
Molecular Mechanisms of Neuroimmune Crosstalk in the Pathogenesis of Stroke


Abstract
:1. Introduction
2. Cascades of Immune Responses after Stroke
2.1. Ischemic Stroke
2.1.1. Leukocyte Adherence after Stroke
2.1.2. Routes of Immune Cells toward the CNS after Stroke
2.1.3. Immune Cascades after Stroke
Microglia
Neutrophils
Astrocytes
Mast Cells
Monocyte-Derived Macrophages
Dendritic Cells (DCs)
Natural Killer (NK) Cells
T-Cells
Regulatory T-Cells (Tregs)
B-Cells
2.2. Hemorrhagic Stroke
3. Brain Edema and the Glymphatic System
3.1. Aquaporins (AQPs)
3.2. AQPs in Stroke
4. Lymphatic System
4.1. General Structure and Roles
4.2. Meningeal Lymphatic Vessels
4.3. Lymphatics near the Cribriform Plate (CP Lymphatics)
5. Lymphangiogenesis
6. Aging
6.1. Inflammaging
6.2. Sex and Aging
6.3. Lymphatic Vessels
7. Communication between the CNS and the Peripheral Immune System
7.1. Cervical Lymph Nodes and Spleen
7.1.1. Peripheral Immunodepression (Lymphopenia)
7.1.2. Systemic Inflammatory Response Syndrome
7.2. Complications in the Periphery
7.2.1. Autoimmunity after Stroke
7.2.2. Complication in Gastrointestinal System after Stroke
8. Novel Treatments Targeting Neuroimmune Communications and Homeostasis in Stroke
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hsu, M.; Rayasam, A.; Kijak, J.A.; Choi, Y.H.; Harding, J.S.; Marcus, S.A.; Karpus, W.J.; Sandor, M.; Fabry, Z. Neuroinflammation-induced lymphangiogenesis near the cribriform plate contributes to drainage of CNS-derived antigens and immune cells. Nat. Commun. 2019, 10, 229. [Google Scholar] [CrossRef]
- Petrova, T.V.; Koh, G.Y. Biological functions of lymphatic vessels. Science 2020, 369, eaax4063. [Google Scholar] [CrossRef]
- Trincot, C.E.; Caron, K.M. Lymphatic Function and Dysfunction in the Context of Sex Differences. ACS Pharmacol. Transl. Sci. 2019, 2, 311–324. [Google Scholar] [CrossRef]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.-L.; Iwasaki, A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337. [Google Scholar] [CrossRef]
- Da Mesquita, S.; Fu, Z.; Kipnis, J. The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuron 2018, 100, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Ahn, J.H.; Cho, H.; Kim, J.-H.; Kim, S.H.; Ham, J.-S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.-H.; Hong, Y.-K.; et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 2019, 572, 62–66. [Google Scholar] [CrossRef]
- Arima, Y.; Kamimura, D.; Sabharwal, L.; Yamada, M.; Bando, H.; Ogura, H.; Atsumi, T.; Murakami, M. Regulation of immune cell infiltration into the CNS by regional neural inputs explained by the gate theory. Mediat. Inflamm. 2013, 2013, 898165. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Gate, D.; Town, T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? J. Neuroimmune Pharmacol. 2009, 4, 462–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarkson, B.D.; Walker, A.; Harris, M.G.; Rayasam, A.; Sandor, M.; Fabry, Z. CCR2-dependent dendritic cell accumulation in the central nervous system during early effector experimental autoimmune encephalomyelitis is essential for effector T cell restimulation in situ and disease progression. J. Immunol. 2015, 194, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-J.; Song, M. Disrupted Ionic Homeostasis in Ischemic Stroke and New Therapeutic Targets. J. Stroke Cerebrovasc. Dis. 2017, 26, 2706–2719. [Google Scholar] [CrossRef]
- Planas, A.M. Role of Immune Cells Migrating to the Ischemic Brain. Stroke 2018, 49, 2261–2267. [Google Scholar] [CrossRef]
- Kim, J.S. tPA Helpers in the Treatment of Acute Ischemic Stroke: Are They Ready for Clinical Use? J. Stroke 2019, 21, 160–174. [Google Scholar] [CrossRef]
- Virani Salim, S.; Alonso, A.; Benjamin Emelia, J.; Bittencourt Marcio, S.; Callaway Clifton, W.; Carson April, P.; Chamberlain Alanna, M.; Chang Alexander, R.; Cheng, S.; Delling Francesca, N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Armstrong, J.R.; Mosher, B.D. Aspiration pneumonia after stroke: Intervention and prevention. Neurohospitalist 2011, 1, 85–93. [Google Scholar] [CrossRef]
- Sellars, C.; Bowie, L.; Bagg, J.; Sweeney, M.P.; Miller, H.; Tilston, J.; Langhorne, P.; Stott David, J. Risk Factors for Chest Infection in Acute Stroke. Stroke 2007, 38, 2284–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poisson Sharon, N.; Johnston, S.C.; Josephson, S.A. Urinary Tract Infections Complicating Stroke. Stroke 2010, 41, e180–e184. [Google Scholar] [CrossRef] [Green Version]
- Demaerschalk Bart, M.; Kleindorfer Dawn, O.; Adeoye Opeolu, M.; Demchuk Andrew, M.; Fugate Jennifer, E.; Grotta James, C.; Khalessi Alexander, A.; Levy Elad, I.; Palesch Yuko, Y.; Prabhakaran, S.; et al. Scientific Rationale for the Inclusion and Exclusion Criteria for Intravenous Alteplase in Acute Ischemic Stroke. Stroke 2016, 47, 581–641. [Google Scholar] [CrossRef] [PubMed]
- Gravanis, I.; Tsirka, S.E. Tissue-type plasminogen activator as a therapeutic target in stroke. Expert Opin. Ther. Targets 2008, 12, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Barber, P.A.; Zhang, J.; Demchuk, A.M.; Hill, M.D.; Buchan, A.M. Why are stroke patients excluded from TPA therapy? Neurology 2001, 56, 1015. [Google Scholar] [CrossRef]
- Miller, D.J.; Simpson, J.R.; Silver, B. Safety of thrombolysis in acute ischemic stroke: A review of complications, risk factors, and newer technologies. Neurohospitalist 2011, 1, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Alberts, M.J. Stroke Treatment with Intravenous Tissue-Type Plasminogen Activator. Circulation 2017, 135, 140–142. [Google Scholar] [CrossRef]
- Chen, S.; Zeng, L.; Hu, Z. Progressing haemorrhagic stroke: Categories, causes, mechanisms and managements. J. Neurol. 2014, 261, 2061–2078. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.-Y.; Pan, S.-Y. The failure of animal models of neuroprotection in acute ischemic stroke to translate to clinical efficacy. Med. Sci. Monit. Basic Res. 2013, 19, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wang, K. The fate of medications evaluated for ischemic stroke pharmacotherapy over the period 1995–2015. Acta Pharm. Sin. B 2016, 6, 522–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.D.; Al-Khoury, L.; Zivin, J.A. Neuroprotection for ischemic stroke: Two decades of success and failure. NeuroRx 2004, 1, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Recommendations for Clinical Trial Evaluation of Acute Stroke Therapies. Stroke 2001, 32, 1598–1606. [CrossRef] [Green Version]
- Iadecola, C.; Buckwalter, M.S.; Anrather, J. Immune responses to stroke: Mechanisms, modulation, and therapeutic potential. J. Clin. Investig. 2020, 130, 2777–2788. [Google Scholar] [CrossRef]
- Shekhar, S.; Liu, Y.; Wang, S.; Zhang, H.; Fang, X.; Zhang, J.; Fan, L.; Zheng, B.; Roman, R.J.; Wang, Z.; et al. Novel Mechanistic Insights and Potential Therapeutic Impact of TRPC6 in Neurovascular Coupling and Ischemic Stroke. Int. J. Mol. Sci. 2021, 22, 2074. [Google Scholar] [CrossRef]
- Tran-Dinh, A.; Levoye, A.; Couret, D.; Galle-Treger, L.; Moreau, M.; Delbosc, S.; Hoteit, C.; Montravers, P.; Amarenco, P.; Huby, T.; et al. High-Density Lipoprotein Therapy in Stroke: Evaluation of Endothelial SR-BI-Dependent Neuroprotective Effects. Int. J. Mol. Sci. 2021, 22, 106. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Shi, J.; Elmadhoun, O.; Duan, Y.; An, H.; Zhang, J.; He, X.; Meng, R.; Liu, X.; Ji, X.; et al. Dihydrocapsaicin (DHC) enhances the hypothermia-induced neuroprotection following ischemic stroke via PI3K/Akt regulation in rat. Brain Res. 2017, 1671, 18–25. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.; Messing, R.O.; Chou, W.-H. Mouse model of middle cerebral artery occlusion. J. Vis. Exp. 2011, 2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grysiewicz, R.A.; Thomas, K.; Pandey, D.K. Epidemiology of ischemic and hemorrhagic stroke: Incidence, prevalence, mortality, and risk factors. Neurol. Clin. 2008, 26, 871–895. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Hayakawa, K.; Miyamoto, N.; Seo, J.H.; Pham, L.D.; Kim, K.W.; Lo, E.H.; Arai, K. High-mobility group box 1 from reactive astrocytes enhances the accumulation of endothelial progenitor cells in damaged white matter. J. Neurochem. 2013, 125, 273–280. [Google Scholar] [CrossRef]
- Rayasam, A.; Hsu, M.; Kijak, J.A.; Kissel, L.; Hernandez, G.; Sandor, M.; Fabry, Z. Immune responses in stroke: How the immune system contributes to damage and healing after stroke and how this knowledge could be translated to better cures? Immunology 2018, 154, 363–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.-M.; Zhang, C.-L.; Chen, A.-Q.; Wang, H.-L.; Zhou, Y.-F.; Li, Y.-N.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418774254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, G.; Granger, D.N. Leukocyte recruitment and ischemic brain injury. Neuromol. Med. 2010, 12, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaie, A.R. Protease-activated receptor signalling by coagulation proteases in endothelial cells. Thromb. Haemost. 2014, 112, 876–882. [Google Scholar] [CrossRef] [Green Version]
- Amara, U.; Rittirsch, D.; Flierl, M.; Bruckner, U.; Klos, A.; Gebhard, F.; Lambris, J.D.; Huber-Lang, M. Interaction between the coagulation and complement system. Adv. Exp. Med. Biol. 2008, 632, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alawieh, A.; Langley, E.F.; Tomlinson, S. Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci. Transl. Med. 2018, 10, eaao6459. [Google Scholar] [CrossRef] [Green Version]
- Mocco, J.; Mack, W.J.; Ducruet, A.F.; Sosunov, S.A.; Sughrue, M.E.; Hassid, B.G.; Nair, M.N.; Laufer, I.; Komotar, R.J.; Claire, M.; et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ. Res. 2006, 99, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Cervera, A.; Planas, A.M.; Justicia, C.; Urra, X.; Jensenius, J.C.; Torres, F.; Lozano, F.; Chamorro, A. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS ONE 2010, 5, e8433. [Google Scholar] [CrossRef] [Green Version]
- Osthoff, M.; Katan, M.; Fluri, F.; Schuetz, P.; Bingisser, R.; Kappos, L.; Steck, A.J.; Engelter, S.T.; Mueller, B.; Christ-Crain, M.; et al. Mannose-binding lectin deficiency is associated with smaller infarction size and favorable outcome in ischemic stroke patients. PLoS ONE 2011, 6, e21338. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Akter, F.; Qin, L.; Cheng, J.; Guo, M.; Yao, S.; Jian, Z.; Liu, R.; Wu, S. Adaptive Immunity Regulation and Cerebral Ischemia. Front. Immunol. 2020, 11, 689. [Google Scholar] [CrossRef] [PubMed]
- Malone, K.; Amu, S.; Moore, A.C.; Waeber, C. The immune system and stroke: From current targets to future therapy. Immunol. Cell Biol. 2019, 97, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanimirovic, D.B.; Wong, J.; Shapiro, A.; Durkin, J.P. Increase in surface expression of ICAM-1, VCAM-1 and E-selectin in human cerebromicrovascular endothelial cells subjected to ischemia-like insults. Acta Neurochir. Suppl. 1997, 70, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chopp, M.; Zhang, Z.; Jiang, N.; Powers, C. The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Res. 1998, 785, 207–214. [Google Scholar] [CrossRef]
- Okada, Y.; Copeland, B.R.; Mori, E.; Tung, M.M.; Thomas, W.S.; del Zoppo, G.J. P-selectin and intercellular adhesion molecule-1 expression after focal brain ischemia and reperfusion. Stroke 1994, 25, 202–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, E.S., Jr.; Winfree, C.J.; Prestigiacomo, C.J.; Kim, S.C.; Choudhri, T.F.; Hoh, B.L.; Naka, Y.; Solomon, R.A.; Pinsky, D.J. Exacerbation of cerebral injury in mice that express the P-selectin gene: Identification of P-selectin blockade as a new target for the treatment of stroke. Circ. Res. 1997, 81, 304–310. [Google Scholar] [CrossRef]
- Frijns, C.J.; Kappelle, L.J.; van Gijn, J.; Nieuwenhuis, H.K.; Sixma, J.J.; Fijnheer, R. Soluble adhesion molecules reflect endothelial cell activation in ischemic stroke and in carotid atherosclerosis. Stroke 1997, 28, 2214–2218. [Google Scholar] [CrossRef]
