
Characteristics of TIMP1, CD63, and β1-Integrin and the Functional Impact of Their Interaction in Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
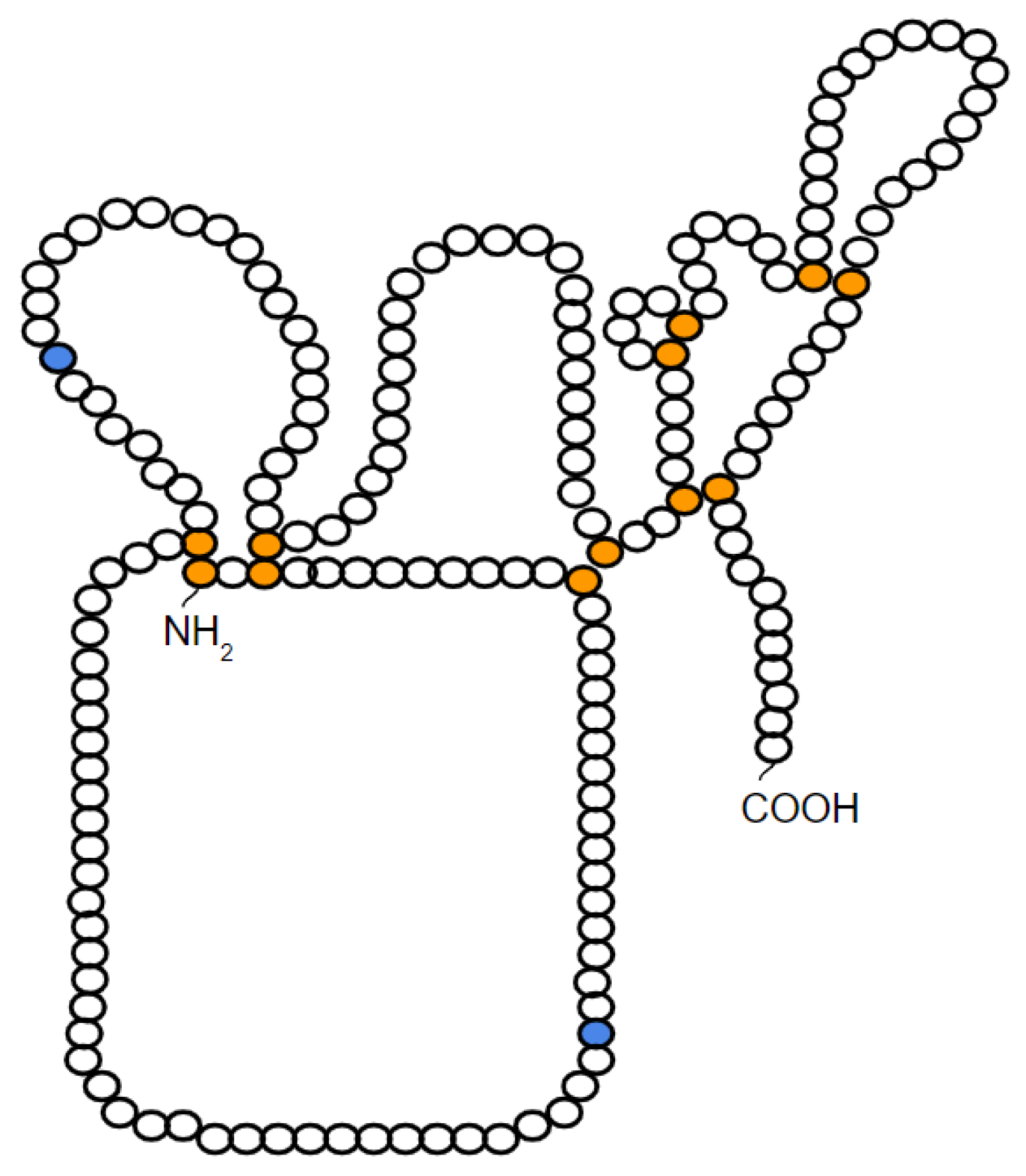
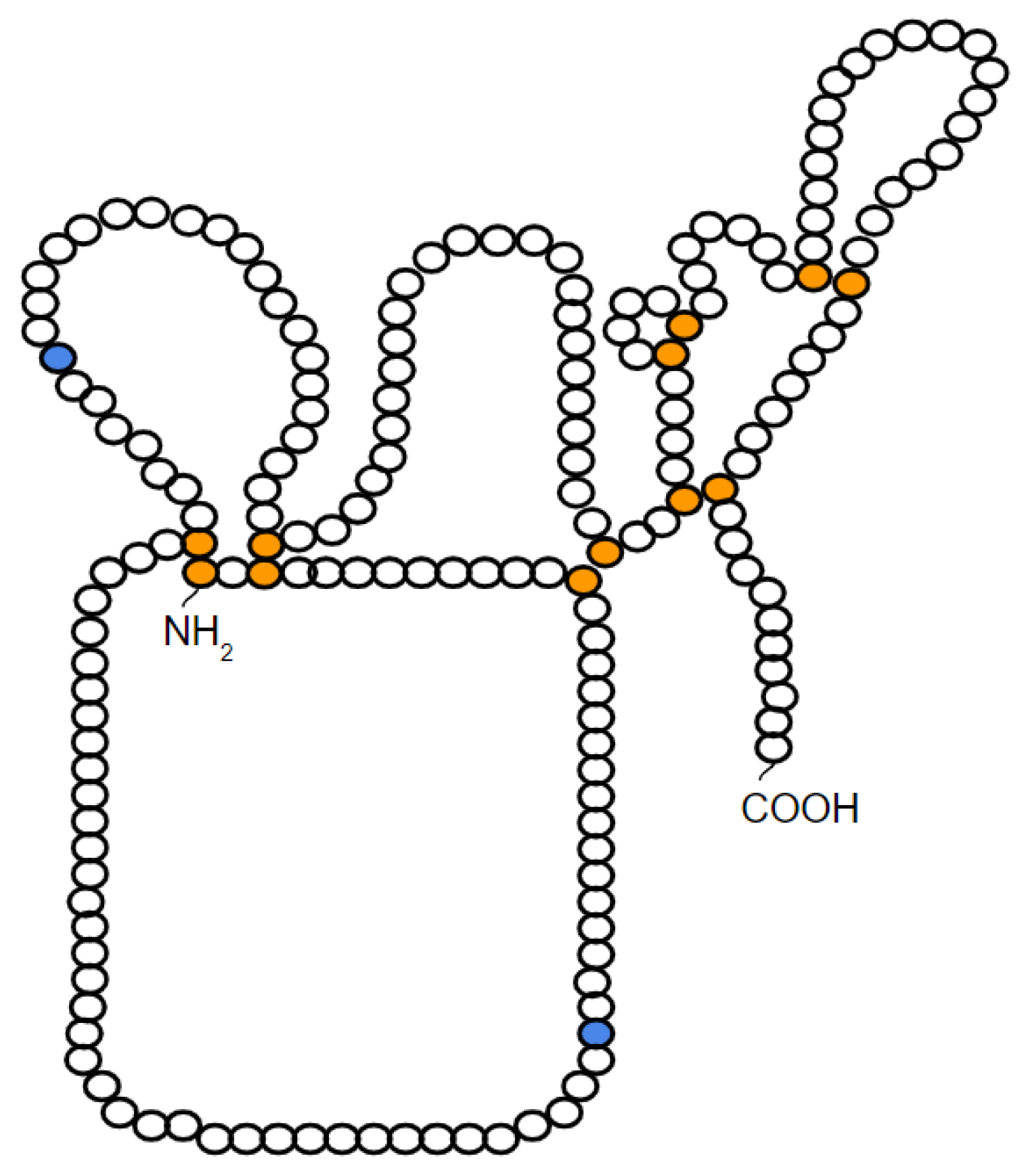
2. Characteristics and Structure of TIMP-1
3. TIMP-1 as a Multifunctional Protein
3.1. Inhibitory Activity of Matrix Metalloproteases (MMPs)
3.2. Activation of Intracellular Signaling Pathways
3.2.1. Cell Growth
3.2.2. Cell Survival and Apoptosis
3.2.3. Cell Proliferation and Differentiation
4. Association between TIMP-1 and CD63
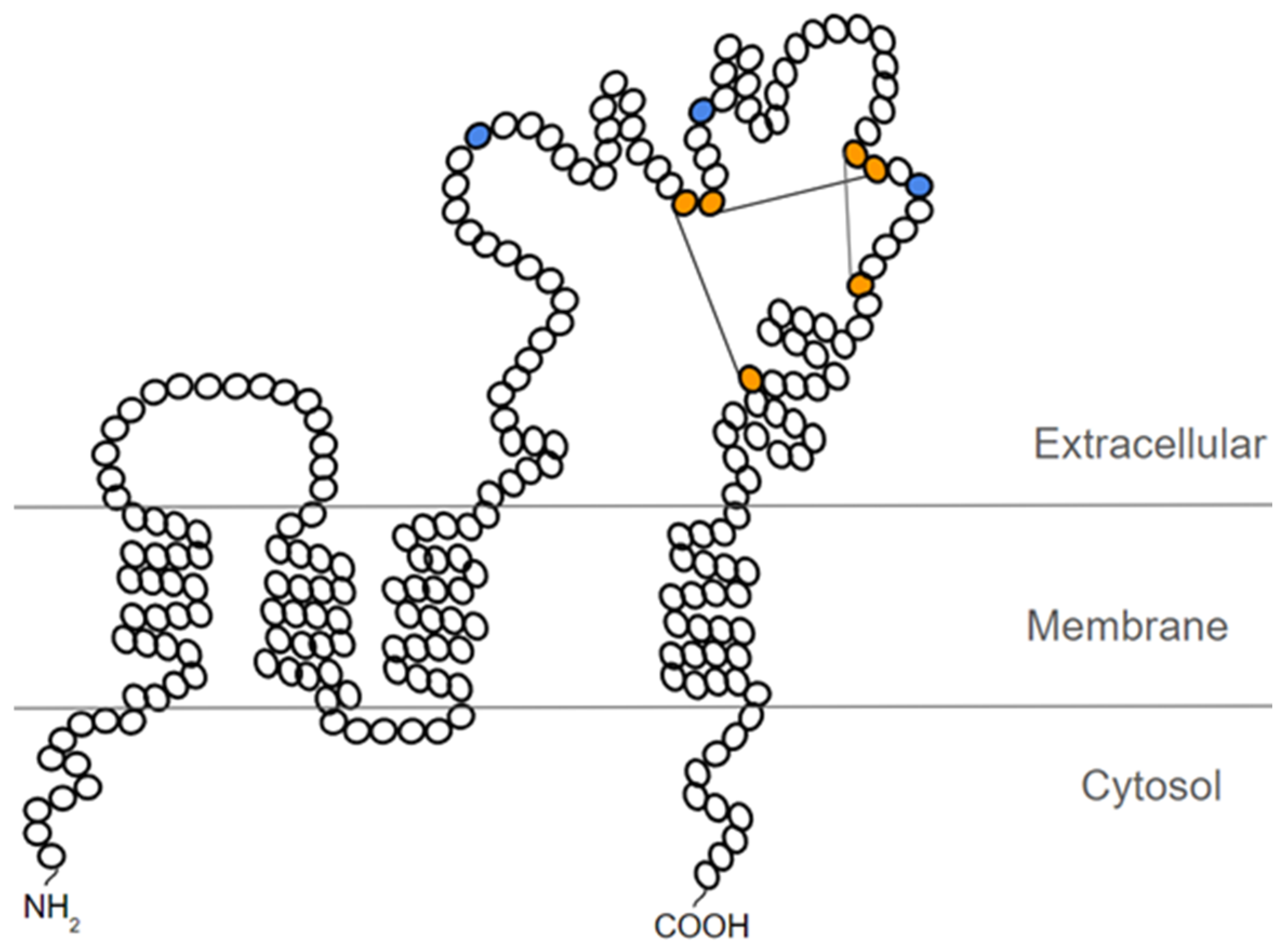
5. Characteristics and Structure of CD63
6. Functions of Tetraspanins and CD63
7. Binding Partners of Tetraspanins and CD63
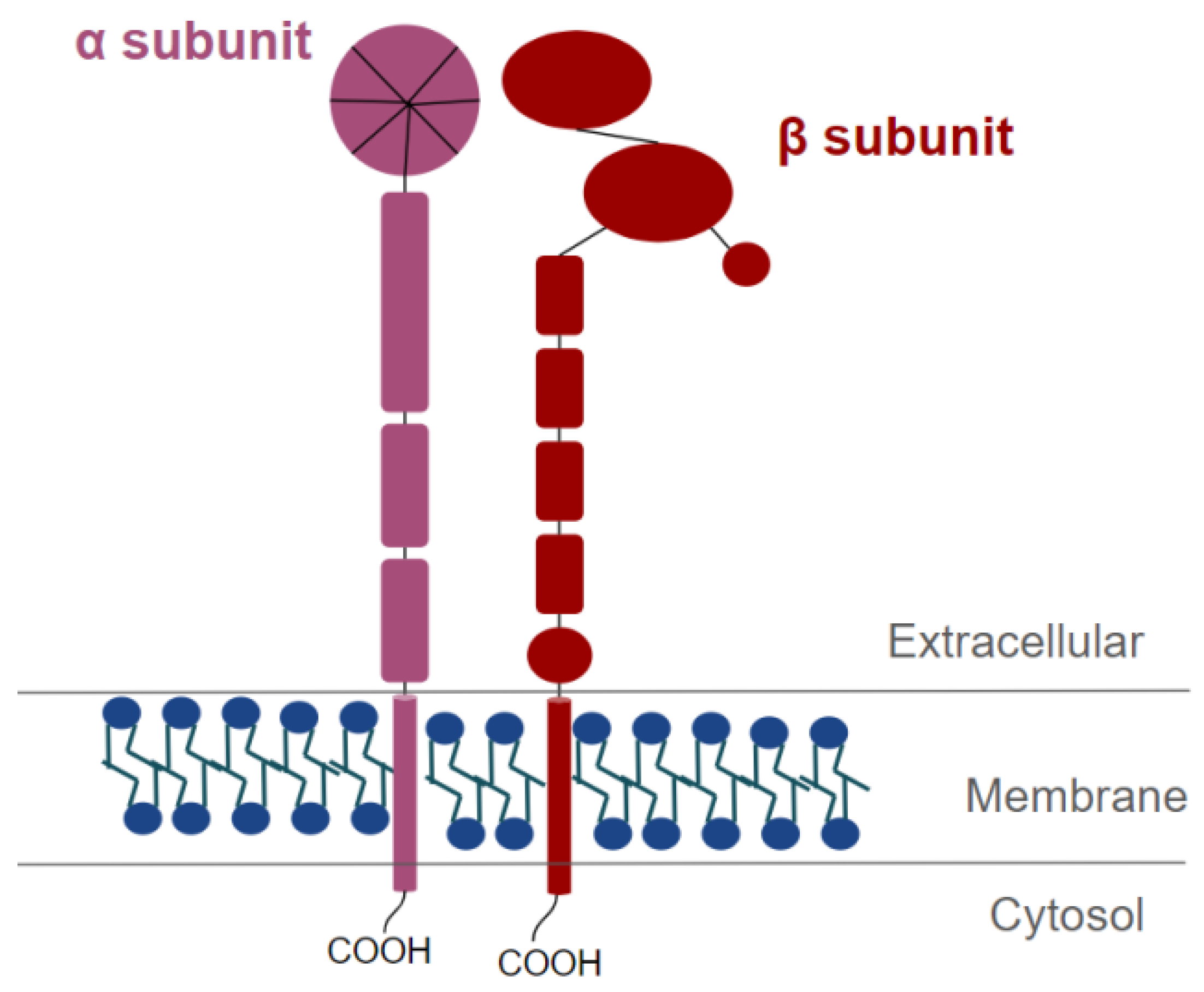
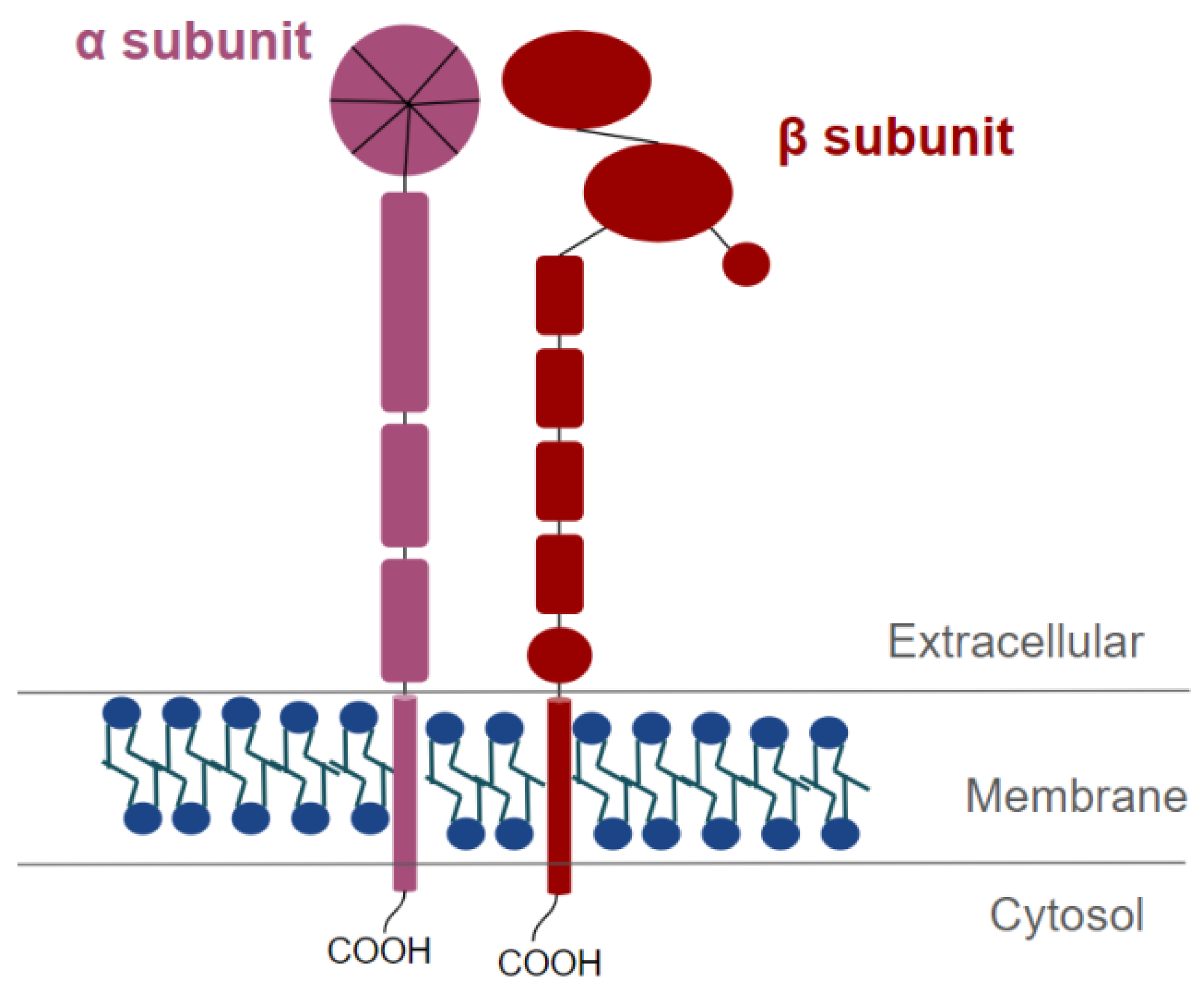
8. Characteristics and Structure of Integrins
9. Functions of Integrins
9.1. Cytoskeleton Rearrangement and Cell Motility
9.2. Integrin-Induced Signal Transduction Pathways
10. Integrin Binding Partners
10.1. Extracellular Binding Partners
10.2. Cytoplasmic Binding Partners
10.3. Cell Membrane Binding Partners
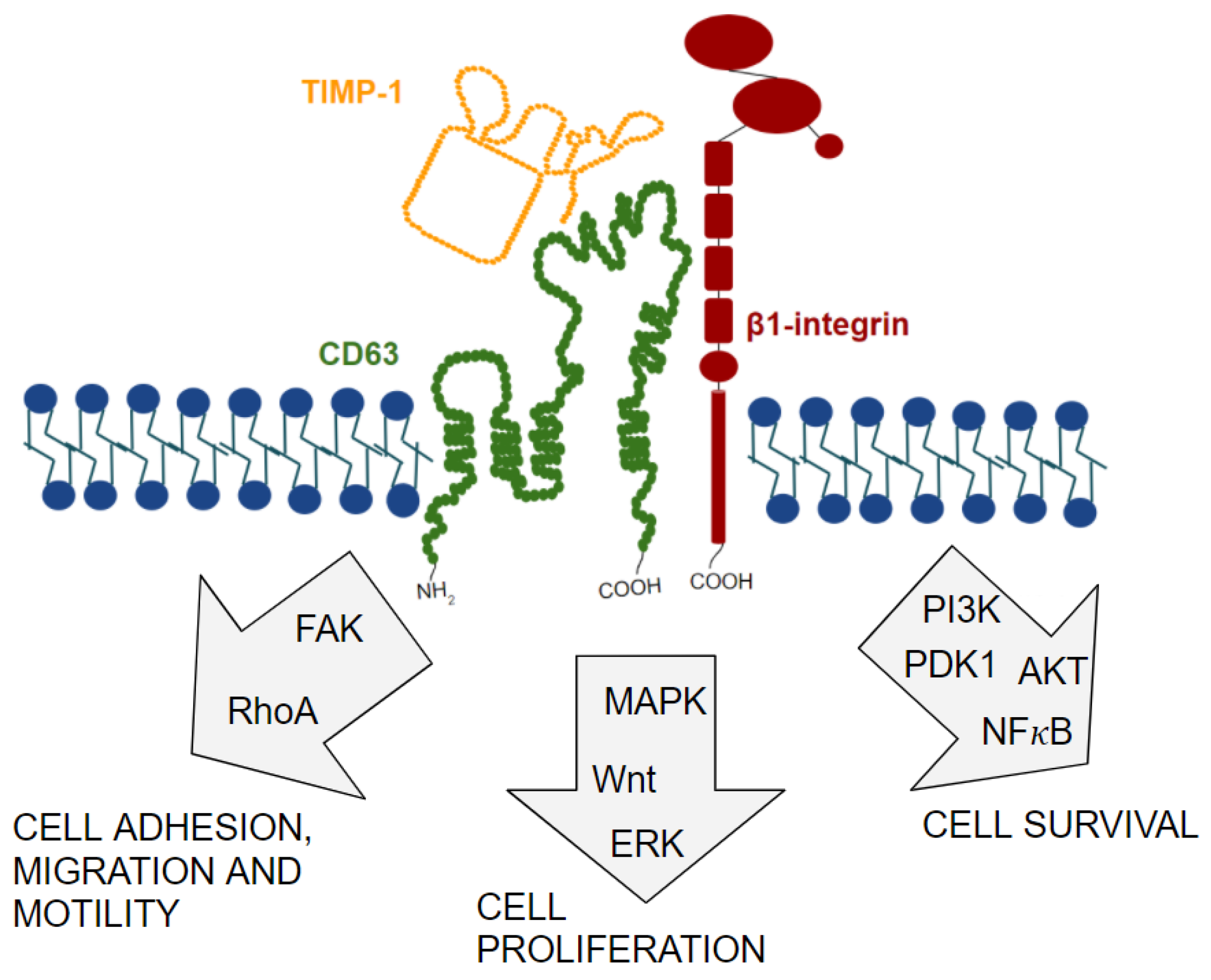
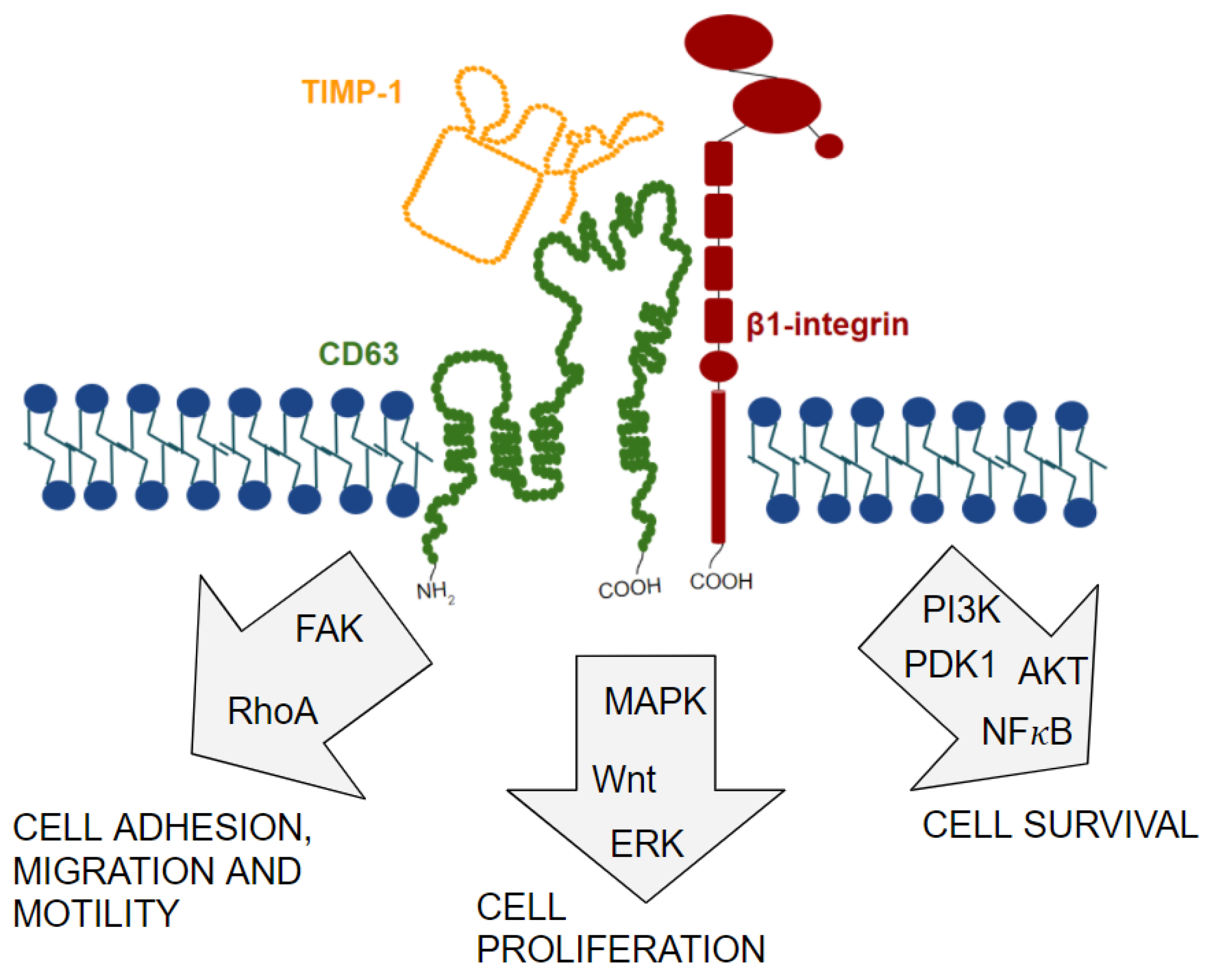
11. TIMP-1/CD63/β1-Integrin Complex Formation and Signal Transduction Pathways Involved
12. Discovery of TIMP-1/CD63/β1-Integrin Complex in Cancer and Its Role in Tumor Progression
13. N-Glycosylation as a Possible Regulator of TIMP-1/CD63/β1-Integrin Complex Formation in Cancer
14. Discussion and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKT (also PKB) | Protein Kinase B |
| BAD | BCL-2 Associated Agonist of Cell Death |
| BCL | B-Cell Lymphoma |
| EGF | Epidermal Growth Factor |
| EPA | Erythroid Potentiating Activity |
| ERK | Extracellular signal-Regulated Kinases |
| FAK | Focal Adhesion Kinase |
| FGF | Fibroblast Growth Factor |
| GlcNAc | N-AcetylGlucosamine |
| HBMECs | Human Brain Microvessel Endothelial Cells |
| hNSCs | Human Neural Stem Cells |
| HSPCs | Hematopoietic Stem and Progenitor Cells |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| ILK | Integrin-Linked Kinase |
| IL | Interleukin |
| JAK | Janus Kinase |
| JNK | c-Jun N-terminal Kinase |
| LEL | Large Extracellular Loop |
| MAPK | Mitogen Activated Protein Kinases |
| GnT-V | β1-6-N-acetylglucosaminyltransferase V |
| MHC | Major Histocompatibility Complex |
| MMPs | Matrix Metalloproteases |
| MPIs | Synthetic Metalloprotease Inhibitors |
| MVBs | Multivesicular Bodies |
| PDGF | Platelet-derived Growth Factor |
| PI3K | Phosphatidylinositol-3-kinase |
| PLC | Phospholipase C |
| SEL | Small Extracellular Loop |
| TAZ | Transcriptional Coactivator with PDZ-binding Motif |
| TCR | T-Cell Receptor |
| TEM | Tetraspanin-Enriched Microdomains |
| TIMP | Tissue Inhibitor of Metalloproteases |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| YAP | Yes-Associated Protein |
| WPB | Weibel-Palade bodies |
References
- Woessner, J.F., Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991, 5, 2145–2154. [Google Scholar] [CrossRef] [Green Version]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys. Acta 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Chambers, A.F.; Matrisian, L.M. Changing views of the role of matrix metalloproteinases in metastasis. J. Natl. Cancer Inst. 1997, 89, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Clerck, Y.A.; Perez, N.; Shimada, H.; Boone, T.C.; Langley, K.E.; Taylor, S.M. Inhibition of invasion and metastasis in cells transfected with an inhibitor of metalloproteinases. Cancer Res. 1992, 52, 701–708. [Google Scholar]
- Wang, M.; Liu, Y.E.; Greene, J.; Sheng, S.; Fuchs, A.; Rosen, E.M.; Shi, Y.E. Inhibition of tumor growth and metastasis of human breast cancer cells transfected with tissue inhibitor of metalloproteinase. Oncogene 1997, 14, 2767–2774. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; So, I.; Lee, S.C.; Lee, J.H.; Lee, C.W.; Kim, W.M.; Park, M.K.; Lee, S.T.; Park, D.Y.; Shin, D.Y.; et al. Suppression of distant pulmonary metastasis of MDA-MB 435 human breast carcinoma established in mammary fat pads of nude mice by retroviral-mediated TIMP-2 gene transfer. J. Gene Med. 2005, 7, 145–157. [Google Scholar] [CrossRef]
