, C.; af Klint, E.; Engstrom, M.; Lampa, J.; Hermansson, Y.; Klareskog, L.; Ulfgren, A.K. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: Extended report. Arthritis Rheum. 2005, 52, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, A.C.; Wildenberg, M.E.; Arijs, I.; Duijvestein, M.; Verhaar, A.P.; de Hertogh, G.; Vermeire, S.; Rutgeerts, P.; van den Brink, G.R.; Hommes, D.W. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflamm. Bowel Dis. 2012, 18, 401–408. [Google Scholar] [CrossRef]
- Aeberli, D.; Kamgang, R.; Balani, D.; Hofstetter, W.; Villiger, P.M.; Seitz, M. Regulation of peripheral classical and non-classical monocytes on infliximab treatment in patients with rheumatoid arthritis and ankylosing spondylitis. RMD Open 2016, 2, e000079. [Google Scholar] [CrossRef] [Green Version]
- Kato, M. New insights into IFN-gamma in rheumatoid arthritis: Role in the era of JAK inhibitors. Immunol. Med. 2020, 43, 72–78. [Google Scholar] [CrossRef]
- Schulert, G.S.; Pickering, A.V.; Do, T.; Dhakal, S.; Fall, N.; Schnell, D.; Medvedovic, M.; Salomonis, N.; Thornton, S.; Grom, A.A. Monocyte and bone marrow macrophage transcriptional phenotypes in systemic juvenile idiopathic arthritis reveal TRIM8 as a mediator of IFN-gamma hyper-responsiveness and risk for macrophage activation syndrome. Ann. Rheum. Dis. 2020, 80, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Macaubas, C.; Wong, E.; Zhang, Y.; Nguyen, K.D.; Lee, J.; Milojevic, D.; Shenoi, S.; Stevens, A.M.; Ilowite, N.; Saper, V.; et al. Altered signaling in systemic juvenile idiopathic arthritis monocytes. Clin. Immunol. 2016, 163, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosten, L.A.; Koenders, M.I.; Smeets, R.L.; Heuvelmans-Jacobs, M.; Helsen, M.M.; Takeda, K.; Akira, S.; Lubberts, E.; van de Loo, F.A.; van den Berg, W.B. Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: Critical role of myeloid differentiation factor 88. J. Immunol. 2003, 171, 6145–6153. [Google Scholar] [CrossRef]
- Myles, A.; Aggarwal, A. Expression of Toll-like receptors 2 and 4 is increased in peripheral blood and synovial fluid monocytes of patients with enthesitis-related arthritis subtype of juvenile idiopathic arthritis. Rheumatology 2011, 50, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fall, N.; Barnes, M.; Thornton, S.; Luyrink, L.; Olson, J.; Ilowite, N.T.; Gottlieb, B.S.; Griffin, T.; Sherry, D.D.; Thompson, S.; et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum. 2007, 56, 3793–3804. [Google Scholar] [CrossRef]
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005, 201, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Ganeva, M.; Fuehner, S.; Kessel, C.; Klotsche, J.; Niewerth, M.; Minden, K.; Foell, D.; Hinze, C.H.; Wittkowski, H. Trajectories of disease courses in the inception cohort of newly diagnosed patients with JIA (ICON-JIA): The potential of serum biomarkers at baseline. Pediatr. Rheumatol. Online J. 2021, 19, 64. [Google Scholar] [CrossRef] [PubMed]
- Schierbeck, H.; Pullerits, R.; Pruunsild, C.; Fischer, M.; Holzinger, D.; Laestadius, A.; Sundberg, E.; Harris, H.E. HMGB1 levels are increased in patients with juvenile idiopathic arthritis, correlate with early onset of disease, and are independent of disease duration. J. Rheumatol. 2013, 40, 1604–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, S.; Singh, A. Study of role of serum amyloid A (SAA) as a marker of disease activity in juvenile idiopathic arthritis. J. Fam. Med. Prim. Care 2019, 8, 2129–2133. [Google Scholar]
- Jung, J.Y.; Kim, J.W.; Suh, C.H.; Kim, H.A. Roles of Interactions Between Toll-Like Receptors and Their Endogenous Ligands in the Pathogenesis of Systemic Juvenile Idiopathic Arthritis and Adult-Onset Still’s Disease. Front. Immunol. 2020, 11, 583513. [Google Scholar] [CrossRef]
- Wang, J.; Vodovotz, Y.; Fan, L.; Li, Y.; Liu, Z.; Namas, R.; Barclay, D.; Zamora, R.; Billiar, T.R.; Wilson, M.A.; et al. Injury-induced MRP8/MRP14 stimulates IP-10/CXCL10 in monocytes/macrophages. FASEB J. 2015, 29, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Van Lent, P.L.; Grevers, L.C.; Schelbergen, R.; Blom, A.; Geurts, J.; Sloetjes, A.; Vogl, T.; Roth, J.; van den Berg, W.B. S100A8 causes a shift toward expression of activatory Fcgamma receptors on macrophages via toll-like receptor 4 and regulates Fcgamma receptor expression in synovium during chronic experimental arthritis. Arthritis Rheum. 2010, 62, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Loser, K.; Vogl, T.; Voskort, M.; Lueken, A.; Kupas, V.; Nacken, W.; Klenner, L.; Kuhn, A.; Foell, D.; Sorokin, L.; et al. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat. Med. 2010, 16, 713–717. [Google Scholar] [CrossRef]