- Zhang, R.L.; Chopp, M.; Zhang, Z.G.; Phillips, M.L.; Rosenbloom, C.L.; Cruz, R.; Manning, A. E-selectin in focal cerebral ischemia and reperfusion in the rat. J. Cereb. Blood Flow Metab. 1996, 16, 1126–1136. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Choudhri, T.F.; Winfree, C.J.; McTaggart, R.A.; Kiss, S.; Mocco, J.; Kim, L.J.; Protopsaltis, T.S.; Zhang, Y.; Pinsky, D.J.; et al. Postischemic cerebrovascular E-selectin expression mediates tissue injury in murine stroke. Stroke 2000, 31, 3047–3053. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Cooper, D.; Russell, J.; Salter, J.W.; Zhang, J.H.; Nanda, A.; Granger, D.N. Molecular determinants of the prothrombogenic and inflammatory phenotype assumed by the postischemic cerebral microcirculation. Stroke 2003, 34, 1777–1782. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Stokes, K.Y.; Zhang, J.H.; Nanda, A.; Granger, D.N. Cerebral microvascular responses to hypercholesterolemia: Roles of NADPH oxidase and P-selectin. Circ. Res. 2004, 94, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Mori, E.; del Zoppo, G.J.; Chambers, J.D.; Copeland, B.R.; Arfors, K.E. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke 1992, 23, 712–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, Z.G.; Zhang, R.L.; Lu, M.; Krams, M.; Chopp, M. Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke 2003, 34, 1790–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, M.; Vowinkel, T.; Stokes, K.Y.; Arumugam, T.V.; Yilmaz, G.; Nanda, A.; Granger, D.N. CD40/CD40 ligand signaling in mouse cerebral microvasculature after focal ischemia/reperfusion. Circulation 2005, 111, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.V.; Chan, S.L.; Jo, D.G.; Yilmaz, G.; Tang, S.C.; Cheng, A.; Gleichmann, M.; Okun, E.; Dixit, V.D.; Chigurupati, S.; et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat. Med. 2006, 12, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Use of anti-ICAM-1 therapy in ischemic stroke: Results of the Enlimomab Acute Stroke Trial. Neurology 2001, 57, 1428–1434. [CrossRef]
- Becker, K.J. Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr. Med. Res. Opin. 2002, 18 (Suppl. 2), s18–s22. [Google Scholar] [CrossRef]
- Yang, Y.; Rosenberg, G.A. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [Green Version]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Steffen, B.J.; Breier, G.; Butcher, E.C.; Schulz, M.; Engelhardt, B. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. Am. J. Pathol. 1996, 148, 1819–1838. [Google Scholar]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Tornero, D.; Hirota, M.; Monni, E.; Laterza, C.; Lindvall, O.; Kokaia, Z. Choroid plexus-cerebrospinal fluid route for monocyte-derived macrophages after stroke. J. Neuroinflamm. 2017, 14, 153. [Google Scholar] [CrossRef]
- Shechter, R.; Miller, O.; Yovel, G.; Rosenzweig, N.; London, A.; Ruckh, J.; Kim, K.W.; Klein, E.; Kalchenko, V.; Bendel, P.; et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 2013, 38, 555–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic-Djergovic, D.; Hyman, M.C.; Ray, J.J.; Bouis, D.; Visovatti, S.H.; Hayasaki, T.; Pinsky, D.J. Tissue-resident ecto-5’ nucleotidase (CD73) regulates leukocyte trafficking in the ischemic brain. J. Immunol. 2012, 188, 2387–2398. [Google Scholar] [CrossRef] [Green Version]
- Chinnery, H.R.; Ruitenberg, M.J.; McMenamin, P.G. Novel characterization of monocyte-derived cell populations in the meninges and choroid plexus and their rates of replenishment in bone marrow chimeric mice. J. Neuropathol. Exp. Neurol. 2010, 69, 896–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef] [Green Version]
- Greter, M.; Lelios, I.; Pelczar, P.; Hoeffel, G.; Price, J.; Leboeuf, M.; Kündig, T.M.; Frei, K.; Ginhoux, F.; Merad, M.; et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 2012, 37, 1050–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Maten, G.; Henck, V.; Wieloch, T.; Ruscher, K. CX(3)C chemokine receptor 1 deficiency modulates microglia morphology but does not affect lesion size and short-term deficits after experimental stroke. BMC Neurosci. 2017, 18, 11. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, X.J.; Zhang, C.; Chen, R.; Li, L.; He, J.; Xie, Y.; Chen, Y. Loss of neuronal CD200 contributed to microglial activation after acute cerebral ischemia in mice. Neurosci. Lett. 2018, 678, 48–54. [Google Scholar] [CrossRef]
- Yanagisawa, D.; Kitamura, Y.; Takata, K.; Hide, I.; Nakata, Y.; Taniguchi, T. Possible involvement of P2X7 receptor activation in microglial neuroprotection against focal cerebral ischemia in rats. Biol. Pharm. Bull. 2008, 31, 1121–1130. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Li, X.; Xu, P.; Huang, L.; Cheng, J.; Huang, X.; Jiang, J.; Wu, L.J.; Tang, Y. TREM2 protects against cerebral ischemia/reperfusion injury. Mol. Brain 2017, 10, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, H.; Zhang, H.; Ye, Q.; Wang, J.; Yang, B.; Mao, L.; Zhu, W.; Leak, R.K.; Xiao, B.; et al. ST2/IL-33-Dependent Microglial Response Limits Acute Ischemic Brain Injury. J. Neurosci. 2017, 37, 4692–4704. [Google Scholar] [CrossRef]
- Li, K.; Yu, W.; Cao, R.; Zhu, Z.; Zhao, G. Microglia-mediated BAFF-BAFFR ligation promotes neuronal survival in brain ischemia injury. Neuroscience 2017, 363, 87–96. [Google Scholar] [CrossRef]
- Jin, W.N.; Shi, S.X.; Li, Z.; Li, M.; Wood, K.; Gonzales, R.J.; Liu, Q. Depletion of microglia exacerbates postischemic inflammation and brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 2224–2236. [Google Scholar] [CrossRef] [Green Version]
- Ritzel, R.M.; Patel, A.R.; Grenier, J.M.; Crapser, J.; Verma, R.; Jellison, E.R.; McCullough, L.D. Functional differences between microglia and monocytes after ischemic stroke. J. Neuroinflamm. 2015, 12, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; An, J.; Haile, W.B.; Echeverry, R.; Strickland, D.K.; Yepes, M. Microglial low-density lipoprotein receptor-related protein 1 mediates the effect of tissue-type plasminogen activator on matrix metalloproteinase-9 activity in the ischemic brain. J. Cereb. Blood Flow Metab. 2009, 29, 1946–1954. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Gelderblom, M.; Weymar, A.; Bernreuther, C.; Velden, J.; Arunachalam, P.; Steinbach, K.; Orthey, E.; Arumugam, T.V.; Leypoldt, F.; Simova, O.; et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 2012, 120, 3793–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otxoa-de-Amezaga, A.; Miró-Mur, F.; Pedragosa, J.; Gallizioli, M.; Justicia, C.; Gaja-Capdevila, N.; Ruíz-Jaen, F.; Salas-Perdomo, A.; Bosch, A.; Calvo, M.; et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 2019, 137, 321–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalancette-Hébert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef] [Green Version]
- Otxoa-de-Amezaga, A.; Gallizioli, M.; Pedragosa, J.; Justicia, C.; Miró-Mur, F.; Salas-Perdomo, A.; Díaz-Marugan, L.; Gunzer, M.; Planas, A.M. Location of Neutrophils in Different Compartments of the Damaged Mouse Brain after Severe Ischemia/Reperfusion. Stroke 2019, 50, 1548–1557. [Google Scholar] [CrossRef]
- Perez-de-Puig, I.; Miró-Mur, F.; Ferrer-Ferrer, M.; Gelpi, E.; Pedragosa, J.; Justicia, C.; Urra, X.; Chamorro, A.; Planas, A.M. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015, 129, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Liu, S.; Hu, M.; Huang, F.; Zhu, Q.; Qiu, W.; Hu, X.; Colello, J.; Zheng, S.G.; Lu, Z. Functional Dynamics of Neutrophils After Ischemic Stroke. Transl. Stroke Res. 2020, 11, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Yu, H.; Yang, X.; Zhu, Y.; Bai, X.; Wang, R.; Cao, Y.; Xu, H.; Luo, H.; Lu, L.; et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat. Commun. 2020, 11, 2488. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Thornton, P.; Denes, A.; McColl, B.W.; Pierozynski, A.; Monestier, M.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 2012, 189, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Stowe, A.M.; Adair-Kirk, T.L.; Gonzales, E.R.; Perez, R.S.; Shah, A.R.; Park, T.S.; Gidday, J.M. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol. Dis. 2009, 35, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gidday, J.M.; Gasche, Y.G.; Copin, J.-C.; Shah, A.R.; Perez, R.S.; Shapiro, S.D.; Chan, P.H.; Park, T.S. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am. J. Physiol.-Heart Circ. Physiol. 2005, 289, H558–H568. [Google Scholar] [CrossRef] [PubMed]
- Justicia, C.; Panés, J.; Solé, S.; Cervera, Á.; Deulofeu, R.; Chamorro, Á.; Planas, A.M. Neutrophil Infiltration Increases Matrix Metalloproteinase-9 in the Ischemic Brain after Occlusion/Reperfusion of the Middle Cerebral Artery in Rats. J. Cereb. Blood Flow Metab. 2003, 23, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- García-Culebras, A.; Durán-Laforet, V.; Peña-Martínez, C.; Moraga, A.; Ballesteros, I.; Cuartero, M.I.; de la Parra, J.; Palma-Tortosa, S.; Hidalgo, A.; Corbí, A.L.; et al. Role of TLR4 (Toll-Like Receptor 4) in N1/N2 Neutrophil Programming After Stroke. Stroke 2019, 50, 2922–2932. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, Y.; Onodera, H.; Shiga, Y.; Nakamura, M.; Ninomiya, M.; Kihara, T.; Kogure, K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke 1994, 25, 1469–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudome, S.; Palmer, C.; Roberts, R.L.; Mauger, D.; Housman, C.; Towfighi, J. The role of neutrophils in the production of hypoxic-ischemic brain injury in the neonatal rat. Pediatr. Res. 1997, 41, 607–616. [Google Scholar] [CrossRef]
- Cauwe, B.; Martens, E.; Proost, P.; Opdenakker, G. Multidimensional degradomics identifies systemic autoantigens and intracellular matrix proteins as novel gelatinase B/MMP-9 substrates. Integr. Biol. 2009, 1, 404–426. [Google Scholar] [CrossRef] [Green Version]
- Christoffersson, G.; Vågesjö, E.; Vandooren, J.; Lidén, M.; Massena, S.; Reinert, R.B.; Brissova, M.; Powers, A.C.; Opdenakker, G.; Phillipson, M. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 2012, 120, 4653–4662. [Google Scholar] [CrossRef] [PubMed]
- Taichman, N.S.; Young, S.; Cruchley, A.T.; Taylor, P.; Paleolog, E. Human neutrophils secrete vascular endothelial growth factor. J. Leukoc. Biol. 1997, 62, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Koh, D.R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2010, 339, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Cuartero, M.I.; Ballesteros, I.; Moraga, A.; Nombela, F.; Vivancos, J.; Hamilton, J.A.; Corbí, Á.L.; Lizasoain, I.; Moro, M.A. N2 neutrophils, novel players in brain inflammation after stroke: Modulation by the PPARγ agonist rosiglitazone. Stroke 2013, 44, 3498–3508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Kohli, P.; Levy, B.D. Resolvins and protectins: Mediating solutions to inflammation. Br. J. Pharmacol. 2009, 158, 960–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tułowiecka, N.; Kotlęga, D.; Prowans, P.; Szczuko, M. The Role of Resolvins: EPA and DHA Derivatives Can Be Useful in the Prevention and Treatment of Ischemic Stroke. Int. J. Mol. Sci. 2020, 21, 7628. [Google Scholar] [CrossRef]
- Dong, X.; Gao, J.; Zhang, C.Y.; Hayworth, C.; Frank, M.; Wang, Z. Neutrophil Membrane-Derived Nanovesicles Alleviate Inflammation To Protect Mouse Brain Injury from Ischemic Stroke. ACS Nano 2019, 13, 1272–1283. [Google Scholar] [CrossRef]
- Yao, C.; Zhang, J.; Chen, F.; Lin, Y. Neuroprotectin D1 attenuates brain damage induced by transient middle cerebral artery occlusion in rats through TRPC6/CREB pathways. Mol. Med. Rep. 2013, 8, 543–550. [Google Scholar] [CrossRef]
- Miao, Z.; Schultzberg, M.; Wang, X.; Zhao, Y. Role of polyunsaturated fatty acids in ischemic stroke—A perspective of specialized pro-resolving mediators. Clin. Nutr. 2021, 40, 2974–2987. [Google Scholar] [CrossRef]
- Becerra-Calixto, A.; Cardona-Gómez, G.P. The Role of Astrocytes in Neuroprotection after Brain Stroke: Potential in Cell Therapy. Front. Mol. Neurosci. 2017, 10, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Ring, A.; Sonnewald, U.; Waagepetersen, H.S. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 2009, 109, 87–93. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, P. Ischemic Cell Death in Brain Neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef]