- Brown, P.D. Matrix metalloproteinase inhibitors. Breast Cancer Res. Treat. 1998, 52, 125–136. [Google Scholar] [CrossRef]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef]
- Lu, X.Q.; Levy, M.; Weinstein, I.B.; Santella, R.M. Immunological Quantitation of Levels of Tissue Inhibitor of Metalloproteinase-1 in Human Colon Cancer. Cancer Res. 1991, 51, 6231–6235. [Google Scholar]
- Yamashita, K.; Suzuki, M.; Iwata, H.; Koike, T.; Hamaguchi, M.; Shinagawa, A.; Noguchi, T.; Hayakawa, T. Tyrosine phosphorylation is crucial for growth signaling by tissue inhibitors of metalloproteinases TIMP-1 and TIMP-2. FEBS Lett. 1996, 396, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.I.; Park, I.C.; Hong, W.S.; Son, Y.S.; Lee, S.H.; Lee, J.I.; Choi, D.W.; Moon, N.M.; Choe, T.B.; Jang, J.J. Overexpression of tissue inhibitors of metalloproteinase-1 and -2 in the stroma of gastric cancer. J. Korean Med. Sci. 1996, 11, 474–479. [Google Scholar] [CrossRef] [Green Version]
- Guedez, L.; Stetler-Stevenson, W.G.; Wolff, L.; Wang, J.; Fukushima, P.; Mansoor, A.; Stetler-Stevenson, M. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J. Clin. Investig. 1998, 102, 2002–2010. [Google Scholar] [CrossRef]
- Li, G.; Fridman, R.; Kim, H.R. Tissue inhibitor of metalloproteinase-1 inhibits apoptosis of human breast epithelial cells. Cancer Res. 1999, 59, 6267–6275. [Google Scholar]
- Wang, T.; Yamashita, K.; Iwata, K.; Hayakawa, T. Both tissue inhibitors of metalloproteinases-1 (TIMP-1) and TIMP-2 activate Ras but through different pathways. Biochem. Biophys. Res. Commun. 2002, 296, 201–205. [Google Scholar] [CrossRef]
- Lee, S.J.; Yoo, H.J.; Bae, Y.S.; Kim, H.J.; Lee, S.T. TIMP-1 inhibits apoptosis in breast carcinoma cells via a pathway involving pertussis toxin-sensitive G protein and c-Src. Biochem. Biophys. Res. Commun. 2003, 312, 1196–1201. [Google Scholar] [CrossRef]
- Ricca, T.I.; Liang, G.; Suenaga, A.P.M.; Han, S.W.; Jones, P.A.; Jasiulionis, M.G. Tissue inhibitor of metalloproteinase 1 expression associated with gene demethylation confers anoikis resistance in early phases of melanocyte malignant transformation. Transl. Oncol. 2009, 2, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Xu, S.; Zhang, H.; Wang, Y.; Xiao, C.; Jiang, T.; Wu, L.; Zhang, T.; Sun, X.; Zhong, L.; et al. TIMP-1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J. Exp. Clin. Cancer Res. 2016, 35, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.K.; Liu, X.W.; Chirco, R.; Fridman, R.; Kim, H.R.C. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 2006, 25, 3934–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caterina, N.C.M.; Windsor, L.J.; Bodden, M.K.; Yermovsky, A.E.; Taylor, K.B.; Birkedal-Hansen, H.; Engler, J.A. Glycosylation and NH2-terminal domain mutants of the tissue inhibitor of metalloproteinases-1 (TIMP-1). Biochim. Biophys. Acta 1998, 1388, 21–34. [Google Scholar] [CrossRef]
- Lambert, E.; Dassé, E.; Haye, B.; Petitfrère, E. TIMPs as multifacial proteins. Crit. Ver. Oncol. Hematol. 2004, 49, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Orucevic, A.; Li, M.; Gorelik, E. Nitric oxide (NO), methylation and TIMP-1 expression in BL6 melanoma cells transfected with MHC class I genes. Clin. Exp. Metastasis 2000, 18, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, C.A.; Gasson, J.C.; Gerber, S.E.; Selsted, M.E.; Golde, D.W. Purification and characterization of human T-lymphocyte-derived erythroid-potentiating activity. J. Biol. Chem. 1984, 259, 9992–9996. [Google Scholar] [CrossRef]