- Cepika, A.M.; Banchereau, R.; Segura, E.; Ohouo, M.; Cantarel, B.; Goller, K.; Cantrell, V.; Ruchaud, E.; Gatewood, E.; Nguyen, P.; et al. A multidimensional blood stimulation assay reveals immune alterations underlying systemic juvenile idiopathic arthritis. J. Exp. Med. 2017, 214, 3449–3466. [Google Scholar] [CrossRef]
- Martini, A.; Ravelli, A.; Avcin, T.; Beresford, M.W.; Burgos-Vargas, R.; Cuttica, R.; Ilowite, N.T.; Khubchandani, R.; Laxer, R.M.; Lovell, D.J.; et al. Toward New Classification Criteria for Juvenile Idiopathic Arthritis: First Steps, Pediatric Rheumatology International Trials Organization International Consensus. J. Rheumatol. 2019, 46, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Yang, H.Y.; Luo, S.F.; Lai, J.H. From Rheumatoid Factor to Anti-Citrullinated Protein Antibodies and Anti-Carbamylated Protein Antibodies for Diagnosis and Prognosis Prediction in Patients with Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 686. [Google Scholar] [CrossRef]
- Wu, C.Y.; Yang, H.Y.; Lai, J.H. Anti-Citrullinated Protein Antibodies in Patients with Rheumatoid Arthritis: Biological Effects and Mechanisms of Immunopathogenesis. Int. J. Mol. Sci. 2020, 21, 4015. [Google Scholar] [CrossRef]
- Bosco, M.C.; Delfino, S.; Ferlito, F.; Puppo, M.; Gregorio, A.; Gambini, C.; Gattorno, M.; Martini, A.; Varesio, L. The hypoxic synovial environment regulates expression of vascular endothelial growth factor and osteopontin in juvenile idiopathic arthritis. J. Rheumatol. 2009, 36, 1318–1329. [Google Scholar] [CrossRef]
- Bosco, M.C.; Delfino, S.; Ferlito, F.; Battaglia, F.; Puppo, M.; Gregorio, A.; Gambini, C.; Gattorno, M.; Martini, A.; Varesio, L. Hypoxic synovial environment and expression of macrophage inflammatory protein 3gamma/CCL20 in juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58, 1833–1838. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Forster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Raggi, F.; Pelassa, S.; Pierobon, D.; Penco, F.; Gattorno, M.; Novelli, F.; Eva, A.; Varesio, L.; Giovarelli, M.; Bosco, M.C. Regulation of Human Macrophage M1-M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front. Immunol. 2017, 8, 1097. [Google Scholar] [CrossRef] [PubMed]
- Shingu, M.; Nagai, Y.; Isayama, T.; Naono, T.; Nobunaga, M.; Nagai, Y. The effects of cytokines on metalloproteinase inhibitors (TIMP) and collagenase production by human chondrocytes and TIMP production by synovial cells and endothelial cells. Clin. Exp. Immunol. 1993, 94, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Do, T.; Tan, R.; Bennett, M.; Medvedovic, M.; Grom, A.A.; Shen, N.; Thornton, S.; Schulert, G.S. MicroRNA networks associated with active systemic juvenile idiopathic arthritis regulate CD163 expression and anti-inflammatory functions in macrophages through two distinct mechanisms. J. Leukoc. Biol. 2018, 103, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.; Schulert, G.S. Functional Regulation of Macrophage Phenotypes by MicroRNAs in Inflammatory Arthritis. Front. Immunol. 2019, 10, 2217. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, H.; Liu, Y.; Song, Y.; Lai, L.; Han, Q.; Cao, X.; Wang, Q. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1beta production in macrophages by targeting STAT3. PLoS ONE 2012, 7, e42971. [Google Scholar]
- Georgopoulos, S.; Plows, D.; Kollias, G. Transmembrane TNF is sufficient to induce localized tissue toxicity and chronic inflammatory arthritis in transgenic mice. J. Inflamm. 1996, 46, 86–97. [Google Scholar]
- Saxena, N.; Aggarwal, A.; Misra, R. Elevated concentrations of monocyte derived cytokines in synovial fluid of children with enthesitis related arthritis and polyarticular types of juvenile idiopathic arthritis. J. Rheumatol. 2005, 32, 1349–1353. [Google Scholar]
- Luo, G.; Li, F.; Li, X.; Wang, Z.G.; Zhang, B. TNFalpha and RANKL promote osteoclastogenesis by upregulating RANK via the NFkappaB pathway. Mol. Med. Rep. 2018, 17, 6605–6611. [Google Scholar] [PubMed] [Green Version]
- Akioka, S. Interleukin-6 in juvenile idiopathic arthritis. Mod. Rheumatol. 2019, 29, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armaroli, G.; Klein, A.; Ganser, G.; Ruehlmann, M.J.; Dressler, F.; Hospach, A.; Minden, K.; Trauzeddel, R.; Foeldvari, I.; Kuemmerle-Deschner, J.; et al. Long-term safety and effectiveness of etanercept in JIA: An 18-year experience from the BiKeR registry. Arthritis Res.Ther. 2020, 22, 258. [Google Scholar] [CrossRef] [PubMed]