- Ito, U.; Hakamata, Y.; Kawakami, E.; Oyanagi, K. Degeneration of Astrocytic Processes and Their Mitochondria in Cerebral Cortical Regions Peripheral to the Cortical Infarction. Stroke 2009, 40, 2173–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassett, C.; Cho, S.M.; Rice, C.; Uchino, K. Is Lactate Dehydrogenase level a Biomarker of Ischemic Stroke in Ventricular Assist Device? (P6.241). Neurology 2018, 90, P6.241. [Google Scholar]
- Anan, M.; Nagai, Y.; Fudaba, H.; Fujiki, M. Lactate and Lactate Dehydrogenase in Cistern as Biomarkers of Early Brain Injury and Delayed Cerebral Ischemia of Subarachnoid Hemorrhage. J. Stroke Cerebrovasc. Dis. 2020, 29, 104765. [Google Scholar] [CrossRef]
- Brouns, R.; Sheorajpanday, R.; Wauters, A.; De Surgeloose, D.; Mariën, P.; De Deyn, P.P. Evaluation of lactate as a marker of metabolic stress and cause of secondary damage in acute ischemic stroke or TIA. Clin. Chim. Acta 2008, 397, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Bellail, A.; Tse, M.C.L.; Yong, V.W.; Hao, C. Human Astrocytes Are Resistant to Fas Ligand and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Induced Apoptosis. J. Neurosci. 2006, 26, 3299. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, Z.; Yao, Y.; Jin, W.-N.; Wood, K.; Liu, Q.; Shi, F.-D.; Hao, J. Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc. Natl. Acad. Sci. USA 2017, 114, E396. [Google Scholar] [CrossRef] [Green Version]
- Haim, L.B.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2017, 18, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.-W.; Duan, C.-L.; Chen, X.-H.; Wang, Y.-Q.; Sun, X.; Zhang, Q.-W.; Cui, H.-R.; Sun, F.-Y. Neurogenic effect of VEGF is related to increase of astrocytes transdifferentiation into new mature neurons in rat brains after stroke. Neuropharmacology 2016, 108, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Lindsberg, P.J.; Strbian, D.; Karjalainen-Lindsberg, M.L. Mast cells as early responders in the regulation of acute blood-brain barrier changes after cerebral ischemia and hemorrhage. J. Cereb. Blood Flow Metab. 2010, 30, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Silverman, A.J.; Vannucci, S.J. Mast cell stabilization limits hypoxic-ischemic brain damage in the immature rat. Dev. Neurosci. 2007, 29, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Parrella, E.; Porrini, V.; Benarese, M.; Pizzi, M. The Role of Mast Cells in Stroke. Cells 2019, 8, 437. [Google Scholar] [CrossRef] [Green Version]
- Winter, M.C.; Shasby, S.S.; Ries, D.R.; Shasby, D.M. Histamine selectively interrupts VE-cadherin adhesion independently of capacitive calcium entry. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, L816–L823. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.C.; Wolters, P.J.; Steinhoff, M.; Bidgol, A.; Blount, J.L.; Caughey, G.H. Mast cell expression of gelatinases A and B is regulated by kit ligand and TGF-beta. J. Immunol. 1999, 162, 5528–5535. [Google Scholar]
- Strbian, D.; Karjalainen-Lindsberg, M.L.; Tatlisumak, T.; Lindsberg, P.J. Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. J. Cereb. Blood Flow Metab. 2006, 26, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Strbian, D.; Karjalainen-Lindsberg, M.L.; Kovanen, P.T.; Tatlisumak, T.; Lindsberg, P.J. Mast cell stabilization reduces hemorrhage formation and mortality after administration of thrombolytics in experimental ischemic stroke. Circulation 2007, 116, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Gliem, M.; Mausberg, A.K.; Lee, J.I.; Simiantonakis, I.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 2012, 71, 743–752. [Google Scholar] [CrossRef]
- Wattananit, S.; Tornero, D.; Graubardt, N.; Memanishvili, T.; Monni, E.; Tatarishvili, J.; Miskinyte, G.; Ge, R.; Ahlenius, H.; Lindvall, O.; et al. Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice. J. Neurosci. 2016, 36, 4182–4195. [Google Scholar] [CrossRef]
- Kim, J.S.; Gautam, S.C.; Chopp, M.; Zaloga, C.; Jones, M.L.; Ward, P.A.; Welch, K.M. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J. Neuroimmunol. 1995, 56, 127–134. [Google Scholar] [CrossRef]
- Tei, N.; Tanaka, J.; Sugimoto, K.; Nishihara, T.; Nishioka, R.; Takahashi, H.; Yano, H.; Matsumoto, S.; Ohue, S.; Watanabe, H.; et al. Expression of MCP-1 and fractalkine on endothelial cells and astrocytes may contribute to the invasion and migration of brain macrophages in ischemic rat brain lesions. J. Neurosci. Res. 2013, 91, 681–693. [Google Scholar] [CrossRef]
- Wang, X.; Yue, T.L.; Barone, F.C.; Feuerstein, G.Z. Monocyte chemoattractant protein-1 messenger RNA expression in rat ischemic cortex. Stroke 1995, 26, 661–665; discussion 665–666. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.M.; Allegrini, P.R.; Rudin, M.; Perry, V.H.; Mir, A.K.; Wiessner, C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J. Cereb. Blood Flow Metab. 2002, 22, 308–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedragosa, J.; Miró-Mur, F.; Otxoa-de-Amezaga, A.; Justicia, C.; Ruíz-Jaén, F.; Ponsaerts, P.; Pasparakis, M.; Planas, A.M. CCR2 deficiency in monocytes impairs angiogenesis and functional recovery after ischemic stroke in mice. J. Cereb. Blood Flow Metab. 2020, 40, S98–S116. [Google Scholar] [CrossRef]
- Garcia-Bonilla, L.; Faraco, G.; Moore, J.; Murphy, M.; Racchumi, G.; Srinivasan, J.; Brea, D.; Iadecola, C.; Anrather, J. Spatio-temporal profile, phenotypic diversity, and fate of recruited monocytes into the post-ischemic brain. J. Neuroinflamm. 2016, 13, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perego, C.; Fumagalli, S.; De Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Ann. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.-C.; Ma, L.-S.; Chu, Z.-H.; Xu, H.; Wu, W.-Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Liu, R.; Zhu, X.; Smerin, D.; Zhong, Y.; Gu, L.; Fang, W.; Xiong, X. The Involvement and Therapy Target of Immune Cells After Ischemic Stroke. Front. Immunol. 2019, 10, 2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedragosa, J.; Salas-Perdomo, A.; Gallizioli, M.; Cugota, R.; Miró-Mur, F.; Briansó, F.; Justicia, C.; Pérez-Asensio, F.; Marquez-Kisinousky, L.; Urra, X.; et al. CNS-border associated macrophages respond to acute ischemic stroke attracting granulocytes and promoting vascular leakage. Acta Neuropathol. Commun. 2018, 6, 76. [Google Scholar] [CrossRef] [PubMed]
- Terao, Y.; Ohta, H.; Oda, A.; Nakagaito, Y.; Kiyota, Y.; Shintani, Y. Macrophage inflammatory protein-3alpha plays a key role in the inflammatory cascade in rat focal cerebral ischemia. Neurosci. Res. 2009, 64, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, P.; Kanninen, K.M.; Lehtonen, Š.; Lemarchant, S.; Puttonen, K.A.; Oksanen, M.; Dhungana, H.; Loppi, S.; Pollari, E.; Wojciechowski, S.; et al. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav. Immun. 2015, 49, 322–336. [Google Scholar] [CrossRef]
- Förster, R.; Schubel, A.; Breitfeld, D.; Kremmer, E.; Renner-Müller, I.; Wolf, E.; Lipp, M. CCR7 Coordinates the Primary Immune Response by Establishing Functional Microenvironments in Secondary Lymphoid Organs. Cell 1999, 99, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Czeloth, N.; Bernhardt, G.; Hofmann, F.; Genth, H.; Förster, R. Sphingosine-1-Phosphate Mediates Migration of Mature Dendritic Cells. J. Immunol. 2005, 175, 2960–2967. [Google Scholar] [CrossRef] [Green Version]
- Faure-André, G.; Vargas, P.; Yuseff, M.-I.; Heuzé, M.; Diaz, J.; Lankar, D.; Steri, V.; Manry, J.; Hugues, S.; Vascotto, F.; et al. Regulation of Dendritic Cell Migration by CD74, the MHC Class II-Associated Invariant Chain. Science 2008, 322, 1705–1710. [Google Scholar] [CrossRef] [Green Version]
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1⁺ macrophages to CD103⁺ dendritic cells. Immunity 2014, 40, 248–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, B.P.; Gee, R.J.; Haberman, A.M.; Shlomchik, M.J.; Mamula, M.J. Antigen presentation and transfer between B cells and macrophages. Eur. J. Immunol. 2007, 37, 1739–1751. [Google Scholar] [CrossRef]
- McMenamin, P.G. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J. Comp. Neurol. 1999, 405, 553–562. [Google Scholar] [CrossRef]
- Felger, J.C.; Abe, T.; Kaunzner, U.W.; Gottfried-Blackmore, A.; Gal-Toth, J.; McEwen, B.S.; Iadecola, C.; Bulloch, K. Brain dendritic cells in ischemic stroke: Time course, activation state, and origin. Brain Behav. Immun. 2010, 24, 724–737. [Google Scholar] [CrossRef] [Green Version]
- Gelderblom, M.; Gallizioli, M.; Ludewig, P.; Thom, V.; Arunachalam, P.; Rissiek, B.; Bernreuther, C.; Glatzel, M.; Korn, T.; Arumugam, T.V.; et al. IL-23 (Interleukin-23)-Producing Conventional Dendritic Cells Control the Detrimental IL-17 (Interleukin-17) Response in Stroke. Stroke 2018, 49, 155–164. [Google Scholar] [CrossRef]
- Kostulas, N.; Li, H.L.; Xiao, B.G.; Huang, Y.M.; Kostulas, V.; Link, H. Dendritic cells are present in ischemic brain after permanent middle cerebral artery occlusion in the rat. Stroke 2002, 33, 1129–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichmann, G.; Schroeter, M.; Jander, S.; Fischer, H.-G. Dendritic cells and dendritic-like microglia in focal cortical ischemia of the mouse brain. J. Neuroimmunol. 2002, 129, 125–132. [Google Scholar] [CrossRef]
- Yilmaz, A.; Fuchs, T.; Dietel, B.; Altendorf, R.; Cicha, I.; Stumpf, C.; Schellinger, P.D.; Blümcke, I.; Schwab, S.; Daniel, W.G.; et al. Transient decrease in circulating dendritic cell precursors after acute stroke: Potential recruitment into the brain. Clin. Sci. 2009, 118, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, Y.; Liu, Q.; Wu, W.; Yin, J.-X.; Bai, X.-F.; Shen, R.; Wang, Y.; Chen, J.; La Cava, A.; Poursine-Laurent, J.; et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc. Natl. Acad. Sci. USA 2014, 111, 2704–2709. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gao, Z.; Wang, D.; Zhang, T.; Sun, B.; Mu, L.; Wang, J.; Liu, Y.; Kong, Q.; Liu, X.; et al. Accumulation of natural killer cells in ischemic brain tissues and the chemotactic effect of IP-10. J. Neuroinflamm. 2014, 11, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef] [Green Version]
- Kleinschnitz, C.; Schwab, N.; Kraft, P.; Hagedorn, I.; Dreykluft, A.; Schwarz, T.; Austinat, M.; Nieswandt, B.; Wiendl, H.; Stoll, G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010, 115, 3835–3842. [Google Scholar] [CrossRef]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.-U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, T.; Olah, L.; Harms, C.; Maul, B.; Bader, M.; Hörtnagl, H.; Schultheiss, H.P.; Mies, G. Ischemic injury in experimental stroke depends on angiotensin II. FASEB J. 2002, 16, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Inaba, S.; Iwai, M.; Tomono, Y.; Senba, I.; Furuno, M.; Kanno, H.; Okayama, H.; Mogi, M.; Higaki, J.; Horiuchi, M. Exaggeration of focal cerebral ischemia in transgenic mice carrying human Renin and human angiotensinogen genes. Stroke 2009, 40, 597–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Filiano, A.J.; Gadani, S.P.; Kipnis, J. How and why do T cells and their derived cytokines affect the injured and healthy brain? Nat. Rev. Neurosci. 2017, 18, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, T.V.; Granger, D.N.; Mattson, M.P. Stroke and T-cells. Neuromol. Med. 2005, 7, 229–242. [Google Scholar] [CrossRef]
- Chu, H.X.; Kim, H.A.; Lee, S.; Moore, J.P.; Chan, C.T.; Vinh, A.; Gelderblom, M.; Arumugam, T.V.; Broughton, B.R.; Drummond, G.R. Immune cell infiltration in malignant middle cerebral artery infarction: Comparison with transient cerebral ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 450–459. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Li, W.; Hersh, J.; Liu, R.; Yang, S.-H. Experimental ischemic stroke induces long-term T cell activation in the brain. J. Cereb. Blood Flow Metab. 2019, 39, 2268–2276. [Google Scholar] [CrossRef]
- Shichita, T.; Sugiyama, Y.; Ooboshi, H.; Sugimori, H.; Nakagawa, R.; Takada, I.; Iwaki, T.; Okada, Y.; Iida, M.; Cua, D.J.; et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat. Med. 2009, 15, 946–950. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, J.C.; Yao, C.Y.; Wu, Y.; Abdelgawad, A.F.; Yao, S.L.; Yuan, S.Y. Critical role of astrocytic interleukin-17 A in post-stroke survival and neuronal differentiation of neural precursor cells in adult mice. Cell Death Dis. 2016, 7, e2273. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, B.D.S.; Ling, C.; Shi, Y.; Harris, M.G.; Rayasam, A.; Sun, D.; Salamat, M.S.; Kuchroo, V.; Lambris, J.D.; Sandor, M.; et al. T cell-derived interleukin (IL)-21 promotes brain injury following stroke in mice. J. Exp. Med. 2014, 211, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef]