- Bertaux, B.; Hornebeck, W.; Eisen, A.Z.; Dubertret, L. Growth stimulation of human keratinocytes by tissue inhibitor of metaloproteinases. J. Investig. Dermatol. 1991, 97, 679–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, T.; Yamashita, K.; Tanzawa, K.; Uchijima, E.; Iwata, K. Growth-promoting activity of tissue inhibitor of metalloproteinases-1 (TIMP-1) for a wide range of cells A possible new growth factor in serum. FEBS Lett. 1992, 298, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Lambert, E.; Boudot, C.; Kadri, Z.; Soula-Rothhut, M.; Sowa, M.L.; Mayeux, P.; Hornebeck, W.; Haye, B.; Petitfrere, E. Tissue inhibitor of metalloproteinases-1 signalling pathway leading to erythroid cell survival. Biochem. J. 2003, 372, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.W.; Bernardo, M.M.; Fridman, R.; Kim, H.R.C. Tissue Inhibitor of Metalloproteinase-1 Protects Human Breast Epithelial Cells Against Intrinsic Apoptotic Cell Death via the Focal Adhesion Kinase/Phosphatidylinositol 3-Kinase and MAPK Signaling Pathway. J. Biol. Chem. 2003, 278, 40364–40372. [Google Scholar] [CrossRef] [Green Version]
- Petitfrère, E.; Kadri, Z.; Boudot, C.; Sowa, M.L.; Mayeux, P.; Haye, B.; Billat, C. Involvement of the p38 mitogen-activated protein kinase pathway in tissue inhibitor of metalloproteinases-1-induced erythroid differentiation. FEBS Lett. 2000, 485, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Warner, R.B.; Najy, A.J.; Jung, Y.S.; Fridman, R.; Kim, S.; Kim, H.R.C. Establishment of Structure-function Relationship of tissue Inhibitor of Metalloproteinase-1 for Its Interaction with CD63: Implication for cancer therapy. Sci. Rep. 2020, 10, 2099. [Google Scholar] [CrossRef]
- Boucheix, C.; Rubinstein, E. Tetraspanins. Cell. Mol. Life Sci. 2001, 58, 1189–1205. [Google Scholar] [CrossRef]
- Wright, M.D.; Tomlinson, M.G. The ins and outs of the transmembrane 4 superfamily. Immunol. Today 1994, 15, 588–594. [Google Scholar] [CrossRef]
- Berditchevski, F. Complexes of tetraspanins with integrins: More than meets the eye. J. Cell Sci. 2001, 114, 4143–4151. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [Green Version]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef]
- Metzelaar, M.J.; Wijngaardg, P.L.J.; Petersll, P.J.; Sixma, J.J.; Nieuwenhuis, H.K.; Clevers, H.C. CD63 Antigen, a novel lysosomal membrane glycoprotein cloned by a screening procedure for intracellular antigens in eukaryotic cells. J. Biol. Chem. 1991, 266, 3239–3245. [Google Scholar] [CrossRef]
- Peters, P.J.; Borst, J.; Oorschot, V.; Fukuda, M.; Krähenbühl, O.; Tschopp, J.; Slot, J.W.; Geuze, H.J. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J. Exp. Med. 1991, 173, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Escola, J.M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.M.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspanin proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef] [Green Version]
- Heijnen, H.F.; Debili, N.; Vainchencker, W.; Breton-Gorius, J.; Geuze, H.J.; Sixma, J.J. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood 1998, 91, 2313–2325. [Google Scholar] [CrossRef] [PubMed]
- Arribas, M.; Cutler, D.F. Weibel-Palade body membrane proteins exhibit differential trafficking after exocytosis in endothelial cells. Traffic 2000, 1, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Marks, M.S.; Cutler, D.F. Lysosome-related organelles: Driving post-Golgi compartments into specialization. Curr. Opin. Cell Biol. 2007, 19, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Stoorvogel, W.; Kleijmeer, M.J.; Geuze, H.J.; Raposo, G. The biogenesis and functions of exosomes. Traffic 2002, 3, 321–330. [Google Scholar] [CrossRef]
- Yunta, M.; Lazo, P.A. Tetraspanin proteins as organisers of membrane microdomains and signalling complexes. Cell. Signal. 2003, 15, 559–564. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdés, M.; Sánchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef]
- Tarrant, J.M.; Robb, L.; Spriel, A.B.; Wright, M.D. Tetraspanins: Molecular organisers of the leukocyte surface. Trends Immunol. 2003, 24, 610–617. [Google Scholar] [CrossRef]
- Ýañes-Mó, M.; Mittelbrunn, M.; Sánchez-Madrid, F. Tetraspanins and intercellular Interactions. Microcirculation 2001, 8, 153–168. [Google Scholar] [CrossRef]