- Lovell, D.J.; Brunner, H.I.; Reiff, A.O.; Jung, L.; Jarosova, K.; Nemcova, D.; Mouy, R.; Sandborg, C.; Bohnsack, J.F.; Elewaut, D.; et al. Long-term outcomes in patients with polyarticular juvenile idiopathic arthritis receiving adalimumab with or without methotrexate. RMD Open 2020, 6, e001208. [Google Scholar] [CrossRef]
- Ruperto, N.; Brunner, H.I.; Pacheco-Tena, C.; Louw, I.; Vega-Cornejo, G.; Spindler, A.J.; Kingsbury, D.J.; Schmeling, H.; Borzutzky, A.; Cuttica, R.; et al. Open-Label Phase 3 Study of Intravenous Golimumab in Patients With Polyarticular Juvenile Idiopathic Arthritis. Rheumatology 2021, 67, 2759–2770. [Google Scholar]
- Wilson, D.C.; Marinov, A.D.; Blair, H.C.; Bushnell, D.S.; Thompson, S.D.; Chaly, Y.; Hirsch, R. Follistatin-like protein 1 is a mesenchyme-derived inflammatory protein and may represent a biomarker for systemic-onset juvenile rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2510–2516. [Google Scholar] [CrossRef] [Green Version]
- Frosch, M.; Ahlmann, M.; Vogl, T.; Wittkowski, H.; Wulffraat, N.; Foell, D.; Roth, J. The myeloid-related proteins 8 and 14 complex, a novel ligand of toll-like receptor 4, and interleukin-1beta form a positive feedback mechanism in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009, 60, 883–891. [Google Scholar] [CrossRef]
- Shiratori, T.; Kyumoto-Nakamura, Y.; Kukita, A.; Uehara, N.; Zhang, J.; Koda, K.; Kamiya, M.; Badawy, T.; Tomoda, E.; Xu, X.; et al. IL-1beta Induces Pathologically Activated Osteoclasts Bearing Extremely High Levels of Resorbing Activity: A Possible Pathological Subpopulation of Osteoclasts, Accompanied by Suppressed Expression of Kindlin-3 and Talin-1. J. Immunol. 2018, 200, 218–228. [Google Scholar] [CrossRef]
- Yang, C.M.; Luo, S.F.; Hsieh, H.L.; Chi, P.L.; Lin, C.C.; Wu, C.C.; Hsiao, L.D. Interleukin-1beta induces ICAM-1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: Involvement of ERK, JNK, AP-1, and NF-kappaB. J. Cell Physiol. 2010, 224, 516–526. [Google Scholar] [CrossRef]
- Nishimura, K.; Hara, R.; Umebayashi, H.; Takei, S.; Iwata, N.; Imagawa, T.; Shimizu, M.; Tomiita, M.; Seko, N.; Kitawaki, T.; et al. Efficacy and safety of canakinumab in systemic juvenile idiopathic arthritis: 48-week results from an open-label phase III study in Japanese patients. Mod. Rheumatol. 2021, 31, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.I.; Quartier, P.; Alexeeva, E.; Constantin, T.; Kone-Paut, I.; Marzan, K.; Schneider, R.; Wulffraat, N.M.; Chasnyk, V.; Tirosh, I.; et al. Efficacy and Safety of Canakinumab in Patients With Systemic Juvenile Idiopathic Arthritis With and Without Fever at Baseline: Results From an Open-Label, Active-Treatment Extension Study. Arthritis Rheumatol. 2020, 72, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, L.A.; Boothby, A.; Gertner, E. Continuous Intravenous Anakinra Infusion to Calm the Cytokine Storm in Macrophage Activation Syndrome. ACR Open Rheumatol. 2020, 2, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Atemnkeng Ntam, V.; Klein, A.; Horneff, G. Safety and efficacy of anakinra as first-line or second-line therapy for systemic onset juvenile idiopathic arthritis—data from the German BIKER registry. Expert Opin. Drug Saf. 2021, 20, 93–100. [Google Scholar] [CrossRef]
- Silacci, P.; Dayer, J.M.; Desgeorges, A.; Peter, R.; Manueddu, C.; Guerne, P.A. Interleukin (IL)-6 and its soluble receptor induce TIMP-1 expression in synoviocytes and chondrocytes, and block IL-1-induced collagenolytic activity. J. Biol. Chem. 1998, 273, 13625–13629. [Google Scholar] [CrossRef] [Green Version]
- Sarma, P.K.; Misra, R.; Aggarwal, A. Elevated serum receptor activator of NFkappaB ligand (RANKL), osteoprotegerin (OPG), matrix metalloproteinase (MMP)3, and ProMMP1 in patients with juvenile idiopathic arthritis. Clin. Rheumatol. 2008, 27, 289–294. [Google Scholar] [CrossRef]
- Dienz, O.; Rincon, M. The effects of IL-6 on CD4 T cell responses. Clin. Immunol. 2009, 130, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Nakagishi, Y.; Yachie, A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine 2013, 61, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Nakagishi, Y.; Inoue, N.; Mizuta, M.; Ko, G.; Saikawa, Y.; Kubota, T.; Yamasaki, Y.; Takei, S.; Yachie, A. Interleukin-18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin. Immunol. 2015, 160, 277–281. [Google Scholar] [CrossRef]