- Li, P.; Wang, L.; Zhou, Y.; Gan, Y.; Zhu, W.; Xia, Y.; Jiang, X.; Watkins, S.; Vazquez, A.; Thomson, A.W.; et al. C-C Chemokine Receptor Type 5 (CCR5)-Mediated Docking of Transferred Tregs Protects Against Early Blood-Brain Barrier Disruption after Stroke. J. Am. Heart Assoc. 2017, 6, e006387. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Kraft, P.; Dreykluft, A.; Hagedorn, I.; Göbel, K.; Schuhmann, M.K.; Langhauser, F.; Helluy, X.; Schwarz, T.; Bittner, S.; et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood 2013, 121, 679–691. [Google Scholar] [CrossRef]
- Voo, K.S.; Wang, Y.H.; Santori, F.R.; Boggiano, C.; Wang, Y.H.; Arima, K.; Bover, L.; Hanabuchi, S.; Khalili, J.; Marinova, E.; et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 4793–4798. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Akiyoshi, K.; Dziennis, S.; Vandenbark, A.A.; Herson, P.S.; Hurn, P.D.; Offner, H. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J. Neurosci. 2011, 31, 8556–8563. [Google Scholar] [CrossRef]
- Ortega, S.B.; Torres, V.O.; Latchney, S.E.; Whoolery, C.W.; Noorbhai, I.Z.; Poinsatte, K.; Selvaraj, U.M.; Benson, M.A.; Meeuwissen, A.J.M.; Plautz, E.J.; et al. B cells migrate into remote brain areas and support neurogenesis and functional recovery after focal stroke in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 4983. [Google Scholar] [CrossRef] [Green Version]
- Doyle, K.P.; Quach, L.N.; Solé, M.; Axtell, R.C.; Nguyen, T.-V.V.; Soler-Llavina, G.J.; Jurado, S.; Han, J.; Steinman, L.; Longo, F.M.; et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J. Neurosci. 2015, 35, 2133–2145. [Google Scholar] [CrossRef]
- Schuhmann, M.K.; Langhauser, F.; Kraft, P.; Kleinschnitz, C. B cells do not have a major pathophysiologic role in acute ischemic stroke in mice. J. Neuroinflamm. 2017, 14, 112. [Google Scholar] [CrossRef] [Green Version]
- Rincon, F.; Mayer, S.A. The epidemiology of intracerebral hemorrhage in the United States from 1979 to 2008. Neurocrit. Care 2013, 19, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Gao, S.; Wang, X.; Cao, Y.; Lu, J.; Chen, S.; Lenahan, C.; Zhang, J.H.; Shao, A.; Zhang, J. Programmed Cell Deaths and Potential Crosstalk with Blood-Brain Barrier Dysfunction after Hemorrhagic Stroke. Front. Cell. Neurosci. 2020, 14, 68. [Google Scholar] [CrossRef]
- Davis, S.M.; Broderick, J.; Hennerici, M.; Brun, N.C.; Diringer, M.N.; Mayer, S.A.; Begtrup, K.; Steiner, T. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology 2006, 66, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Leira, R.; Dávalos, A.; Silva, Y.; Gil-Peralta, A.; Tejada, J.; Garcia, M.; Castillo, J. Early neurologic deterioration in intracerebral hemorrhage: Predictors and associated factors. Neurology 2004, 63, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Jickling, G.C.; Liu, D.; Stamova, B.; Ander, B.P.; Zhan, X.; Lu, A.; Sharp, F.R. Hemorrhagic transformation after ischemic stroke in animals and humans. J. Cereb. Blood Flow Metab. 2014, 34, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saand, A.R.; Yu, F.; Chen, J.; Chou, S.H.Y. Systemic inflammation in hemorrhagic strokes—A novel neurological sign and therapeutic target? J. Cereb. Blood Flow Metab. 2019, 39, 959–988. [Google Scholar] [CrossRef]
- Frontera, J.A.; Provencio, J.J.; Sehba, F.A.; McIntyre, T.M.; Nowacki, A.S.; Gordon, E.; Weimer, J.M.; Aledort, L. The Role of Platelet Activation and Inflammation in Early Brain Injury Following Subarachnoid Hemorrhage. Neurocrit. Care 2017, 26, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, A.; Mancha, F.; Macher, H.C.; García-Berrocoso, T.; Giralt, D.; Ribó, M.; Guerrero, J.M.; Montaner, J. Circulating cell-free DNA is a predictor of short-term neurological outcome in stroke patients treated with intravenous thrombolysis. J. Circ. Biomark. 2016, 5, 1849454416668791. [Google Scholar] [CrossRef]
- Rainer, T.H.; Wong, L.K.; Lam, W.; Yuen, E.; Lam, N.Y.; Metreweli, C.; Lo, Y.M. Prognostic use of circulating plasma nucleic acid concentrations in patients with acute stroke. Clin. Chem. 2003, 49, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.C.; Yang, T.M.; Lin, W.C.; Lin, Y.J.; Tsai, N.W.; Liou, C.W.; Kwan, A.L.; Lu, C.H. The value of serial plasma and cerebrospinal fluid nuclear and mitochondrial deoxyribonucleic acid levels in aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2013, 118, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Yao, X.; Li, J.; He, B.; Liu, Q.; Ren, H.; Liang, F.; Li, M.; Lin, H.; Peng, J.; et al. Paravascular pathways contribute to vasculitis and neuroinflammation after subarachnoid hemorrhage independently of glymphatic control. Cell Death Dis. 2016, 7, e2160. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.D.; Chen, J.S.; Zhou, F.; Liu, Q.C.; Chen, G.; Zhang, J.M. Relationship between plasma high mobility group box-1 protein levels and clinical outcomes of aneurysmal subarachnoid hemorrhage. J. Neuroinflamm. 2012, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, N.; Dewan, V.; Grace, P.M.; Gunn, R.J.; Tamura, R.; Tzarum, N.; Watkins, L.R.; Wilson, I.A.; Yin, H. HMGB1 Activates Proinflammatory Signaling via TLR5 Leading to Allodynia. Cell Rep. 2016, 17, 1128–1140. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Han, X.; Liu, X.; Wang, J. Inflammatory responses after intracerebral hemorrhage: From cellular function to therapeutic targets. J. Cereb. Blood Flow Metab. 2019, 39, 184–186. [Google Scholar] [CrossRef]
- Quan, W.; Zhang, Z.; Li, P.; Tian, Q.; Huang, J.; Qian, Y.; Gao, C.; Su, W.; Wang, Z.; Zhang, J.; et al. Role of Regulatory T cells in Atorvastatin Induced Absorption of Chronic Subdural Hematoma in Rats. Aging Dis. 2019, 10, 992–1002. [Google Scholar] [CrossRef] [Green Version]
- Plog, B.A.; Nedergaard, M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. 2018, 13, 379–394. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Wang, M.; Zeppenfeld, D.M.; Venkataraman, A.; Plog, B.A.; Liao, Y.; Deane, R.; Nedergaard, M. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci. 2013, 33, 18190–18199. [Google Scholar] [CrossRef] [Green Version]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The glymphatic system: A beginner’s guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Wang, M.H.; Liao, Y.H.; .Plogg, B.A.; Peng, W.G.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [Green Version]
- Goulay, R.; Flament, J.; Gauberti, M.; Naveau, M.; Pasquet, N.; Gakuba, C.; Emery, E.; Hantraye, P.; Vivien, D.; Aron-Badin, R.; et al. Subarachnoid Hemorrhage Severely Impairs Brain Parenchymal Cerebrospinal Fluid Circulation in Nonhuman Primate. Stroke 2017, 48, 2301–2305. [Google Scholar] [CrossRef] [PubMed]
- Gaberel, T.; Gakuba, C.; Goulay, R.; Lizarrondo, S.M.D.; Hanouz, J.-L.; Emery, E.; Touze, E.; Vivien, D.; Gauberti, M. Impaired Glymphatic Perfusion After Strokes Revealed by Contrast-Enhanced MRI. Stroke 2014, 45, 3092–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef] [PubMed]
- Back, D.B.; Kwon, K.J.; Choi, D.H.; Shin, C.Y.; Lee, J.; Han, S.H.; Kim, H.Y. Chronic cerebral hypoperfusion induces post-stroke dementia following acute ischemic stroke in rats. J. Neuroinflamm. 2017, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhang, T.; Wu, G.; McBride, D.W.; Xu, N.; Klebe, D.W.; Zhang, Y.; Li, Q.; Tang, J.; Zhang, J.H. Astrogliosis inhibition attenuates hydrocephalus by increasing cerebrospinal fluid reabsorption through the glymphatic system after germinal matrix hemorrhage. Exp. Neurol. 2019, 320, 113003. [Google Scholar] [CrossRef]
- Wang, W.W.; Xie, C.L.; Zhou, L.L.; Wang, G.S. The function of aquaporin4 in ischemic brain edema. Clin. Neurol. Neurosurg. 2014, 127, 5–9. [Google Scholar] [CrossRef]
- Dinet, V.; Petry, K.G.; Badaut, J. Brain-Immune Interactions and Neuroinflammation After Traumatic Brain Injury. Front. Neurosci. 2019, 13, 1178. [Google Scholar] [CrossRef] [Green Version]
- Golanov, E.V.; Bovshik, E.I.; Wong, K.K.; Pautler, R.G.; Foster, C.H.; Federley, R.G.; Zhang, J.Y.; Mancuso, J.; Wong, S.T.; Britz, G.W. Subarachnoid hemorrhage—Induced block of cerebrospinal fluid flow: Role of brain coagulation factor III (tissue factor). J. Cereb. Blood Flow Metab. 2018, 38, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Simard, J.M.; Staley, K.J.; Nahed, B.V.; Jones, P.S.; Sun, D. Molecular mechanisms of ischemic cerebral edema: Role of electroneutral ion transport. Physiology 2009, 24, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin-4 and brain edema. Pediatr. Nephrol. 2007, 22, 778–784. [Google Scholar] [CrossRef] [Green Version]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta BBA-Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef]
- Verkman, A.S.; Mitra, A.K. Structure and function of aquaporin water channels. Am. J. Physiol.-Ren. Physiol. 2000, 278, F13–F28. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Pickel, S.U.; Jennings, J.; Törnroth-Horsefield, S.; Conner, M.T.; Bill, R.M.; Conner, A.C. Water channel pore size determines exclusion properties but not solute selectivity. Sci. Rep. 2019, 9, 20369. [Google Scholar] [CrossRef] [Green Version]
- Hara-Chikuma, M.; Verkman, A.S. Physiological roles of glycerol-transporting aquaporins: The aquaglyceroporins. Cell. Mol. Life Sci. CMLS 2006, 63, 1386–1392. [Google Scholar] [CrossRef]
- Kitchen, P.; Day, R.E.; Salman, M.M.; Conner, M.T.; Bill, R.M.; Conner, A.C. Beyond water homeostasis: Diverse functional roles of mammalian aquaporins. Biochim. Biophys. Acta BBA-Gen. Subj. 2015, 1850, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Tsukaguchi, H.; Shayakul, C.; Berger, U.V.; Mackenzie, B.; Devidas, S.; Guggino, W.B.; van Hoek, A.N.; Hediger, M.A. Molecular characterization of a broad selectivity neutral solute channel. J. Biol. Chem. 1998, 273, 24737–24743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakhoul, N.L.; Davis, B.A.; Romero, M.F.; Boron, W.F. Effect of expressing the water channel aquaporin-1 on the CO2 permeability of Xenopus oocytes. Am. J. Physiol.-Cell Physiol. 1998, 274, C543–C548. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.J.; Boron, W.F. Effect of PCMBS on CO2 permeability of Xenopus oocytes expressing aquaporin 1 or its C189S mutant. Am. J. Physiol.-Cell Physiol. 1998, 275, C1481–C1486. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e719. [Google Scholar] [CrossRef]
- Haj-Yasein, N.N.; Vindedal, G.F.; Eilert-Olsen, M.; Gundersen, G.A.; Skare, Ø.; Laake, P.; Klungland, A.; Thorén, A.E.; Burkhardt, J.M.; Ottersen, O.P. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood–brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. USA 2011, 108, 17815–17820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Goulay, R.; Mena Romo, L.; Hol, E.M.; Dijkhuizen, R.M. From Stroke to Dementia: A Comprehensive Review Exposing Tight Interactions Between Stroke and Amyloid-β Formation. Transl. Stroke Res. 2020, 11, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Vella, J.; Zammit, C.; Di Giovanni, G.; Muscat, R.; Valentino, M. The central role of aquaporins in the pathophysiology of ischemic stroke. Front. Cell. Neurosci. 2015, 9, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirt, L.; Price, M.; Benakis, C.; Badaut, J. Aquaporins in neurological disorders. Clin. Transl. Neurosci. 2018, 2. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Zador, Z.; Verkman, A.S. Glial cell aquaporin-4 overexpression in transgenic mice accelerates cytotoxic brain swelling. J. Biol. Chem. 2008, 283, 15280–15286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirt, L.; Fukuda, A.M.; Ambadipudi, K.; Rashid, F.; Binder, D.; Verkman, A.; Ashwal, S.; Obenaus, A.; Badaut, J. Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. J. Cereb. Blood Flow Metab. 2017, 37, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Reulen, H.J.; Graham, R.; Spatz, M.; Klatzo, I. Role of pressure gradients and bulk flow in dynamics of vasogenic brain edema. J. Neurosurg. 1977, 46, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badaut, J.; Hirt, L.; Granziera, C.; Bogousslavsky, J.; Magistretti, P.J.; Regli, L. Astrocyte-Specific Expression of Aquaporin-9 in Mouse Brain is Increased after Transient Focal Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2001, 21, 477–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirt, L.; Price, M.; Mastour, N.; Brunet, J.-F.; Barrière, G.; Friscourt, F.; Badaut, J. Increase of aquaporin 9 expression in astrocytes participates in astrogliosis. J. Neurosci. Res. 2018, 96, 194–206. [Google Scholar] [CrossRef]