- Azorsa, D.O.; Hyman, J.A.; Hildreth, J.E. CD63/Pltgp40: A platelet activation antigen identical to the stage-specific, melanoma-associated antigen ME491. Blood 1991, 78, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildreth, J.E.; Derr, D.; Azorsa, D.O. Characterization of a novel self-associating Mr 40,000 platelet glycoprotein. Blood 1991, 77, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israels, S.J.; McMillan-Ward, E.M. CD63 modulates spreading and tyrosine phosphorylation of platelets on immobilized fibrinogen. Thromb. Haemost. 2005, 93, 311–318. [Google Scholar] [CrossRef]
- Doyle, E.L.; Ridger, V.; Ferraro, F.; Turmaine, M.; Saftig, P.; Cutler, D.F. CD63 is an essential cofactor to leukocyte recruitment by endothelial P-selectin. Blood 2011, 118, 4265–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tugues, S.; Honjo, S.; König, C.; Padhan, N.; Kroon, J.; Gualandi, L.; Li, X.; Barkefors, I.; Thijssen, V.L.; Griffioen, A.W.; et al. Tetraspanin CD63 promotes vascular endothelial growth factor receptor 2- β1-integrin complex formation, thereby regulating activation and downstream signaling in endothelial cells in vitro and in vivo. J. Biol. Chem. 2013, 288, 19060–19071. [Google Scholar] [CrossRef] [Green Version]
- Pfistershammer, K.; Majdic, O.; Stöckl, J.; Zlabinger, G.; Kirchberger, S.; Steinberger, P.; Knapp, W. CD63 as an activation-linked T cell costimulatory element. J. Immunol. 2004, 173, 6000–6008. [Google Scholar] [CrossRef] [Green Version]
- Mannion, B.A.; Berditchevski, F.; Kraeft, S.K.; Chen, L.B.; Hemler, M.E. Transmembrane-4 superfamily proteins CD81 (TAPA-1), CD82, CD63 and CD53 specifically associate with α4β1 integrin. J. Immunol. 1996, 157, 2039–2047. [Google Scholar]
- Serru, V.; Naour, F.L.; Billard, M.; Azorsa, D.O.; Lanza, F.; Boucheix, C.; Rubinstein, E. Selective tetraspan-integrin complexes (CD81/α4β1, CD151/α3β1, D151/α6β1) under conditions disrupting tetraspan interactions. Biochem. J. 1999, 340, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Lozahic, S.; Christiansen, D.; Manié, S.; Gerlier, D.; Billard, M.; Boucheix, C.; Rubinstein, E. CD46 (membrane cofactor protein) associates with multiple beta1 integrins and tetraspans. Eur. J. Immunol. 2000, 30, 900–907. [Google Scholar] [CrossRef]
- Park, K.R.; Inoue, T.; Ueda, M.; Hirano, T.; Higuchi, T.; Maeda, M.; Konishi, I.; Fujiwara, H.; Fujii, S. CD9 is expressed on human endometrial epithelial cells in association with integrins alpha(6), alpha(3) and beta(1). Mol. Hum. Reprod. 2000, 6, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Israels, S.J.; McMillan-Ward, E.M.; Easton, J.; Robertson, C.; McNicol, A. CD63 associates with the alphaIIb beta3 integrin-CD9 complex on the surface of activated platelets. Thromb. Haemost. 2001, 85, 134–141. [Google Scholar]
- Berditchevski, F.; Zutter, M.M.; Hemler, M.E. Characterization of Novel Complexes on the Cell Surface between Integrins and Proteins with 4 Transmembrane Domains (TM4 proteins). Mol. Biol. Cell 1996, 7, 193–207. [Google Scholar] [CrossRef]
- Jones, P.H.; Bishop, L.A.; Watt, F.M. Functional significance of CD9 association with beta 1 integrins in human epidermal keratinocytes. Cell Adhes. Commun. 1996, 4, 297–305. [Google Scholar] [CrossRef]
- Shaw, A.R.; Domanska, A.; Mak, A.; Gilchrist, A.; Dobler, K.; Visser, L.; Poppema, S.; Fliegel, L.; Letarte, M.; Willett, B.J. Ectopic expression of human and feline CD9 in a human B cell line confers β1 integrin-dependent motility on fibronectin and laminin substrates and enhanced tyrosine phosphorylation. J. Biol. Chem. 1995, 270, 24092–24099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domanico, S.Z.; Pelletier, A.J.; Havran, W.L.; Quaranta, V. Integrin alpha 6A beta 1 induces CD81-dependent cell motility without engaging the extracellular matrix migration substrate. Mol. Biol. Cell 1997, 8, 2253–2265. [Google Scholar] [CrossRef]
- Baudoux, B.; Castanares-Zapatero, D.; Leclercq-Smekens, M.; Berna, N.; Poumay, Y. The tetraspanin CD9 associates with the integrin alpha6beta4 in cultured human epidermal keratinocytes and is involved in cell motility. Eur. J. Cell Biol. 2000, 79, 41–51. [Google Scholar] [CrossRef]
- Skubitz, K.M.; Campbell, K.D.; Iida, J.; Skubitz, A.P. CD63 associates with tyrosine kinase activity and CD11/CD18, and transmits an activation signal in neutrophils. J. Immunol. 1996, 157, 3617–3626. [Google Scholar]