- Boe, A.; Baiocchi, M.; Carbonatto, M.; Papoian, R.; Serlupi-Crescenzi, O. Interleukin 6 knock-out mice are resistant to antigen-induced experimental arthritis. Cytokine 1999, 11, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Brunner, H.I.; Ruperto, N.; Kenwright, A.; Wright, S.; Calvo, I.; Cuttica, R.; Ravelli, A.; Schneider, R.; Woo, P.; et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2385–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, H.I.; Ruperto, N.; Zuber, Z.; Cuttica, R.; Keltsev, V.; Xavier, R.M.; Burgos-Vargas, R.; Penades, I.C.; Silverman, E.D.; Espada, G.; et al. Efficacy and Safety of Tocilizumab for Polyarticular-Course Juvenile Idiopathic Arthritis in the Open-Label Two-Year Extension of a Phase III Trial. Arthritis Rheumatol. 2021, 73, 530–541. [Google Scholar] [CrossRef]
- Mizuta, M.; Shimizu, M.; Inoue, N.; Ikawa, Y.; Nakagishi, Y.; Yasuoka, R.; Iwata, N.; Yachie, A. Clinical significance of interleukin-18 for the diagnosis and prediction of disease course in systemic juvenile idiopathic arthritis. Rheumatology 2021, 60, 2421–2426. [Google Scholar] [CrossRef]
- Volin, M.V.; Koch, A.E. Interleukin-18: A mediator of inflammation and angiogenesis in rheumatoid arthritis. J. Interferon Cytokine Res. 2011, 31, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Kaplanski, G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.W.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 cytokines: From discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef]
- Bridgewood, C.; Sharif, K.; Sherlock, J.; Watad, A.; McGonagle, D. Interleukin-23 pathway at the enthesis: The emerging story of enthesitis in spondyloarthropathy. Immunol. Rev. 2020, 294, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Tsukazaki, H.; Kaito, T. The Role of the IL-23/IL-17 Pathway in the Pathogenesis of Spondyloarthritis. Int. J. Mol. Sci. 2020, 21, 6401. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Mazzoni, A.; Cimaz, R.; Liotta, F.; Annunziato, F.; Cosmi, L. Th17 and Th1 Lymphocytes in Oligoarticular Juvenile Idiopathic Arthritis. Front. Immunol. 2019, 10, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenders, M.I.; Lubberts, E.; van de Loo, F.A.; Oppers-Walgreen, B.; van den Bersselaar, L.; Helsen, M.M.; Kolls, J.K.; Di Padova, F.E.; Joosten, L.A.; van den Berg, W.B. Interleukin-17 acts independently of TNF-alpha under arthritic conditions. J. Immunol. 2006, 176, 6262–6269. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef]
- Mahendra, A.; Misra, R.; Aggarwal, A. Th1 and Th17 Predominance in the Enthesitis-related Arthritis Form of Juvenile Idiopathic Arthritis. J. Rheumatol. 2009, 36, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Beringer, A.; Miossec, P. Systemic effects of IL-17 in inflammatory arthritis. Nat. Rev. Rheumatol. 2019, 15, 491–501. [Google Scholar] [CrossRef]
- Griffin, G.K.; Newton, G.; Tarrio, M.L.; Bu, D.X.; Maganto-Garcia, E.; Azcutia, V.; Alcaide, P.; Grabie, N.; Luscinskas, F.W.; Croce, K.J.; et al. IL-17 and TNF-alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J. Immunol. 2012, 188, 6287–6299. [Google Scholar] [CrossRef]
- De Waal Malefyt, R.; Abrams, J.; Bennett, B.; Figdor, C.G.; de Vries, J.E. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991, 174, 1209–1220. [Google Scholar] [CrossRef] [Green Version]
- Harsini, S.; Saghazadeh, A.; Nedjat, S.; Rezaei, N. Associations between interleukin-10 polymorphisms and susceptibility to juvenile idiopathic arthritis: A systematic review and meta-analysis. Eur. Cytokine Netw. 2018, 29, 16–26. [Google Scholar] [CrossRef]
- Imbrechts, M.; Avau, A.; Vandenhaute, J.; Malengier-Devlies, B.; Put, K.; Mitera, T.; Berghmans, N.; Burton, O.; Junius, S.; Liston, A.; et al. Insufficient IL-10 Production as a Mechanism Underlying the Pathogenesis of Systemic Juvenile Idiopathic Arthritis. J. Immunol. 2018, 201, 2654–2663. [Google Scholar] [CrossRef]
- Brescia, A.C.; Simonds, M.M.; Sullivan, K.E.; Rose, C.D. Secretion of pro-inflammatory cytokines and chemokines and loss of regulatory signals by fibroblast-like synoviocytes in juvenile idiopathic arthritis. Proteom. Clin. Appl. 2017, 11, 1600088. [Google Scholar] [CrossRef] [Green Version]
- Pavkova Goldbergova, M.; Lipkova, J.; Pavek, N.; Gatterova, J.; Vasku, A.; Soucek, M.; Nemec, P. RANTES, MCP-1 chemokines and factors describing rheumatoid arthritis. Mol. Immunol. 2012, 52, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Schraufstatter, I.U.; Zhao, M.; Khaldoyanidi, S.K.; Discipio, R.G. The chemokine CCL18 causes maturation of cultured monocytes to macrophages in the M2 spectrum. Immunology 2012, 135, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, Y.; Takei, S.; Imanaka, H.; Nerome, Y.; Kubota, T.; Nonaka, Y.; Akaike, H.; Takezaki, T.; Kawano, Y. Prediction of long-term remission of oligo/polyarticular juvenile idiopathic arthritis with S100A12 and vascular endothelial growth factor. Mod. Rheumatol. 2016, 26, 551–556. [Google Scholar] [CrossRef]