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.; Perry, V.H.; Weller, R.O. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: Significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef]
- Laman, J.D.; Weller, R.O. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J. Neuroimmune Pharmacol. 2013, 8, 840–856. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Du, T.; Sweeney, A.M.; Liu, G.; Samson, A.J.; Peng, W.; Mortensen, K.N.; Stæger, F.F.; Bork, P.A.R.; Bashford, L.; et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 2020, 367, eaax7171. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.N.; Bacskai, B.J.; Arbel-Ornath, M.; Aldea, R.; Bedussi, B.; Morris, A.W.; Weller, R.O.; Carare, R.O. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2016, 36, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshuhri, M.S.; Gallagher, L.; Work, L.M.; Holmes, W.M. Direct imaging of glymphatic transport using H217O MRI. JCI Insight 2021, 6, e141159. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.J.; Tsujita, M.; Nakada, T. Identification of aquaporin 4 inhibitors using in vitro and in silico methods. Bioorg. Med. Chem. 2009, 17, 411–417. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, H.; Verkman, A.S. Lack of aquaporin-4 water transport inhibition by antiepileptics and arylsulfonamides. Bioorg. Med. Chem. 2008, 16, 7489–7493. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.M.; Kitchen, P.; Iliff, J.J.; Bill, R.M. Aquaporin 4 and glymphatic flow have central roles in brain fluid homeostasis. Nat. Rev. Neurosci. 2021. [Google Scholar] [CrossRef]
- Igarashi, H.; Huber, V.J.; Tsujita, M.; Nakada, T. Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol. Sci. 2011, 32, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Pirici, I.; Balsanu, T.A.; Bogdan, C.; Margaritescu, C.; Divan, T.; Vitalie, V.; Mogoanta, L.; Pirici, D.; Carare, R.O.; Muresanu, D.F. Inhibition of Aquaporin-4 Improves the Outcome of Ischaemic Stroke and Modulates Brain Paravascular Drainage Pathways. Int. J. Mol. Sci. 2018, 19, 46. [Google Scholar] [CrossRef] [Green Version]
- Toft-Bertelsen, T.L.; Larsen, B.R.; Christensen, S.K.; Khandelia, H.; Waagepetersen, H.S.; MacAulay, N. Clearance of activity-evoked K+ transients and associated glia cell swelling occur independently of AQP4: A study with an isoform-selective AQP4 inhibitor. Glia 2021, 69, 28–41. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N. Molecular mechanisms of brain water transport. Nat. Rev. Neurosci. 2021, 22, 326–344. [Google Scholar] [CrossRef]
- Li, J.; Jia, Z.; Xu, W.; Guo, W.; Zhang, M.; Bi, J.; Cao, Y.; Fan, Z.; Li, G. TGN-020 alleviates edema and inhibits astrocyte activation and glial scar formation after spinal cord compression injury in rats. Life Sci. 2019, 222, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liu, J.; Wang, X.; Li, W.; Chen, J.; Sun, H. Pretreatment with AQP4 and NKCC1 Inhibitors Concurrently Attenuated Spinal Cord Edema and Tissue Damage after Spinal Cord Injury in Rats. Front. Physiol. 2018, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Suzuki, Y.; Tsujita, M.; Huber, V.J.; Yamada, K.; Nakada, T. Development of a Novel Ligand, [C]TGN-020, for Aquaporin 4 Positron Emission Tomography Imaging. ACS Chem. Neurosci. 2011, 2, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Brady, M.; Rahman, A.; Combs, A.; Venkatraman, C.; Kasper, R.T.; McQuaid, C.; Kwok, W.-C.E.; Wood, R.W.; Deane, R. Cerebrospinal fluid drainage kinetics across the cribriform plate are reduced with aging. Fluids Barriers CNS 2020, 17, 71. [Google Scholar] [CrossRef]
- Földi, M.; Gellért, A.; Kozma, M.; Poberai, M.; Zoltán, O.T.; Csanda, E. New contributions to the anatomical connections of the brain and the lymphatic system. Cells Tissues Organs 1966, 64, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.A.; Valera, V.A.; Takahashi, S.; Ushiki, T. The olfactory route for cerebrospinal fluid drainage into the peripheral lymphatic system. Neuropathol. Appl. Neurobiol. 2006, 32, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.; Zakharov, A.; Papaiconomou, C.; Salmasi, G.; Armstrong, D. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cerebrospinal Fluid Res. 2004, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Leon, M.J.; Li, Y.; Okamura, N.; Tsui, W.H.; Saint-Louis, L.A.; Glodzik, L.; Osorio, R.S.; Fortea, J.; Butler, T.; Pirraglia, E.; et al. Cerebrospinal Fluid Clearance in Alzheimer Disease Measured with Dynamic PET. J. Nucl. Med. 2017, 58, 1471–1476. [Google Scholar] [CrossRef] [Green Version]
- Mollanji, R.; Bozanovic-Sosic, R.; Silver, I.; Li, B.; Kim, C.; Midha, R.; Johnston, M. Intracranial pressure accommodation is impaired by blocking pathways leading to extracranial lymphatics. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2001, 280, R1573–R1581. [Google Scholar] [CrossRef] [Green Version]
- Földi, M.; Csanda, E.; Simon, M.; Obál, F.; Schneider, I.; Dobranovics, I.; Zoltán, O.T.; Kozma, M.; Poberai, M. Lymphogenic haemangiopathy. “Prelymphatic” pathways in the wall of cerebral and cervical blood vessels. Angiologica 1968, 5, 250–262. [Google Scholar]
- Qiuhang, Z.; Zhenlin, W.; Yan, Q.; Jun, H.; Yongfeng, S.; Bo, H. Lymphatic drainage of the skull base: Comparative anatomic and advanced imaging studies in the rabbit and human with implications for spread of nasopharyngeal carcinoma. Lymphology 2010, 43, 98–109. [Google Scholar]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018, 21, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef] [Green Version]
- Shibata-Germanos, S.; Goodman, J.R.; Grieg, A.; Trivedi, C.A.; Benson, B.C.; Foti, S.C.; Faro, A.; Castellan, R.F.P.; Correra, R.M.; Barber, M.; et al. Structural and functional conservation of non-lumenized lymphatic endothelial cells in the mammalian leptomeninges. Acta Neuropathol. 2020, 139, 383–401. [Google Scholar] [CrossRef] [Green Version]
- Alitalo, K. The lymphatic vasculature in disease. Nat. Med. 2011, 17, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; García-Verdugo, J.M.; Soriano-Navarro, M.; Srinivasan, R.S.; Scallan, J.P.; Singh, M.K.; Epstein, J.A.; Oliver, G. Lymphatic endothelial progenitors bud from the cardinal vein and intersomitic vessels in mammalian embryos. Blood 2012, 120, 2340–2348. [Google Scholar] [CrossRef]
- Butler, M.G.; Isogai, S.; Weinstein, B.M. Lymphatic development. Birth Defects Res. Part C Embryo Today Rev. 2009, 87, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Breslin, J.W.; Yang, Y.; Scallan, J.P.; Sweat, R.S.; Adderley, S.P.; Murfee, W.L. Lymphatic Vessel Network Structure and Physiology. Compr. Physiol. 2018, 9, 207–299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Oliver, G. Development of the mammalian lymphatic vasculature. J. Clin. Investig. 2014, 124, 888–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkkainen, M.J.; Petrova, T.V. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene 2000, 19, 5598–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antila, S.; Karaman, S.; Nurmi, H.; Airavaara, M.; Voutilainen, M.H.; Mathivet, T.; Chilov, D.; Li, Z.; Koppinen, T.; Park, J.-H.; et al. Development and plasticity of meningeal lymphatic vessels. J. Exp. Med. 2017, 214, 3645–3667. [Google Scholar] [CrossRef]
- Wang, X.N.; McGovern, N.; Gunawan, M.; Richardson, C.; Windebank, M.; Siah, T.W.; Lim, H.Y.; Fink, K.; Yao Li, J.L.; Ng, L.G.; et al. A three-dimensional atlas of human dermal leukocytes, lymphatics, and blood vessels. J. Investig. Dermatol. 2014, 134, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Dackor, R.T.; Fritz-Six, K.; Dunworth, W.P.; Gibbons, C.L.; Smithies, O.; Caron, K.M. Hydrops fetalis, cardiovascular defects, and embryonic lethality in mice lacking the calcitonin receptor-like receptor gene. Mol. Cell. Biol. 2006, 26, 2511–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerji, S.; Ni, J.; Wang, S.X.; Clasper, S.; Su, J.; Tammi, R.; Jones, M.; Jackson, D.G. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J. Cell Biol. 1999, 144, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.G. Immunological functions of hyaluronan and its receptors in the lymphatics. Immunol. Rev. 2009, 230, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Bos, F.L.; Caunt, M.; Peterson-Maduro, J.; Planas-Paz, L.; Kowalski, J.; Karpanen, T.; van Impel, A.; Tong, R.; Ernst, J.A.; Korving, J.; et al. CCBE1 is essential for mammalian lymphatic vascular development and enhances the lymphangiogenic effect of vascular endothelial growth factor-C in vivo. Circ. Res. 2011, 109, 486–491. [Google Scholar] [CrossRef]
- Janssen, L.; Dupont, L.; Bekhouche, M.; Noel, A.; Leduc, C.; Voz, M.; Peers, B.; Cataldo, D.; Apte, S.S.; Dubail, J.; et al. ADAMTS3 activity is mandatory for embryonic lymphangiogenesis and regulates placental angiogenesis. Angiogenesis 2016, 19, 53–65. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, G.; Korhonen, E.A.; Waltari, M.; Saharinen, P.; Laakkonen, P.; Alitalo, K. Loss of endothelial Tie1 receptor impairs lymphatic vessel development-brief report. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 207–209. [Google Scholar] [CrossRef] [Green Version]
- Dellinger, M.; Hunter, R.; Bernas, M.; Gale, N.; Yancopoulos, G.; Erickson, R.; Witte, M. Defective remodeling and maturation of the lymphatic vasculature in Angiopoietin-2 deficient mice. Dev. Biol. 2008, 319, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Petrova, T.V.; Karpanen, T.; Norrmén, C.; Mellor, R.; Tamakoshi, T.; Finegold, D.; Ferrell, R.; Kerjaschki, D.; Mortimer, P.; Ylä-Herttuala, S.; et al. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 2004, 10, 974–981. [Google Scholar] [CrossRef]
- Fatima, A.; Wang, Y.; Uchida, Y.; Norden, P.; Liu, T.; Culver, A.; Dietz, W.H.; Culver, F.; Millay, M.; Mukouyama, Y.-S.; et al. Foxc1 and Foxc2 deletion causes abnormal lymphangiogenesis and correlates with ERK hyperactivation. J. Clin. Investig. 2016, 126, 2437–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Shepro, D. Microvascular Research: Biology and Pathology, Two-Volume Set; Elsevier: Amsterdam, The Netherlands, 2005; Volume 1. [Google Scholar]
- Hirosue, S.; Vokali, E.; Raghavan, V.R.; Rincon-Restrepo, M.; Lund, A.W.; Corthésy-Henrioud, P.; Capotosti, F.; Halin Winter, C.; Hugues, S.; Swartz, M.A. Steady-state antigen scavenging, cross-presentation, and CD8+ T cell priming: A new role for lymphatic endothelial cells. J. Immunol. 2014, 192, 5002–5011. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, B.A.; Burchill, M.A.; Kedl, R.M. Antigen capture and archiving by lymphatic endothelial cells following vaccination or viral infection. Nat. Commun. 2014, 5, 3989. [Google Scholar] [CrossRef] [PubMed]
- Kedl, R.M.; Lindsay, R.S.; Finlon, J.M.; Lucas, E.D.; Friedman, R.S.; Tamburini, B.A.J. Migratory dendritic cells acquire and present lymphatic endothelial cell-archived antigens during lymph node contraction. Nat. Commun. 2017, 8, 2034. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Ineichen, B.V.; Detmar, M.; Proulx, S.T. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat. Commun. 2017, 8, 1434. [Google Scholar] [CrossRef] [Green Version]
- Absinta, M.; Ha, S.K.; Nair, G.; Sati, P.; Luciano, N.J.; Palisoc, M.; Louveau, A.; Zaghloul, K.A.; Pittaluga, S.; Kipnis, J.; et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife 2017, 6, e29738. [Google Scholar] [CrossRef]
- Planas, A.M.; Gómez-Choco, M.; Urra, X.; Gorina, R.; Caballero, M.; Chamorro, Á. Brain-Derived Antigens in Lymphoid Tissue of Patients with Acute Stroke. J. Immunol. 2012, 188, 2156–2163. [Google Scholar] [CrossRef]