- Sincock, P.M.; Mayrhofer, G.; Ashman, L.K. Localization of the transmembrane 4 superfamily (TM4SF) member PETA-3 (CD151) in normal human tissues: Comparison with CD9, CD63, and alpha5beta1 integrin. J. Histochem. Cytochem. 1997, 45, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, E.; Naour, F.L.; Lagaudriere-Gesbert, C.; Billard, M.; Conjeaud, H.; Boucheix, C. CD9, CD63, CD81, and CD82 are components of a surface tetraspan network connected to HLA-DR and VLA integrins. Eur. J. Immunol. 1996, 26, 2657–2665. [Google Scholar] [CrossRef]
- Hirst, J.; Bright, N.A.; Rous, B.; Robinson, M.S. Characterization of a fourth adaptor-related protein complex. Mol. Biol. Cell 1999, 10, 2787–2802. [Google Scholar] [CrossRef] [Green Version]
- Latysheva, N.; Muratov, G.; Rajesh, S.; Padgett, M.; Hotchin, N.A.; Overduin, M.; Berditchevski, F. Syntenin-1 is a new component of tetraspanin-enriched microdomains: Mechanisms and consequences of the interaction of syntenin-1 with CD63. Mol. Cell. Biol. 2006, 26, 7707–7718. [Google Scholar] [CrossRef] [Green Version]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, I.; Korhonen, M.; Kariniemi, A.L.; Gould, V.E.; Laitinen, L.; Ylänne, J. Integrins in human cells and tumors. Cell. Differ. Dev. 1990, 32, 215–227. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, D.A.; Zent, R.; Grant, R.; Rees, D.J.; Hynes, R.O.; Ginsberg, M.H. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 1999, 274, 28071–28074. [Google Scholar] [CrossRef] [Green Version]
- Dedhar, S. Integrins and signal transduction. Curr. Opin. Hematol. 1999, 6, 37–43. [Google Scholar] [CrossRef]
- Miyamoto, S.; Teramoto, H.; Coso, A.O.; Gutkind, J.S.; Burbelo, P.D.; Akiyama, S.K.; Yamada, K.M. Integrin function: Molecular hierarchies of cytoskeletal and signaling molecules. J. Cell Biol. 1995, 131, 791–805. [Google Scholar] [CrossRef] [Green Version]
- Flier, A.; Sonnenberg, A. Function and interactions of integrins. Cell Tissue Res. 2001, 305, 285–298. [Google Scholar] [CrossRef]
- Wary, K.K.; Mainiero, F.; Isakoff, S.J.; Marcantonio, E.E.; Giancotti, F.G. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 1996, 87, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Frisch, S.M.; Vuori, K.; Ruoslahtit, E.; Chan-Hui, P.Y. Control of Adhesion-dependent Cell Survival by Focal Adhesion Kinase. J. Cell Biol. 1996, 134, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Vuori, K.; Reed, J.C.; Ruoslahti, E. The alpha 5 beta 1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc. Natl. Acad. Sci. USA 1995, 92, 6161–6165. [Google Scholar] [CrossRef] [Green Version]
- Frisch, S.M.; Ruoslahtit, E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997, 9, 701–706. [Google Scholar] [CrossRef]
- Ruoslahtit, E.; Reed, J.C. Anchorage Dependence, Integrins, and Apoptosis. Cell 1994, 77, 477–478. [Google Scholar] [CrossRef]
- Tong, C.F.; Zhang, Y.; Lü, S.Q.; Li, N.; Gong, Y.X.; Yang, H.; Feng, S.L.; Du, Y.; Huang, D.D.; Long, M. Binding of intercellular adhesion molecule 1 to β 2-integrin regulates distinct cell adhesion processes on hepatic and cerebral endothelium. Am. J. Physiol. Cell Physiol. 2018, 315, C409–C421. [Google Scholar] [CrossRef]
- Parsons, J.T.; Martin, K.H.; Slack, J.K.; Taylor, J.M.; Weed, S.A. Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene 2000, 19, 5606–5613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemler, M.E. Integrin associated proteins. Curr. Opin. Cell Biol. 1998, 10, 578–585. [Google Scholar] [CrossRef]
- Sincock, P.M.; Fitter, S.; Parton, R.G.; Berndt, M.; Gamble, J.R.; Ashman, L.K. PETA-3/CD151, a member of the transmembrane 4 superfamily, is localised to the plasma membrane and endocytic system of endothelial cells, associates with multiple integrins and modulates cell function. J. Cell Sci. 1999, 112, 833–844. [Google Scholar] [CrossRef]
- Klein-Soyer, C.; Azorsa, D.O.; Cazenave, J.P.; Lanza, F. CD9 participates in endothelial cell migration during in vitro wound repair. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Bassani, S.; Cingolani, L.A. Tetraspanins: Interactions and interplay with integrins. Int. J. Biochem. Cell Biol. 2012, 44, 703–708. [Google Scholar] [CrossRef]