- Tsianakas, A.; Varga, G.; Barczyk, K.; Bode, G.; Nippe, N.; Kran, N.; Roth, J.; Luger, T.A.; Ehrchen, J.; Sunderkoetter, C. Induction of an anti-inflammatory human monocyte subtype is a unique property of glucocorticoids, but can be modified by IL-6 and IL-10. Immunobiology 2012, 217, 329–335. [Google Scholar] [CrossRef]
- Swidrowska-Jaros, J.; Smolewska, E. A fresh look at angiogenesis in juvenile idiopathic arthritis. Cent. Eur. J. Immunol. 2018, 43, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, K.W.; Kim, B.M.; Cho, M.L.; Lee, S.H. The effect of vascular endothelial growth factor on osteoclastogenesis in rheumatoid arthritis. PLoS ONE 2015, 10, e0124909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignola, S.; Picco, P.; Falcini, F.; Sabatini, F.; Buoncompagni, A.; Gattorno, M. Serum and synovial fluid concentration of vascular endothelial growth factor in juvenile idiopathic arthritides. Rheumatology 2002, 41, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.E. Angiogenesis as a target in rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Siebelt, M.; Korthagen, N.; Wei, W.; Groen, H.; Bastiaansen-Jenniskens, Y.; Muller, C.; Waarsing, J.H.; de Jong, M.; Weinans, H. Triamcinolone acetonide activates an anti-inflammatory and folate receptor-positive macrophage that prevents osteophytosis in vivo. Arthritis Res. Ther. 2015, 17, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Municio, C.; Soler Palacios, B.; Estrada-Capetillo, L.; Benguria, A.; Dopazo, A.; Garcia-Lorenzo, E.; Fernandez-Arroyo, S.; Joven, J.; Miranda-Carus, M.E.; Gonzalez-Alvaro, I.; et al. Methotrexate selectively targets human proinflammatory macrophages through a thymidylate synthase/p53 axis. Ann. Rheum. Dis. 2016, 75, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Municio, C.; Dominguez-Soto, A.; Fuentelsaz-Romero, S.; Lamana, A.; Montes, N.; Cuevas, V.D.; Campos, R.G.; Pablos, J.L.; Gonzalez-Alvaro, I.; Puig-Kroger, A. Methotrexate limits inflammation through an A20-dependent cross-tolerance mechanism. Ann. Rheum. Dis. 2018, 77, 752–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gremese, E.; Alivernini, S.; Tolusso, B.; Zeidler, M.P.; Ferraccioli, G. JAK inhibition by methotrexate (and csDMARDs) may explain clinical efficacy as monotherapy and combination therapy. J. Leukoc. Biol. 2019, 106, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, R.; Vink, J.; Dijkmans, B.A.; Assaraf, Y.G.; van Miltenburg, M.; van der Heijden, J.; Ifergan, I.; Lems, W.F.; Scheper, R.J.; Kaspers, G.J.; et al. Sulfasalazine sensitises human monocytic/macrophage cells for glucocorticoids by upregulation of glucocorticoid receptor alpha and glucocorticoid induced apoptosis. Ann. Rheum. Dis. 2007, 66, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Giancane, G.; Alongi, A.; Ravelli, A. Update on the pathogenesis and treatment of juvenile idiopathic arthritis. Curr. Opin. Rheumatol. 2017, 29, 523–529. [Google Scholar] [CrossRef]
- Onuora, S. Experimental arthritis: Anti-TNF kills the macrophage response. Nat. Rev. Rheumatol. 2018, 14, 64. [Google Scholar]
- Huang, Q.Q.; Birkett, R.; Doyle, R.; Shi, B.; Roberts, E.L.; Mao, Q.; Pope, R.M. The Role of Macrophages in the Response to TNF Inhibition in Experimental Arthritis. J. Immunol. 2018, 200, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Tono, T.; Aihara, S.; Hoshiyama, T.; Arinuma, Y.; Nagai, T.; Hirohata, S. Effects of anti-IL-6 receptor antibody on human monocytes. Mod. Rheumatol. 2015, 25, 79–84. [Google Scholar] [CrossRef]
- Obeng, J.A.; Amoruso, A.; Camaschella, G.L.; Sola, D.; Brunelleschi, S.; Fresu, L.G. Modulation of human monocyte/macrophage activity by tocilizumab, abatacept and etanercept: An in vitro study. Eur. J. Pharmacol. 2016, 780, 33–37. [Google Scholar] [CrossRef]
- Ruperto, N.; Synoverska, O.; Ting, T.; Abud-Mendoza, C.; Spindler, A.; Vyzhga, Y.; Marzan, K.; Keltsev, V.; Tirosh, I.; Imundo, L.; et al. OP0291TOFACITINIB for the treatment of polyarticular course juvenile idiopathic arthritis: Results of a phase 3, randomised, double-blind, placebo-controlled withdrawal study. Ann. Rheum. Dis. 2020, 79, 180–181. [Google Scholar] [CrossRef]