- Hu, X.; Deng, Q.; Ma, L.; Li, Q.; Chen, Y.; Liao, Y.; Zhou, F.; Zhang, C.; Shao, L.; Feng, J.; et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell Res. 2020, 30, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Chamorro, A.; Meisel, A.; Planas, A.M.; Urra, X.; Van De Beek, D.; Veltkamp, R. The immunology of acute stroke. Nat. Rev. Neurol. 2012, 8, 401–410. [Google Scholar] [CrossRef]
- Engelhardt, B.; Carare, R.O.; Bechmann, I.; Flügel, A.; Laman, J.D.; Weller, R.O. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016, 132, 317–338. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, L.; Xu, H.; Xing, L.; Zhuang, Z.; Zheng, Y.; Li, X.; Wang, C.; Chen, S.; Guo, Z.; et al. Meningeal lymphatics clear erythrocytes that arise from subarachnoid hemorrhage. Nat. Commun. 2020, 11, 3159. [Google Scholar] [CrossRef]
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-R.; Yang, J.-H.; Wang, X.; Yao, Z.-B. Induced dural lymphangiogenesis facilities soluble amyloid-beta clearance from brain in a transgenic mouse model of Alzheimer’s disease. Neural Regener. Res. 2018, 13, 709–716. [Google Scholar] [CrossRef]
- Norwood, J.N.; Zhang, Q.; Card, D.; Craine, A.; Ryan, T.M.; Drew, P.J. Anatomical basis and physiological role of cerebrospinal fluid transport through the murine cribriform plate. eLife 2019, 8, e44278. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, A.; Papaiconomou, C.; Johnston, M. Lymphatic vessels gain access to cerebrospinal fluid through unique association with olfactory nerves. Lymphat. Res. Biol. 2004, 2, 139–146. [Google Scholar] [CrossRef]
- Koh, L.; Zakharov, A.; Nagra, G.; Armstrong, D.; Friendship, R.; Johnston, M. Development of cerebrospinal fluid absorption sites in the pig and rat: Connections between the subarachnoid space and lymphatic vessels in the olfactory turbinates. Anat. Embryol. 2006, 211, 335–344. [Google Scholar] [CrossRef]
- Weller, R.O.; Sharp, M.M.; Christodoulides, M.; Carare, R.O.; Møllgård, K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol. 2018, 135, 363–385. [Google Scholar] [CrossRef] [Green Version]
- Johnston, M. The importance of lymphatics in cerebrospinal fluid transport. Lymphat. Res. Biol. 2003, 1, 41–44; discussion 45. [Google Scholar] [CrossRef]
- Johnston, M.; Papaiconomou, C. Cerebrospinal fluid transport: A lymphatic perspective. News Physiol. Sci. 2002, 17, 227–230. [Google Scholar] [CrossRef]
- Osaka, K.; Handa, H.; Matsumoto, S.; Yasuda, M. Development of the cerebrospinal fluid pathway in the normal and abnormal human embryos. Childs Brain 1980, 6, 26–38. [Google Scholar] [CrossRef]
- Mollanji, R.; Papaiconomou, C.; Boulton, M.; Midha, R.; Johnston, M. Comparison of cerebrospinal fluid transport in fetal and adult sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R1215–R1223. [Google Scholar] [CrossRef]
- Zakharov, A.; Papaiconomou, C.; Koh, L.; Djenic, J.; Bozanovic-Sosic, R.; Johnston, M. Integrating the roles of extracranial lymphatics and intracranial veins in cerebrospinal fluid absorption in sheep. Microvasc. Res. 2004, 67, 96–104. [Google Scholar] [CrossRef]
- Erlich, S.S.; McComb, J.G.; Hyman, S.; Weiss, M.H. Ultrastructural morphology of the olfactory pathway for cerebrospinal fluid drainage in the rabbit. J. Neurosurg. 1986, 64, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Weller, R.O.; Kida, S.; Zhang, E.T. Pathways of fluid drainage from the brain—Morphological aspects and immunological significance in rat and man. Brain Pathol. 1992, 2, 277–284. [Google Scholar] [CrossRef]
- Kida, S.; Pantazis, A.; Weller, R.O. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol. Appl. Neurobiol. 1993, 19, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Young, A.; Hay, J.; Armstrong, D.; Flessner, M.; Schwartz, M.; Johnston, M. Drainage of CSF through lymphatic pathways and arachnoid villi in sheep: Measurement of 125I-albumin clearance. Neuropathol. Appl. Neurobiol. 1996, 22, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Armstrong, D.; Flessner, M.; Hay, J.; Szalai, J.P.; Johnston, M. Raised intracranial pressure increases CSF drainage through arachnoid villi and extracranial lymphatics. Am. J. Physiol. 1998, 275, R889–R896. [Google Scholar] [CrossRef]
- Goldmann, J.; Kwidzinski, E.; Brandt, C.; Mahlo, J.; Richter, D.; Bechmann, I. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J. Leukoc. Biol. 2006, 80, 797–801. [Google Scholar] [CrossRef]
- Zakharov, A.; Papaiconomou, C.; Djenic, J.; Midha, R.; Johnston, M. Lymphatic cerebrospinal fluid absorption pathways in neonatal sheep revealed by subarachnoid injection of Microfil. Neuropathol. Appl. Neurobiol. 2003, 29, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Melin, E.; Eide, P.K.; Ringstad, G. In vivo assessment of cerebrospinal fluid efflux to nasal mucosa in humans. Sci. Rep. 2020, 10, 14974. [Google Scholar] [CrossRef] [PubMed]
- Silver, I.; Kim, C.; Mollanji, R.; Johnston, M. Cerebrospinal fluid outflow resistance in sheep: Impact of blocking cerebrospinal fluid transport through the cribriform plate. Neuropathol. Appl. Neurobiol. 2002, 28, 67–74. [Google Scholar] [CrossRef]
- Mollanji, R.; Bozanovic-Sosic, R.; Zakharov, A.; Makarian, L.; Johnston, M.G. Blocking cerebrospinal fluid absorption through the cribriform plate increases resting intracranial pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R1593–R1599. [Google Scholar] [CrossRef] [Green Version]
- Brea, D.; Poon, C.; Murphy, M.; Lubitz, G.; Iadecola, C.; Anrather, J. Ablation of nasal-associated lymphoid tissue does not affect focal ischemic brain injury in mice. PLoS ONE 2018, 13, e0205470. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, L.; Xu, H.; Wang, Y.; Liang, Q. The lymphatic drainage system of the CNS plays a role in lymphatic drainage, immunity, and neuroinflammation in stroke. J. Leukoc. Biol. 2021, 110, 283–291. [Google Scholar] [CrossRef]
- Karpanen, T.; Mäkinen, T. Regulation of lymphangiogenesis—From cell fate determination to vessel remodeling. Exp. Cell Res. 2006, 312, 575–583. [Google Scholar] [CrossRef]
- Stacker, S.A.; Achen, M.G.; Jussila, L.; Baldwin, M.E.; Alitalo, K. Lymphangiogenesis and cancer metastasis. Nat. Rev. Cancer 2002, 2, 573–583. [Google Scholar] [CrossRef]
- Randolph, G.J.; Ivanov, S.; Zinselmeyer, B.H.; Scallan, J.P. The Lymphatic System: Integral Roles in Immunity. Annu. Rev. Immunol. 2017, 35, 31–52. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, J.M.; Boardman, K.C.; Swartz, M.A. Characterization of lymphangiogenesis in a model of adult skin regeneration. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1402–H1410. [Google Scholar] [CrossRef]
- Oliver, G. Lymphatic vasculature development. Nat. Rev. Immunol. 2004, 4, 35–45. [Google Scholar] [CrossRef]
- Baluk, P.; Tammela, T.; Ator, E.; Lyubynska, N.; Achen, M.G.; Hicklin, D.J.; Jeltsch, M.; Petrova, T.V.; Pytowski, B.; Stacker, S.A.; et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Investig. 2005, 115, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benest, A.V.; Harper, S.J.; Herttuala, S.Y.; Alitalo, K.; Bates, D.O. VEGF-C induced angiogenesis preferentially occurs at a distance from lymphangiogenesis. Cardiovasc. Res. 2008, 78, 315–323. [Google Scholar] [CrossRef]
- Mauri, C.; Wang, G.; Schulte-Merker, S. From fish embryos to human patients: Lymphangiogenesis in development and disease. Curr. Opin. Immunol. 2018, 53, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Rauniyar, K.; Chronowska, E.; Mattonet, K.; Maina, E.W.; Koistinen, H.; Stenman, U.H.; Alitalo, K.; Jeltsch, M. KLK3/PSA and cathepsin D activate VEGF-C and VEGF-D. Elife 2019, 8, e44478. [Google Scholar] [CrossRef]
- Xu, Y.; Yuan, L.; Mak, J.; Pardanaud, L.; Caunt, M.; Kasman, I.; Larrivée, B.; Del Toro, R.; Suchting, S.; Medvinsky, A.; et al. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J. Cell Biol. 2010, 188, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ulvmar, M.H.; Stanczuk, L.; Martinez-Corral, I.; Frye, M.; Alitalo, K.; Mäkinen, T. Heterogeneity in VEGFR3 levels drives lymphatic vessel hyperplasia through cell-autonomous and non-cell-autonomous mechanisms. Nat. Commun. 2018, 9, 1296. [Google Scholar] [CrossRef]
- Esposito, E.; Ahn, B.J.; Shi, J.; Nakamura, Y.; Park, J.H.; Mandeville, E.T.; Yu, Z.; Chan, S.J.; Desai, R.; Hayakawa, A.; et al. Brain-to-cervical lymph node signaling after stroke. Nat. Commun. 2019, 10, 5306. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.L.; Xia, Z.L.; Yan, Z.W.; Chen, Y.S.; Yang, M.F. Effects of blockade of cerebral lymphatic drainage on cerebral ischemia after middle cerebral artery occlusion in rats. Clin. Hemorheol. Microcirc. 2000, 23, 321–325. [Google Scholar] [PubMed]
- Yanev, P.; Poinsatte, K.; Hominick, D.; Khurana, N.; Zuurbier, K.R.; Berndt, M.; Plautz, E.J.; Dellinger, M.T.; Stowe, A.M. Impaired meningeal lymphatic vessel development worsens stroke outcome. J. Cereb. Blood Flow Metab. 2020, 40, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Gordon, E.J.; Rao, S.; Pollard, J.W.; Nutt, S.L.; Lang, R.A.; Harvey, N.L. Macrophages define dermal lymphatic vessel calibre during development by regulating lymphatic endothelial cell proliferation. Development 2010, 137, 3899–3910. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, S.; Scallan, J.P.; Kim, K.W.; Werth, K.; Johnson, M.W.; Saunders, B.T.; Wang, P.L.; Kuan, E.L.; Straub, A.C.; Ouhachi, M.; et al. CCR7 and IRF4-dependent dendritic cells regulate lymphatic collecting vessel permeability. J. Clin. Investig. 2016, 126, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.; Flores-Borja, F.; Nassiri, S.; Miranda, E.; Lawler, K.; Grigoriadis, A.; Monypenny, J.; Gillet, C.; Owen, J.; Gordon, P.; et al. Integrin-Mediated Macrophage Adhesion Promotes Lymphovascular Dissemination in Breast Cancer. Cell Rep. 2019, 27, 1967–1978.e1964. [Google Scholar] [CrossRef] [Green Version]
- Bieniasz-Krzywiec, P.; Martín-Pérez, R.; Ehling, M.; García-Caballero, M.; Pinioti, S.; Pretto, S.; Kroes, R.; Aldeni, C.; Di Matteo, M.; Prenen, H.; et al. Podoplanin-Expressing Macrophages Promote Lymphangiogenesis and Lymphoinvasion in Breast Cancer. Cell Metab. 2019, 30, 917–936.e910. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.W.; Chong, S.Z.; Wong, F.H.S.; Evrard, M.; Tan, S.M.-L.; Keeble, J.; Kemeny, D.M.; Ng, L.G.; Abastado, J.-P.; Angeli, V. Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood 2013, 122, 3666–3677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.; Choi, J.S.; Rho, C.R.; Joo, C.K.; Lee, S.K. MicroRNA miR-466 inhibits Lymphangiogenesis by targeting prospero-related homeobox 1 in the alkali burn corneal injury model. J. Biomed. Sci. 2015, 22, 3. [Google Scholar] [CrossRef] [Green Version]
- Kazenwadel, J.; Michael, M.Z.; Harvey, N.L. Prox1 expression is negatively regulated by miR-181 in endothelial cells. Blood 2010, 116, 2395–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrioli, D.M.; Karpanen, T.; Dabouras, V.; Jurisic, G.; van de Hoek, G.; Shin, J.W.; Marino, D.; Kälin, R.E.; Leidel, S.; Cinelli, P.; et al. miR-31 functions as a negative regulator of lymphatic vascular lineage-specific differentiation in vitro and vascular development in vivo. Mol. Cell. Biol. 2010, 30, 3620–3634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunworth, W.P.; Cardona-Costa, J.; Bozkulak, E.C.; Kim, J.D.; Meadows, S.; Fischer, J.C.; Wang, Y.; Cleaver, O.; Qyang, Y.; Ober, E.A.; et al. Bone morphogenetic protein 2 signaling negatively modulates lymphatic development in vertebrate embryos. Circ. Res. 2014, 114, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, Á.; Molnár, K.; Nógrádi, B.; Hernádi, Z.; Nyúl-Tóth, Á.; Wilhelm, I.; Krizbai, I.A. Neurovascular Inflammaging in Health and Disease. Cells 2020, 9, 1614. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Ham, P.B., 3rd; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef]