- Berditchevski, F.; Bazzoni, G.; Hemler, M.E. Specific association of CD63 with the VLA-3 and VLA-6 integrins. J. Biol. Chem. 1995, 270, 17784–17790. [Google Scholar] [CrossRef] [Green Version]
- Wilk, C.M.; Schildberg, F.A.; Lauterbach, M.A.; Cadeddu, R.P.; Fröbel, J.; Westphal, V.; Tolba, R.H.; Hell, S.W.; Czibere, A.; Bruns, I.; et al. The tissue inhibitor of metalloproteinases-1 improves migration and adhesion of hematopoietic stem and progenitor cells. Exp. Hematol. 2013, 41, 823–831. [Google Scholar] [CrossRef]
- Nicaise, A.M.; Johnson, K.M.; Willis, C.M.; Guzzo, R.M.; Crocker, S.J. TIMP-1 Promotes Oligodendrocyte Differentiation Through Receptor-Mediated Signaling. Mol. Neurobiol. 2019, 56, 3380–3392. [Google Scholar] [CrossRef] [PubMed]
- Ólafsson, E.B.; Ross, E.C.; Varas-Godoy, M.; Barragan, A. Correction: TIMP-1 promotes hypermigration of Toxoplasma-infected primary dendritic cells via CD63-ITGB1-FAK signaling. J. Cell Sci. 2019, 132, jcs230920. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Kang, Y.; Huang, L.; Wu, L.; Peng, Y. TIMP1 preserves the blood-brain barrier through interacting with CD63/integrin β 1 complex and regulating downstream FAK/RhoA signaling. Acta Pharm. Sin. B 2020, 10, 987–1003. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.S.; Cohen, A.M.; Zhang, Z.F.; Stetler-Stevenson, W.; Guillem, J.G. Elevated Tissue Inhibitor of Metalloproteinase 1 RNA in Colorectal Cancer Stroma Correlates with Lymph Node and Distant Metastases. Clin. Cancer Res. 1995, 1, 899–906. [Google Scholar] [PubMed]
- Yoshiji, H.; Gomez, D.E.; Thorgeirsso, U.P. Enhanced RNA expression of tissue inhibitor of metaloproteinases-1 (TIMP-1) in human breast cancer. Int. J. Cancer 1996, 69, 131–134. [Google Scholar] [CrossRef]
- Fong, K.M.; Kida, Y.; Zimmerman, P.V.; Smith, P.J. TIMP-1 and Adverse Prognosis in Non-Small Cell Lung Cancer. Clin. Cancer Res. 1996, 2, 1369–1372. [Google Scholar]
- Joo, Y.E.; Seo, K.S.; Kim, H.S.; Rew, J.S.; Park, C.S.; Kim, S.J. Expression of Tissue Inhibitors of Metalloproteinases (TIMPs) in Gastric Cancer. Dig. Dis. Sci. 2000, 45, 114–121. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, J.M.; Cho, S.Y.; Kim, H.S.; Shin, H.S.; Jeon, J.Y.; Kausar, R.; Jeong, S.Y.; Lee, Y.S.; Lee, M.A. TIMP-1 modulates chemotaxis of human neural stem cells through CD63 and integrin signalling. Biochem. J. 2014, 459, 565–576. [Google Scholar] [CrossRef]
- Ando, T.; Charindra, D.; Shrestha, M.; Umehara, H.; Ogawa, I.; Miyauchi, M.; Takata, T. Tissue inhibitor of metalloproteinase-1 promotes cell proliferation through YAP/TAZ activation in cancer. Oncogene 2018, 37, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Toricelli, M. (UNIFESP, São Paulo, SP, Brazil). Association between TIMP-1, β1-Integrins and CD63 throughout the Genesis of Melanoma. Unpublished article. 2010. [Google Scholar]
- Toricelli, M.; Melo, F.H.M.; Peres, G.B.; Silva, D.C.P.; Jasiulionis, M.G. Timp1 interacts with beta-1 integrin and CD63 along melanoma genesis and confers Anoikis resistance by activating PI3-K signaling pathway independently of Akt phosphorylation. Mol. Cancer 2013, 12, 1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Liu, J.; Xiao, X.; Sun, S.; Zhang, H.; Zhang, Y.; Zhou, W.; Zhang, B.; Roy, M.; Liu, H.; et al. A Novel AptamerLL4A Specifically Targets Vemurafenib-Resistant Melanoma through Binding to the CD63 Protein. Mol. Ther. Nucleic Acids 2019, 18, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Cherepanova, N.; Shrimal, S.; Gilmore, R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 2016, 41, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Lis, H.; Sharon, N. Protein glycosylation. Structural and functional aspects. Eur. J. Biochem. 1993, 218, 1–27. [Google Scholar] [CrossRef]
- Eichler, J. Protein glycosylation. Curr. Biol. 2019, 29, R229–R231. [Google Scholar] [CrossRef] [Green Version]
- Hakomori, S. Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv. Cancer Res. 1989, 52, 257–331. [Google Scholar]