- Boyle, D.L.; Soma, K.; Hodge, J.; Kavanaugh, A.; Mandel, D.; Mease, P.; Shurmur, R.; Singhal, A.K.; Wei, N.; Rosengren, S.; et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1311–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, J.F.; Baron, M.; Constantin, A.; Degboe, Y.; Cantagrel, A.; Davignon, J.L. Anti-TNF certolizumab pegol induces antioxidant response in human monocytes via reverse signaling. Arthritis Res. Ther. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gupta, S.; Ilstad-Minnihan, A.; Ayyangar, S.; Hay, A.D.; Pascual, V.; Ilowite, N.T.; Macaubas, C.; Mellins, E.D. Interleukin-1 in monocyte activation phenotypes in systemic juvenile idiopathic arthritis: Observations from a clinical trial of rilonacept, an interleukin-1 inhibitor. Clin. Immunol. 2018, 194, 9–18. [Google Scholar] [CrossRef]
- Benedetti, F.D.; Penadés, I.C.; Pérez, N.E.R.; Maschan, A.; Quartier, P.; Żuber, Z.; Stanislav, M.; Barria, R.; Garulo, D.C.; Cornejo, G.V.; et al. FRI0549SARILUMAB, a human monoclonal antibody to the interleukin-6 (il-6) receptor, in polyarticular-course juvenile idiopathic arthritis (pcjia): A 12-week multinational open-label dose-finding study. Ann. Rheum. Dis. 2019, 78, 969–970. [Google Scholar]
- Reich, K.; Yasothan, U.; Kirkpatrick, P. Ustekinumab. Nat. Rev. Drug Discov. 2009, 8, 355–356. [Google Scholar] [CrossRef]
- Kaaij, M.H.; Helder, B.; van Mens, L.J.J.; van de Sande, M.G.H.; Baeten, D.L.P.; Tas, S.W. Anti-IL-17A treatment reduces serum inflammatory, angiogenic and tissue remodeling biomarkers accompanied by less synovial high endothelial venules in peripheral spondyloarthritis. Sci. Rep. 2020, 10, 21094. [Google Scholar] [CrossRef]
- Mease, P.J.; van der Heijde, D.; Ritchlin, C.T.; Okada, M.; Cuchacovich, R.S.; Shuler, C.L.; Lin, C.Y.; Braun, D.K.; Lee, C.H.; Gladman, D.D.; et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: Results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann. Rheum. Dis. 2017, 76, 79–87. [Google Scholar]
- Al-Salama, Z.T. Emapalumab: First Global Approval. Drugs 2019, 79, 99–103. [Google Scholar] [CrossRef]
- Bozec, A.; Luo, Y.; Engdahl, C.; Figueiredo, C.; Bang, H.; Schett, G. Abatacept blocks anti-citrullinated protein antibody and rheumatoid factor mediated cytokine production in human macrophages in IDO-dependent manner. Arthritis Res. Ther. 2018, 20, 24. [Google Scholar] [CrossRef] [Green Version]
- Wenink, M.H.; Santegoets, K.C.; Platt, A.M.; van den Berg, W.B.; van Riel, P.L.; Garside, P.; Radstake, T.R.; McInnes, I.B. Abatacept modulates proinflammatory macrophage responses upon cytokine-activated T cell and Toll-like receptor ligand stimulation. Ann. Rheum. Dis. 2012, 71, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Magnol, M.; Rauwel, B.; Sayegh, S.; Diallo, K.; Baron, M.; Lacassagne, E.; Boyer, J.-F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J.-L.; et al. AB0040JAK inhibitors—Baricitinib and tofacitinib—Modulate the in vitro inflammatory and alternative polarizations of macrophages. Ann. Rheum. Dis. 2019, 78, 1486–1487. [Google Scholar]
- Parmentier, J.M.; Voss, J.; Graff, C.; Schwartz, A.; Argiriadi, M.; Friedman, M.; Camp, H.S.; Padley, R.J.; George, J.S.; Hyland, D.; et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, T.; Tanaka, Y.; Close, D.; Godwood, A.; Wu, C.Y.; Saurigny, D. Efficacy and safety of mavrilimumab in Japanese subjects with rheumatoid arthritis: Findings from a Phase IIa study. Mod. Rheumatol. 2015, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Greven, D.E.; Cohen, E.S.; Gerlag, D.M.; Campbell, J.; Woods, J.; Davis, N.; van Nieuwenhuijze, A.; Lewis, A.; Heasmen, S.; McCourt, M.; et al. Preclinical characterisation of the GM-CSF receptor as a therapeutic target in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1924–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, F.; Fossati, G.; Lewis, E.C.; Lee, J.K.; Porro, G.; Pagani, P.; Modena, D.; Moras, M.L.; Pozzi, P.; Reznikov, L.L.; et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol. Med. 2005, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.S.; Sun, H.L.; Lii, C.K.; Chen, H.W.; Chen, P.Y.; Liu, K.L. Gamma-linolenic acid inhibits inflammatory responses by regulating NF-kappaB and AP-1 activation in lipopolysaccharide-induced RAW 264.7 macrophages. Inflammation 2010, 33, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Y.; Zhu, W.; Ma, C.; Ruan, J.; Long, H.; Wang, Y. Sinomenine Inhibits the Progression of Rheumatoid Arthritis by Regulating the Secretion of Inflammatory Cytokines and Monocyte/Macrophage Subsets. Front. Immunol. 2018, 9, 2228. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, H.; Wang, H.; Chiu, Y.G.; Wang, M.; Ritchlin, C.T.; Kiernan, A.; Boyce, B.F.; Xing, L. Targeting Notch-Activated M1 Macrophages Attenuates Joint Tissue Damage in a Mouse Model of Inflammatory Arthritis. J. Bone Miner. Res. 2017, 32, 1469–1480. [Google Scholar] [CrossRef]