- He, H.; Lam, M.; McCormick, T.S.; Distelhorst, C.W. Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J. Cell Biol. 1997, 138, 1219–1228. [Google Scholar] [CrossRef]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Fleming, I.; Busse, R. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Zhang, K.; Li, P.; Zhu, L.; Xu, J.; Yang, B.; Hu, X.; Lu, Z.; Chen, J. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Res. Rev. 2017, 34, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurata, T.; Lukic, V.; Kozuki, M.; Wada, D.; Miyazaki, K.; Morimoto, N.; Ohta, Y.; Deguchi, K.; Ikeda, Y.; Kamiya, T.; et al. Telmisartan reduces progressive accumulation of cellular amyloid beta and phosphorylated tau with inflammatory responses in aged spontaneously hypertensive stroke resistant rat. J. Stroke Cerebrovasc. Dis. 2014, 23, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Rutten, B.P.; Schmitz, C.; Gerlach, O.H.; Oyen, H.M.; de Mesquita, E.B.; Steinbusch, H.W.; Korr, H. The aging brain: Accumulation of DNA damage or neuron loss? Neurobiol. Aging 2007, 28, 91–98. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; de Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-aging: An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Lourbopoulos, A.; Ertürk, A.; Hellal, F. Microglia in action: How aging and injury can change the brain’s guardians. Front. Cell. Neurosci. 2015, 9, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suenaga, J.; Hu, X.; Pu, H.; Shi, Y.; Hassan, S.H.; Xu, M.; Leak, R.K.; Stetler, R.A.; Gao, Y.; Chen, J. White matter injury and microglia/macrophage polarization are strongly linked with age-related long-term deficits in neurological function after stroke. Exp. Neurol. 2015, 272, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 2011, 34, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sun, L.; Wang, H.; Ma, H.; Liu, G.; Zhao, Y. Changes of CD4+CD25+Foxp3+ regulatory T cells in aged Balb/c mice. J. Leukoc. Biol. 2007, 81, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, M.; Shah, I.M. Neuroinflammation and cerebrovascular disease in old age: A translational medicine perspective. J. Aging Res. 2011, 2011, 857484. [Google Scholar] [CrossRef] [Green Version]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Golomb, M.R.; Fullerton, H.J.; Nowak-Gottl, U.; Deveber, G. Male predominance in childhood ischemic stroke: Findings from the international pediatric stroke study. Stroke 2009, 40, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Yuan, R.; Benashski, S.E.; McCullough, L.D. Changes in experimental stroke outcome across the life span. J. Cereb. Blood Flow Metab. 2009, 29, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, P.P.; Feng, X.; Barker, J.L.; Smith, S.V.; Rubinow, D.R. Sex-related differences in neuronal cell survival and signaling in rats. Neurosci. Lett. 2003, 337, 65–68. [Google Scholar] [CrossRef]
- Du, L.; Bayir, H.; Lai, Y.; Zhang, X.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Clark, R.S. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J. Biol. Chem. 2004, 279, 38563–38570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Hurn, P.D.; Roselli, C.E.; Alkayed, N.J. Role of P450 aromatase in sex-specific astrocytic cell death. J. Cereb. Blood Flow Metab. 2007, 27, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Oyarzabal, E.A.; Yang, R.; Murphy, S.J.; Hurn, P.D. A novel method for assessing sex-specific and genotype-specific response to injury in astrocyte culture. J. Neurosci. Methods 2008, 171, 214–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, J.T.; McCullough, L.D. Pathways to ischemic neuronal cell death: Are sex differences relevant? J. Transl. Med. 2008, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairweather, D. Sex differences in inflammation during atherosclerosis. Clin. Med. Insights Cardiol. 2014, 8, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhang, L.; Ding, M.; Luo, Z.; Yuan, S.; Bansal, M.B.; Gilkeson, G.; Lang, R.; Jiang, W. Estrogen decreases tight junction protein ZO-1 expression in human primary gut tissues. Clin. Immunol. 2017, 183, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisamoto, K.; Ohmichi, M.; Kanda, Y.; Adachi, K.; Nishio, Y.; Hayakawa, J.; Mabuchi, S.; Takahashi, K.; Tasaka, K.; Miyamoto, Y.; et al. Induction of endothelial nitric-oxide synthase phosphorylation by the raloxifene analog LY117018 is differentially mediated by Akt and extracellular signal-regulated protein kinase in vascular endothelial cells. J. Biol. Chem. 2001, 276, 47642–47649. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Takahashi, E.; Kobayashi, M.; Goto, M.; Krust, A.; Chambon, P.; Iguchi, T. The estrogen-responsive adrenomedullin and receptor-modifying protein 3 gene identified by DNA microarray analysis are directly regulated by estrogen receptor. J. Mol. Endocrinol. 2006, 36, 81–89. [Google Scholar] [CrossRef]
- Ikeda, K.; Arao, Y.; Otsuka, H.; Kikuchi, A.; Kayama, F. Estrogen and phytoestrogen regulate the mRNA expression of adrenomedullin and adrenomedullin receptor components in the rat uterus. Mol. Cell. Endocrinol. 2004, 223, 27–34. [Google Scholar] [CrossRef]
- Huang, A.; Kaley, G. Gender-specific regulation of cardiovascular function: Estrogen as key player. Microcirculation 2004, 11, 9–38. [Google Scholar] [CrossRef]
- Li, L.; Hisamoto, K.; Kim, K.H.; Haynes, M.P.; Bauer, P.M.; Sanjay, A.; Collinge, M.; Baron, R.; Sessa, W.C.; Bender, J.R. Variant estrogen receptor-c-Src molecular interdependence and c-Src structural requirements for endothelial NO synthase activation. Proc. Natl. Acad. Sci. USA 2007, 104, 16468–16473. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.L.; Limacher, M.; Assaf, A.R.; Bassford, T.; Beresford, S.A.; Black, H.; Bonds, D.; Brunner, R.; Brzyski, R.; Caan, B.; et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA 2004, 291, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 2017, 8, 33. [Google Scholar] [CrossRef]
- Charest-Marcotte, A.; Dufour, C.R.; Wilson, B.J.; Tremblay, A.M.; Eichner, L.J.; Arlow, D.H.; Mootha, V.K.; Giguère, V. The homeobox protein Prox1 is a negative modulator of ERR{alpha}/PGC-1{alpha} bioenergetic functions. Genes Dev. 2010, 24, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Morfoisse, F.; Tatin, F.; Chaput, B.; Therville, N.; Vaysse, C.; Métivier, R.; Malloizel-Delaunay, J.; Pujol, F.; Godet, A.C.; De Toni, F.; et al. Lymphatic Vasculature Requires Estrogen Receptor-α Signaling to Protect From Lymphedema. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1346–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silha, J.V.; Krsek, M.; Sucharda, P.; Murphy, L.J. Angiogenic factors are elevated in overweight and obese individuals. Int. J. Obes. 2005, 29, 1308–1314. [Google Scholar] [CrossRef] [Green Version]
- Greene, R.; Fowler, R. Physical therapy management of primary lymphedema in the lower extremities: A case report. Physiother. Theory Pract. 2010, 26, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Torre, O.; Elia, D.; Caminati, A.; Harari, S. New insights in lymphangioleiomyomatosis and pulmonary Langerhans cell histiocytosis. Eur. Respir. Rev. 2017, 26, 170042. [Google Scholar] [CrossRef] [PubMed]
- Bridenbaugh, E.A.; Nizamutdinova, I.T.; Jupiter, D.; Nagai, T.; Thangaswamy, S.; Chatterjee, V.; Gashev, A.A. Lymphatic muscle cells in rat mesenteric lymphatic vessels of various ages. Lymphat. Res. Biol. 2013, 11, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.J.; Rahbar, E.; Gashev, A.A.; Zawieja, D.C.; Moore, J.E., Jr. Determinants of valve gating in collecting lymphatic vessels from rat mesentery. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H48–H60. [Google Scholar] [CrossRef] [Green Version]
- Zolla, V.; Nizamutdinova, I.T.; Scharf, B.; Clement, C.C.; Maejima, D.; Akl, T.; Nagai, T.; Luciani, P.; Leroux, J.C.; Halin, C.; et al. Aging-related anatomical and biochemical changes in lymphatic collectors impair lymph transport, fluid homeostasis, and pathogen clearance. Aging Cell 2015, 14, 582–594. [Google Scholar] [CrossRef]
- Gasheva, O.Y.; Knippa, K.; Nepiushchikh, Z.V.; Muthuchamy, M.; Gashev, A.A. Age-related alterations of active pumping mechanisms in rat thoracic duct. Microcirculation 2007, 14, 827–839. [Google Scholar] [CrossRef]
- Gasheva, O.Y.; Zawieja, D.C.; Gashev, A.A. Contraction-initiated NO-dependent lymphatic relaxation: A self-regulatory mechanism in rat thoracic duct. J. Physiol. 2006, 575, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Shang, T.; Liang, J.; Kapron, C.M.; Liu, J. Pathophysiology of aged lymphatic vessels. Aging 2019, 11, 6602–6613. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.; Tharakan, B. Vascular hyperpermeability and aging. Aging Dis. 2014, 5, 114–125. [Google Scholar] [CrossRef]
- Pal, S.; Meininger, C.J.; Gashev, A.A. Aged Lymphatic Vessels and Mast Cells in Perilymphatic Tissues. Int. J. Mol. Sci. 2017, 18, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaman, S.; Buschle, D.; Luciani, P.; Leroux, J.C.; Detmar, M.; Proulx, S.T. Decline of lymphatic vessel density and function in murine skin during aging. Angiogenesis 2015, 18, 489–498. [Google Scholar] [CrossRef]
- Zhou, Y.; Cai, J.; Zhang, W.; Gong, X.; Yan, S.; Zhang, K.; Luo, Z.; Sun, J.; Jiang, Q.; Lou, M. Impairment of the Glymphatic Pathway and Putative Meningeal Lymphatic Vessels in the Aging Human. Ann. Neurol. 2020, 87, 357–369. [Google Scholar] [CrossRef]
- Akl, T.J.; Nagai, T.; Coté, G.L.; Gashev, A.A. Mesenteric lymph flow in adult and aged rats. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1828–H1840. [Google Scholar] [CrossRef] [Green Version]
- Nagai, T.; Bridenbaugh, E.A.; Gashev, A.A. Aging-associated alterations in contractility of rat mesenteric lymphatic vessels. Microcirculation 2011, 18, 463–473. [Google Scholar] [CrossRef]
- Thangaswamy, S.; Bridenbaugh, E.A.; Gashev, A.A. Evidence of increased oxidative stress in aged mesenteric lymphatic vessels. Lymphat. Res. Biol. 2012, 10, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, N.; Chalasani, M.L.S.; Li, T.M.; Feng, Z.; Shipman, W.D.; Lu, T.T. Lymphatic Function in Autoimmune Diseases. Front. Immunol. 2019, 10, 519. [Google Scholar] [CrossRef] [Green Version]
- Da Mesquita, S.; Herz, J.; Wall, M.; Dykstra, T.; de Lima, K.A.; Norris, G.T.; Dabhi, N.; Kennedy, T.; Baker, W.; Kipnis, J. Aging-associated deficit in CCR7 is linked to worsened glymphatic function, cognition, neuroinflammation, and β-amyloid pathology. Sci. Adv. 2021, 7, eabe4601. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, Z.-Y.; Huang, T.-T.; Zhou, Y.-X.; Wang, X.; Yang, L.-Q.; Chen, Z.-A.; Yu, W.-F.; Li, P.-Y. The peripheral immune response after stroke-A double edge sword for blood-brain barrier integrity. CNS Neurosci. Ther. 2018, 24, 1115–1128. [Google Scholar] [CrossRef]
- Liu, Q.; Jin, W.-N.; Liu, Y.; Shi, K.; Sun, H.; Zhang, F.; Zhang, C.; Gonzales, R.J.; Sheth, K.N.; La Cava, A. Brain ischemia suppresses immunity in the periphery and brain via different neurogenic innervations. Immunity 2017, 46, 474–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, S.; Vannberg, F.O.; Dixon, J.B. Lymphatic transport of exosomes as a rapid route of information dissemination to the lymph node. Sci. Rep. 2016, 6, 24436. [Google Scholar] [CrossRef] [Green Version]
- Seifert, H.A.; Pennypacker, K.R. Molecular and cellular immune responses to ischemic brain injury. Transl. Stroke Res. 2014, 5, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Petrovic-Djergovic, D.; Goonewardena, S.N.; Pinsky, D.J. Inflammatory Disequilibrium in Stroke. Circ. Res. 2016, 119, 142–158. [Google Scholar] [CrossRef] [Green Version]
- Jauch, E.C.; Lindsell, C.; Broderick, J.; Fagan, S.C.; Tilley, B.C.; Levine, S.R. Association of Serial Biochemical Markers with Acute Ischemic Stroke. Stroke 2006, 37, 2508–2513. [Google Scholar] [CrossRef] [Green Version]
- Gelderblom, M.; Daehn, T.; Schattling, B.; Ludewig, P.; Bernreuther, C.; Arunachalam, P.; Matschke, J.; Glatzel, M.; Gerloff, C.; Friese, M.A.; et al. Plasma levels of neuron specific enolase quantify the extent of neuronal injury in murine models of ischemic stroke and multiple sclerosis. Neurobiol. Dis. 2013, 59, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; He, D.; Zeng, Y.-Y.; Zhu, L.; Yang, C.; Lu, Y.-J.; Huang, J.-Q.; Cheng, X.-Y.; Huang, X.-H.; Tan, X.-J. The spleen may be an important target of stem cell therapy for stroke. J. Neuroinflamm. 2019, 16, 20. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Choco, M.; Doucerain, C.; Urra, X.; Planas, A.M.; Chamorro, Á. Presence of heat shock protein 70 in secondary lymphoid tissue correlates with stroke prognosis. J. Neuroimmunol. 2014, 270, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauvillain, C.; Cunin, P.; Doni, A.; Scotet, M.; Jaillon, S.; Loiry, M.L.; Magistrelli, G.; Masternak, K.; Chevailler, A.; Delneste, Y.; et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood 2011, 117, 1196–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pösel, C.; Uri, A.; Schulz, I.; Boltze, J.; Weise, G.; Wagner, D.-C. Flow cytometric characterization of brain dendritic cell subsets after murine stroke. Exp. Transl. Stroke Med. 2014, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Pu, T.; Zou, W.; Feng, W.; Zhang, Y.; Wang, L.; Wang, H.; Xiao, M. Persistent Malfunction of Glymphatic and Meningeal Lymphatic Drainage in a Mouse Model of Subarachnoid Hemorrhage. Exp. Neurobiol. 2019, 28, 104–118. [Google Scholar] [CrossRef]