- Singhal, A.; Hakomori, S. Molecular changes in carbohydrate antigens associated with cancer. Bioessays 1990, 12, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Dabelsteen, E.; Clausen, H.; Mandel, U. Aberrant glycosylation in oral malignant and premalignant lesions. J. Oral Pathol. Med. 1991, 20, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Dabelsteen, E.; Clausen, H.; Mandel, U. Carbohydrate changes in squamous cell carcinomas. APMIS Suppl. 1992, 27, 130–138. [Google Scholar]
- Prystas, E.M.; Parker, C.J.; Holguin, M.H.; Bohnsack, J.F. Aberrant glycosylation of L-selectin on the lymphocytes of chronic lymphocytic leukemia. Leukemia 1993, 7, 1355–1362. [Google Scholar]
- Hiraizumi, S.; Takasaki, S.; Ohuchi, N.; Harada, Y.; Nose, M.; Mori, S.; Kobata, A. Altered glycosylation of membrane glycoproteins associated with human mammary carcinoma. Jpn. J. Cancer Res. 1992, 83, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Chen, R.; Tamura, Y.; Crispin, D.A.; Lai, L.A.; May, D.H.; McIntosh, M.W.; Goodlett, D.R.; Brentnall, T.A. Quantitative glycoproteomics analysis reveals changes in N-Glycosylation level associated with pancreatic ductal adenocarcinoma. J. Proteome Res. 2014, 13, 1293–1306. [Google Scholar] [CrossRef]
- Drake, R.R.; Jones, E.E.; Powers, T.W.; Nyalwidhe, J.O. Altered glycosylation in prostate cancer. Adv. Cancer Res. 2015, 126, 345–382. [Google Scholar]
- Very, N.; Lefebvre, T.; Yazidi-Belkoura, I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget 2017, 9, 1380–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veillon, L.; Fakih, C.; Abou-El-Hassan, H.; Kobeissy, F.; Mechref, Y. Glycosylation Changes in Brain Cancer. ACS Chem. Neurosci. 2018, 9, 51–72. [Google Scholar] [CrossRef]
- Dennis, J.W.; Laferté, S.; Waghorne, C.; Breitman, M.L.; Kerbel, R.S. β1-6 Branching of Asn-Linked Oligosaccharides Is Directly Associated with Metastasis. Science 1987, 236, 582–585. [Google Scholar] [CrossRef]
- Dennis, J.W.; Laferté, S. Oncodevelopmental Expression of –GlcAcβ1-6Manα1-6Manβ1- Branched Asparagine-linked Oligosaccharides in Murine Tissues and Human Breast Carcinomas. Cancer Res. 1989, 49, 945–950. [Google Scholar]
- Rye, P.D.; Fodstad, O.; Emilsen, E.; Bryne, M. Invasion Potential and N-Acetylgalactosamine Expression In A Human Melanoma Model. Int. J. Cancer 1998, 75, 609–614. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, J.; Lubman, D.M.; Gao, C. Aberrant glycosylation and cancer biomarker discovery: A promising and thorny journey. Clin. Chem. Lab. Med. 2019, 57, 407–416. [Google Scholar] [CrossRef]
- Granovsky, M.; Fata, J.; Pawling, J.; Muller, W.J.; Khokha, R.; Dennis, J.W. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat. Med. 2000, 6, 306–312. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.; Wu, X.; Li, Q.; Yu, J.; Gen, J.; Zhang, X.L. Knockdown of Mgat5 inhibits breast cancer cell growth with activation of CD4+ T cells and macrophages. J. Immunol. 2008, 180, 3158–3165. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Chen, H.; Wang, Q.; Zhang, L.; Zhao, J. Knockdown of Mgat5 inhibits CD133+ human pulmonary adenocarcinoma cell growth in vitro and in vivo. Clin. Investig. Med. 2011, 34, E155–E162. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, H.; Zhang, W.; Wu, Q.; Liu, W.; Liu, Y.; Pan, D.; Xu, J.; Gu, J. N-acetylglucosaminyltransferase V confers hepatoma cells with resistance to anoikis through EGFR/PAK1 activation. Glycobiology 2013, 23, 1097–1109. [Google Scholar] [CrossRef]
- Srivastava, O.P.; Hindsgaul, O.; Shoreibah, M.; Pierce, M. Recognition of oligosaccharide substrates by N-acetyl-glucosaminyltransferase-V. Carbohydr Res. 1988, 179, 137–161. [Google Scholar] [CrossRef]
- Seelentag, W.K.; Li, W.P.; Schmitz, S.F.; Metzger, U.; Aeberhard, P.; Heitz, P.U.; Roth, J. Prognostic value of beta1,6-branched oligosaccharides in human colorectal carcinoma. Cancer Res. 1998, 58, 5559–5564. [Google Scholar]
- Handerson, T.; Camp, R.; Harigopal, M.; Rimm, D.; Pawelek, J. Beta1,6-branched oligosaccharides are increased in lymph node metastases and predict poor outcome in breast carcinoma. Clin. Cancer Res. 2005, 11, 2969–2973. [Google Scholar] [CrossRef] [Green Version]