- Sultana, F.; Neog, M.K.; Rasool, M. Withaferin-A, a steroidal lactone encapsulated mannose decorated liposomes ameliorates rheumatoid arthritis by intriguing the macrophage repolarization in adjuvant-induced arthritic rats. Colloids Surf. B. Biointerfaces 2017, 155, 349–365. [Google Scholar] [CrossRef]
- Yu, Y.; Cai, W.; Zhou, J.; Lu, H.; Wang, Y.; Song, Y.; He, R.; Pei, F.; Wang, X.; Zhang, R.; et al. Anti-arthritis effect of berberine associated with regulating energy metabolism of macrophages through AMPK/ HIF-1alpha pathway. Int. Immunopharmacol. 2020, 87, 106830. [Google Scholar] [CrossRef]
- Abdel-Maged, A.E.; Gad, A.M.; Wahdan, S.A.; Azab, S.S. Efficacy and safety of Ramucirumab and methotrexate co-therapy in rheumatoid arthritis experimental model: Involvement of angiogenic and immunomodulatory signaling. Toxicol. Appl. Pharmacol. 2019, 380, 114702. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Maged, A.E.; Gad, A.M.; Abdel-Aziz, A.K.; Aboulwafa, M.M.; Azab, S.S. Comparative study of anti-VEGF Ranibizumab and Interleukin-6 receptor antagonist Tocilizumab in Adjuvant-induced Arthritis. Toxicol. Appl. Pharmacol. 2018, 356, 65–75. [Google Scholar] [CrossRef]
- Shankar, J.; Thippegowda, P.B.; Kanum, S.A. Inhibition of HIF-1alpha activity by BP-1 ameliorates adjuvant induced arthritis in rats. Biochem. Biophys. Res. Commun. 2009, 387, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Feng, Z.; Chen, S.; Zhu, J.; Wu, X.; Chen, X.; Li, J. Taxol alleviates collagen-induced arthritis in mice by inhibiting the formation of microvessels. Clin. Rheumatol. 2019, 38, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Zhang, T.; Chen, J.; Cheng, W.; Chen, J.; Zheng, Z.; Lin, J.; Zhu, G.; Zhang, Y.; Bai, X.; et al. Downregulation of Hypoxia-Inducible Factor-1alpha by RNA Interference Alleviates the Development of Collagen-Induced Arthritis in Rats. Mol. Ther. Nucleic. Acids 2020, 19, 1330–1342. [Google Scholar] [CrossRef]
- Barrera, P.; Blom, A.; van Lent, P.L.; van Bloois, L.; Beijnen, J.H.; van Rooijen, N.; de Waal Malefijt, M.C.; van de Putte, L.B.; Storm, G.; van den Berg, W.B. Synovial macrophage depletion with clodronate-containing liposomes in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- Shin, T.H.; Kim, H.S.; Kang, T.W.; Lee, B.C.; Lee, H.Y.; Kim, Y.J.; Shin, J.H.; Seo, Y.; Won Choi, S.; Lee, S.; et al. Human umbilical cord blood-stem cells direct macrophage polarization and block inflammasome activation to alleviate rheumatoid arthritis. Cell Death Dis. 2016, 7, e2524. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Macrophages | Classically Activated (M1) | Alternatively Activated (M2) | ||
|---|---|---|---|---|
| M2a | M2b | M2c | ||
| Mediators of polarization | LPS, IFNγ, TNFα, GM-CSF | IL-4, IL-13 | ICs + TLR/IL-1β | IL-10, TGFβ, steroid |
| Surface markers | CD68, CD80, CD86, IL-1R, TLR2, TLR4, iNOs, IFNγR, MHC-IIhigh | CD200R, CD206/MMR, IL-1RII, Dectin-1, MHC-IIlow | CD86, MHC-IIlow | CD163, TLR1, TLR8 |
| Transcription factors and cellular markers | NFκB, STAT1, STAT5, IRF3, IRF5 | IRF4, PPARγ, STAT6 | IRF4, SOCS3 | IRF4, SOCS3 |
| Produced cytokines | IL-1α, IL-1β, IL-6, IL-12, IL-18, TNFα, M-CSF | IL-10, TGFβ, IL-1Ra | IL-1β, IL-6, IL-10, TNFα | IL-10, TGFβ |
| Produced chemokines | CXCL9, CXCL10, CXCL11 | CCL17, CCL18, CCL22, CCL24 | CCL1, CCL20, CXCL1, CXCL2, CXCL3 | CCL16, CCL18 |
| Features | Proinflammatory, microbicidal, and tumoricidal | endocytic activity, tissue remodeling, and repair | immunoregulation | immunoregulation |
| Agent | Targets | Actions | Developmental Stage | Ref. |
|---|---|---|---|---|
| Etanercept | TNF receptor | Blocks TNFα signaling; shifts Mos/Mφs toward an anti-inflammatory phenotype and induces apoptosis of Mos/Mφs | JIA–marketing | [61,62] |
| Adalimumab | TNF | Blocks TNFα signaling; shifts Mos/Mφs toward an anti-inflammatory phenotype; induces apoptosis of Mos/Mφs and reduces Mo migration into the joint | polyarticular JIA–marketing | [63,149,154] |
| Infliximab | TNF | Blocks TNFα signaling; shifts Mos/Mφs toward an anti-inflammatory phenotype and induces apoptosis of Mos/Mφs; increases circulating nonclassical Mos and decreases circulating classical Mos; reduces CCR2 and CXCR4 expression on the nonclassical Mo subpopulation | JIA–marketing | [62,63,64] |
| Certolizumab | TNF | Blocks TNFα signaling; induces HO-1 mRNA and protein production in Mos; inhibits IL-1β production at the mRNA and protein level upon LPS stimulation | polyarticular JIA–phase III | [154] |
| Anakinra | IL-1β receptor | Blocks IL-1β signaling | sJIA–marketing | [109] |