- Przybyło, M.; Martuszewska, D.; Pocheć, E.; Hoja-Łukowicz, D.; Lityńska, A. Identification of proteins bearing beta1-6 branched N-glycans in human melanoma cell lines from different progression stages by tandem mass spectrometry analysis. Biochim. Biophys. Acta 2007, 1770, 1427–1435. [Google Scholar] [CrossRef]
- Kim, Y.S.; Hwang, S.Y.; Kang, H.Y.; Sohn, H.; Oh, S.; Kim, J.Y.; Yoo, J.S.; Kim, Y.H.; Kim, S.H.; Jeon, J.H.; et al. Functional Proteomics Study Reveals That N-Acetylglucosaminyltransferase V Reinforces the Invasive/Metastatic Potential of Colon Cancer through Aberrant Glycosylation on Tissue Inhibitor of Metalloproteinase-1. Mol. Cell. Proteom. 2008, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, N.; Hagiwara, K.; Kosaka, N.; Honma, K.; Nakagama, H.; Ochiya, T. RPN2-mediated glycosylation of tetraspanin CD63 regulates breast cancer cell malignancy. Mol. Cancer 2014, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Huang, W.; Wu, B.; Jin, J.; Jing, L.; Shi, W.P.; Liu, Z.Y.; Yuan, L.; Luo, D.; Li, L.; et al. N-Glycosylation by N-acetylglucosaminyltransferase V enhances the interaction of CD147/basigin with integrin β1 and promotes HCC metastasis. J. Pathol. 2018, 245, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janik, M.E.; Lityńska, A.; Vereecken, P. Cell migration—The role of integrin glycosylation. Biochim. Biophys. Acta 2010, 1800, 545–555. [Google Scholar] [CrossRef]
- Cai, X.; Thinn, A.M.; Wang, Z.; Shan, H.; Zhu, J. The importance of N-Glycosylation on β3 integrin ligand binding and conformational regulation. Sci. Rep. 2017, 7, 4656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Isaji, T.; Sato, Y.; Kariya, Y.; Fukuda, T. Importance of N-Glycosylation on a5b1 Integrin for Its Biological Functions. Biol. Pharm. Bull. 2009, 32, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Jasiulionis, M.G.; Chammas, R.; Ventura, A.M.; Travassos, L.R.; Brentani, R.R. Alpha6Beta1 Integrin, a major cell surface carrier of beta1-6-branched oligosaccharides, mediates migration of EJ-ras-transformed fibroblasts on laminin-1 independently of its glycosylation state. Cancer Res. 1996, 56, 1682–1689. [Google Scholar]
- Guo, H.B.; Lee, I.; Kamar, M.; Akiyama, S.K.; Pierce, M. Aberrant N-Glycosylation of beta1 integrin causes reduced alpha5beta1 integrin clustering and stimulates cell migration. Cancer Res. 2002, 62, 6837–6845. [Google Scholar] [PubMed]
- Pocheć, E.; Lityńska, A.; Amoresano, A.; Casbarra, A. Glycosylation profile of integrin α3β1 changes with melanoma progression. Biochim. Biophys. Acta 2003, 1643, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liang, Y.; Li, Z.; Cai, X.; Zhang, W.; Wu, G.; Jin, J.; Fang, Z.; Yang, Y.; Zha, X. Increase in beta1-6 GlcNAc branching caused by N-acetylglucosaminyltransferase V directs integrin beta1 stability in human hepatocellular carcinoma cell line SMMC-7721. J. Cell. Biochem. 2007, 100, 230–241. [Google Scholar] [CrossRef]
- Kim, Y.S.; Ahn, Y.H.; Song, K.J.; Kang, J.G.; Lee, J.H.; Jeon, S.K.; Kim, H.C.; Yoo, J.S.; Ko, J.H. Overexpression and β-1,6-N-Acetylglucosaminylation-initiated Aberrant Glycosylation of TIMP-1. J. Biol. Chem. 2012, 287, 32467–32478. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Justo, B.L.; Jasiulionis, M.G. Characteristics of TIMP1, CD63, and β1-Integrin and the Functional Impact of Their Interaction in Cancer. Int. J. Mol. Sci. 2021, 22, 9319. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179319
Justo BL, Jasiulionis MG. Characteristics of TIMP1, CD63, and β1-Integrin and the Functional Impact of Their Interaction in Cancer. International Journal of Molecular Sciences. 2021; 22(17):9319. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179319
Chicago/Turabian StyleJusto, Beatriz Laís, and Miriam Galvonas Jasiulionis. 2021. "Characteristics of TIMP1, CD63, and β1-Integrin and the Functional Impact of Their Interaction in Cancer" International Journal of Molecular Sciences 22, no. 17: 9319. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179319