| Canakinumab | IL-1β | Blocks IL-1β signaling | sJIA–marketing | [106,107] |
| Rilonacept | IL-1β/IL-1α | Blocks IL-1 signaling; skews Mos toward an alternatively activated phenotype | sJIA–phase II | [155] |
| Tocilizumab | IL-6 receptor | Blocks IL-6 signaling; shifts Mos/Mφs toward an anti-inflammatory phenotype and induces apoptosis of Mos | sJIA/polyarticular JIA–marketing | [150,151] |
| Sarilumab | IL-6 receptor | Blocks IL-6 signaling | polyarticular JIA–phase II | [156] |
| Ustekinumab | IL-12/IL-23 | Blocks IL-12/IL-23 signaling | psoriatic arthritis–marketing | [157] |
| Secukinumab | IL-17A | Blocks IL-17 signaling; decreases serum IL-6, S100A8, S100A9, VEGF, TNFα, osteopontin, and MMP | ERA/juvenile psoriatic arthritis –phase III | [158] |
| Ixekizumab | IL-17A | Blocks IL-17 signaling | ERA/juvenile psoriatic arthritis –phase III | [159] |
| Emapalumab | IFNγ | Blocks IFNγ signaling | sJIA–phase II | [160] |
| Abatacept | CTLA-4 | Blocks ACPA and RF mediated cytokine production in human Mφs; modulates proinflammatory Mφ responses upon cytokine-activated T cell and TLR stimulation | Polyarticular JIA–marketing | [161,162] |
| Tofacitinib | JAK1/JAK3 | Small molecule that abrogates TNF- induced STAT1 activation; inhibits proinflammatory mediator production | polyarticular JIA–marketing | [153] |
| Baricitinib | JAK1/JAK2 | Decreases expression of the inflammatory IP-10 and increases IL-10 production | JIA–phase III | [163] |
| Upadacitinib | JAK1 | Selectively targets JAK1 dependent disease drivers such as IL-6 and IFNγ | Polyarticular JIA–phase I | [164] |
| Mavrilimumab (CAM-3001) | GM-CSF receptor α | Blocks GM-CSF signaling and classically activated polarization | RA–phase IIb | [165] |
| Otilimab (MOR103) | GM-CSF | Blocks GM-CSF signaling and classically activated polarization | RA–phase III | [166] |
| Givinostat (ITF2357) | histone deacetylase inhibitor | Prevents LPS-induced TNFα gene transcription and secretion of IL-1β | JIA–phase II | [167] |
| Gamma-linolenic acid | n-6 polyunsaturated fatty acids | Inhibits inflammatory responses through inactivation of NFκB and AP-1 by suppressing oxidative stress and the ERK and JNK signal transduction pathways in LPS-induced Mφs | JIA-phase I | [168] |
| Sinomenine | plant alkaloid | Attenuates CD11b+F4/80+CD64+ resident Mφs in the synovial tissue and reduces number of CD14+CD16+ circulating Mos | Herbal medicine | [169] |
| Thapsigargin | inhibitor of SERCA | Decreases the number of TNF-induced classically activated Mφs and increases the number of alternatively activated Mφs | preclinical | [170] |
| Withaferin-A | steroidal lactone | Promotes classically activated Mφ to alternatively activated Mφ repolarization | preclinical | [171] |
| Berberine | antimicrobial agent | Increases the proportion of alternatively activated Mφs and decreases the proportion of classically activated Mφs, downregulates HIF-1α expression in synovial Mφs | Preclinical | [172] |
| Ramucirumab | VEGF | Blocks VEGF signaling | preclinical | [173] |
| Ranibizumab | VEGF | Blocks VEGF signaling | preclinical | [174] |
| 2-benzoyl-phenoxy acetamide | benzophenone analog | Targets VEGF and HIF-1α | preclinical | [175] |
| Paclitaxel (PTX) | tubulin, chemotherapy | Targets VEGF and HIF-1α | preclinical | [176] |
| pLVX-shRNA-HIF-1α | shRNA targeting HIF-1α | Inhibits HIF-1α and VEGF expression, leading to decreased proinflammatory cytokine expression | Preclinical | [177] |
| Clodronate liposomes | release chlorophosphate | Mφ depletion | Preclinical | [178] |
| Human umbilical cord blood-derived mesenchymal stem cells | stem cells | Polarizes naive Mφs toward an alternatively activated phenotype | preclinical | [179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-Y.; Yang, H.-Y.; Huang, J.-L.; Lai, J.-H. Signals and Mechanisms Regulating Monocyte and Macrophage Activation in the Pathogenesis of Juvenile Idiopathic Arthritis. Int. J. Mol. Sci. 2021, 22, 7960. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157960
Wu C-Y, Yang H-Y, Huang J-L, Lai J-H. Signals and Mechanisms Regulating Monocyte and Macrophage Activation in the Pathogenesis of Juvenile Idiopathic Arthritis. International Journal of Molecular Sciences. 2021; 22(15):7960. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157960
Chicago/Turabian StyleWu, Chao-Yi, Huang-Yu Yang, Jing-Long Huang, and Jenn-Haung Lai. 2021. "Signals and Mechanisms Regulating Monocyte and Macrophage Activation in the Pathogenesis of Juvenile Idiopathic Arthritis" International Journal of Molecular Sciences 22, no. 15: 7960. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157960