- Sun, B.L.; Xia, Z.L.; Wang, J.R.; Yuan, H.; Li, W.X.; Chen, Y.S.; Yang, M.F.; Zhang, S.M. Effects of blockade of cerebral lymphatic drainage on regional cerebral blood flow and brain edema after subarachnoid hemorrhage. Clin. Hemorheol. Microcirc. 2006, 34, 227–232. [Google Scholar]
- Vahidy, F.S.; Parsha, K.N.; Rahbar, M.H.; Lee, M.; Bui, T.T.; Nguyen, C.; Barreto, A.D.; Bambhroliya, A.B.; Sahota, P.; Yang, B.; et al. Acute splenic responses in patients with ischemic stroke and intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2016, 36, 1012–1021. [Google Scholar] [CrossRef]
- Ajmo, C.T., Jr.; Vernon, D.O.; Collier, L.; Hall, A.A.; Garbuzova-Davis, S.; Willing, A.; Pennypacker, K.R. The spleen contributes to stroke-induced neurodegeneration. J. Neurosci. Res. 2008, 86, 2227–2234. [Google Scholar] [CrossRef] [Green Version]
- Offner, H. Subramanianm, S.; Parker, S.M.; Afentoulis, M.E.; Vandenbark, A.A.; Hurn, P.D. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb. Blood Flow. Metab. 2006, 26, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, K.Z.; Dale, V.Q.; Dénes, A.; Bennett, G.; Rothwell, N.J.; Allan, S.M.; McColl, B.W. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J. Cereb. Blood Flow Metab. 2009, 29, 1764–1768. [Google Scholar] [CrossRef] [Green Version]
- Ferrarese, C.; Mascarucci, P.; Zoia, C.; Cavarretta, R.; Frigo, M.; Begni, B.; Sarinella, F.; Frattola, L.; De Simoni, M.G. Increased cytokine release from peripheral blood cells after acute stroke. J. Cereb. Blood Flow Metab. 1999, 19, 1004–1009. [Google Scholar] [CrossRef] [Green Version]
- Seifert, H.A.; Hall, A.A.; Chapman, C.B.; Collier, L.A.; Willing, A.E.; Pennypacker, K.R. A transient decrease in spleen size following stroke corresponds to splenocyte release into systemic circulation. J. Neuroimmune Pharmacol. 2012, 7, 1017–1024. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, L.; Alfieri, A.; McColl, B.W. Experimental Stroke Differentially Affects Discrete Subpopulations of Splenic Macrophages. Front. Immunol. 2018, 9, 1108. [Google Scholar] [CrossRef]
- Ran, Y.; Liu, Z.; Huang, S.; Shen, J.; Li, F.; Zhang, W.; Chen, C.; Geng, X.; Ji, Z.; Du, H.; et al. Splenectomy Fails to Provide Long-Term Protection Against Ischemic Stroke. Aging Dis. 2018, 9, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Miró-Mur, F.; Urra, X.; Gallizioli, M.; Chamorro, A.; Planas, A.M. Antigen Presentation After Stroke. Neurotherapeutics 2016, 13, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Członkowska, A.; Cyrta, B.; Korlak, J. Immunological observations on patients with acute cerebral vascular disease. J. Neurol. Sci. 1979, 43, 455–464. [Google Scholar] [CrossRef]
- Prass, K.; Meisel, C.; Höflich, C.; Braun, J.; Halle, E.; Wolf, T.; Ruscher, K.; Victorov, I.V.; Priller, J.; Dirnagl, U.; et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J. Exp. Med. 2003, 198, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Liesz, A.; Hu, X.; Kleinschnitz, C.; Offner, H. Functional role of regulatory lymphocytes in stroke: Facts and controversies. Stroke 2015, 46, 1422–1430. [Google Scholar] [CrossRef]
- Chamorro, Á.; Amaro, S.; Vargas, M.; Obach, V.; Cervera, Á.; Gómez-Choco, M.; Torres, F.; Planas, A.M. Catecholamines, infection, and death in acute ischemic stroke. J. Neurol. Sci. 2007, 252, 29–35. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, K.; Li, Z.; Li, M.; Han, Y.; Wang, L.; Zhang, Z.; Yu, C.; Zhang, F.; Song, L. Organ-and cell-specific immune responses are associated with the outcomes of intracerebral hemorrhage. FASEB J. 2018, 32, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Neidert, S.; Katan, M.; Schuetz, P.; Fluri, F.; Ernst, A.; Bingisser, R.; Kappos, L.; Engelter, S.; Steck, A.; Müller, B. Anterior pituitary axis hormones and outcome in acute ischaemic stroke. J. Intern. Med. 2011, 269, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, K.; Hosoi, J.; Torii, H.; Ozawa, H.; Ding, W.; Campton, K.; Wagner, J.A.; Granstein, R.D. Catecholamines Inhibit the Antigen-Presenting Capability of Epidermal Langerhans Cells. J. Immunol. 2002, 168, 6128–6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, E.C.; Vieira, P.L.; Kalinski, P.; Kapsenberg, M.L. Corticosteroids inhibit the production of inflammatory mediators in immature monocyte-derived DC and induce the development of tolerogenic DC3. J. Leukoc. Biol. 1999, 66, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Wilson, S.; Dzielak, D.; Yang, W.-H.; Viveros, O. Opioid peptides and noradrenaline co-exist in large dense-cored vesicles from sympathetic nerve. Neuroscience 1982, 7, 2255–2261. [Google Scholar] [CrossRef]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve—An integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar]
- Offner, H.; Subramanian, S.; Parker, S.M.; Wang, C.; Afentoulis, M.E.; Lewis, A.; Vandenbark, A.A.; Hurn, P.D. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J. Immunol. 2006, 176, 6523–6531. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.H.; Jenne, C.N.; Lee, W.Y.; Léger, C.; Kubes, P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science 2011, 334, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Römer, C.; Engel, O.; Winek, K.; Hochmeister, S.; Zhang, T.; Royl, G.; Klehmet, J.; Dirnagl, U.; Meisel, C.; Meisel, A. Blocking stroke-induced immunodeficiency increases CNS antigen-specific autoreactivity but does not worsen functional outcome after experimental stroke. J. Neurosci. 2015, 35, 7777–7794. [Google Scholar] [CrossRef]
- Wang, J.; Yu, L.; Jiang, C.; Fu, X.; Liu, X.; Wang, M.; Ou, C.; Cui, X.; Zhou, C.; Wang, J. Cerebral ischemia increases bone marrow CD4+ CD25+ FoxP3+ regulatory T cells in mice via signals from sympathetic nervous system. Brain Behav. Immun. 2015, 43, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Li, L.; Wang, L.; Degos, V.; Maze, M.; Su, H. Alpha-7 nicotinic acetylcholine receptor agonist treatment reduces neuroinflammation, oxidative stress, and brain injury in mice with ischemic stroke and bone fracture. J. Neurochem. 2014, 131, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-T.; Chu, K.; Jung, K.-H.; Kang, K.-M.; Kim, J.-H.; Bahn, J.-J.; Jeon, D.; Kim, M.; Lee, S.K.; Roh, J.-K. Cholinergic anti-inflammatory pathway in intracerebral hemorrhage. Brain Res. 2010, 1309, 164–171. [Google Scholar] [CrossRef]
- Illanes, S.; Liesz, A.; Sun, L.; Dalpke, A.; Zorn, M.; Veltkamp, R. Hematoma size as major modulator of the cellular immune system after experimental intracerebral hemorrhage. Neurosci. Lett. 2011, 490, 170–174. [Google Scholar] [CrossRef]
- Lee, S.-T.; Chu, K.; Jung, K.-H.; Kim, S.-J.; Kim, D.-H.; Kang, K.-M.; Hong, N.H.; Kim, J.-H.; Ban, J.-J.; Park, H.-K. Anti-inflammatory mechanism of intravascular neural stem cell transplantation in haemorrhagic stroke. Brain 2008, 131, 616–629. [Google Scholar] [CrossRef] [Green Version]
- Sahota, P.; Vahidy, F.; Nguyen, C.; Bui, T.-T.; Yang, B.; Parsha, K.; Garrett, J.; Bambhroliya, A.; Barreto, A.; Grotta, J.C. Changes in spleen size in patients with acute ischemic stroke: A pilot observational study. Int. J. Stroke 2013, 8, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Chiu, N.L.; Kaiser, B.; Nguyen, Y.V.; Welbourne, S.; Lall, C.; Cramer, S.C. The volume of the spleen and its correlates after acute stroke. J. Stroke Cerebrovasc. Dis. 2016, 25, 2958–2961. [Google Scholar] [CrossRef] [Green Version]
- Boehme, A.K.; Kapoor, N.; Albright, K.C.; Lyerly, M.J.; Rawal, P.V.; Bavarsad Shahripour, R.; Alvi, M.; Houston, J.T.; Sisson, A.; Beasley, T.M. Systemic inflammatory response syndrome in tissue-type plasminogen activator–treated patients is associated with worse short-term functional outcome. Stroke 2013, 44, 2321–2323. [Google Scholar] [CrossRef] [Green Version]
- Sarrafzadeh, A.; Schlenk, F.; Meisel, A.; Dreier, J.; Vajkoczy, P.; Meisel, C. Immunodepression after aneurysmal subarachnoid hemorrhage. Stroke 2011, 42, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Sykora, M.; Diedler, J.; Rupp, A.; Turcani, P.; Rocco, A.; Steiner, T. Impaired baroreflex sensitivity predicts outcome of acute intracerebral hemorrhage. Crit. Care Med. 2008, 36, 3074–3079. [Google Scholar] [CrossRef]
- Sykora, M.; Steiner, T.; Poli, S.; Rocco, A.; Turcani, P.; Diedler, J. Autonomic effects of intraventricular extension in intracerebral hemorrhage. Neurocrit. Care 2012, 16, 102–108. [Google Scholar] [CrossRef]
- Sykora, M.; Diedler, J.; Poli, S.; Rizos, T.; Turcani, P.; Veltkamp, R.; Steiner, T. Autonomic shift and increased susceptibility to infections after acute intracerebral hemorrhage. Stroke 2011, 42, 1218–1223. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Selim, M.H.; Caplan, L.R. Medical complications after stroke. Lancet Neurol. 2010, 9, 105–118. [Google Scholar] [CrossRef]
- Westendorp, W.F.; Nederkoorn, P.J.; Vermeij, J.-D.; Dijkgraaf, M.G.; van de Beek, D. Post-stroke infection: A systematic review and meta-analysis. BMC Neurol. 2011, 11, 110. [Google Scholar] [CrossRef] [Green Version]
- Chamorro, A.; Urra, X.; Planas, A.M. Infection after acute ischemic stroke: A manifestation of brain-induced immunodepression. Stroke 2007, 38, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Otite, F.O.; Khandelwal, P.; Malik, A.M.; Chaturvedi, S.; Sacco, R.L.; Romano, J.G. Ten-year temporal trends in medical complications after acute intracerebral hemorrhage in the United States. Stroke 2017, 48, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Lord, A.S.; Lewis, A.; Czeisler, B.; Ishida, K.; Torres, J.; Kamel, H.; Woo, D.; Elkind, M.S.; Boden-Albala, B. Majority of 30-day readmissions after intracerebral hemorrhage are related to infections. Stroke 2016, 47, 1768–1771. [Google Scholar] [CrossRef] [Green Version]
- Shim, R.; Wong, C.H. Ischemia, immunosuppression and infection—Tackling the predicaments of post-stroke complications. Int. J. Mol. Sci. 2016, 17, 64. [Google Scholar] [CrossRef]
- Laban, K.G.; Rinkel, G.J.; Vergouwen, M.D. Nosocomial infections after aneurysmal subarachnoid hemorrhage: Time course and causative pathogens. Int. J. Stroke 2015, 10, 763–766. [Google Scholar] [CrossRef] [Green Version]
- Busl, K.M. Nosocomial infections in the neurointensive care unit. Neurol. Clin. 2017, 35, 785–807. [Google Scholar] [CrossRef]
- Emsley, H.C.; Hopkins, S.J. Acute ischaemic stroke and infection: Recent and emerging concepts. Lancet Neurol. 2008, 7, 341–353. [Google Scholar] [CrossRef]
- Elkind, M.S.; Carty, C.L.; O’Meara, E.S.; Lumley, T.; Lefkowitz, D.; Kronmal, R.A.; Longstreth Jr, W. Hospitalization for infection and risk of acute ischemic stroke: The Cardiovascular Health Study. Stroke 2011, 42, 1851–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heikinheimo, T.; Broman, J.; Haapaniemi, E.; Kaste, M.; Tatlisumak, T.; Putaala, J. Preceding and poststroke infections in young adults with first-ever ischemic stroke: Effect on short-term and long-term outcomes. Stroke 2013, 44, 3331–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeller, J.A.; Lenz, A.; Eschenfelder, C.C.; Zunker, P.; Deuschl, G. Platelet-Leukocyte Interaction and Platelet Activation in Acute Stroke with and without Preceding Infection. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1519–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tascilar, N.; Irkorucu, O.; Tascilar, O.; Comert, F.; Eroglu, O.; Bahadir, B.; Cakmak, G.K.; Ankarali, H.; Sayan, H. Bacterial translocation in experimental stroke: What happens to the gut barrier? Bratisl. Lek. Listy 2010, 111, 194–199. [Google Scholar]
- Dirnagl, U.; Klehmet, J.; Braun, J.S.; Harms, H.; Meisel, C.; Ziemssen, T.; Prass, K.; Meisel, A. Stroke-Induced Immunodepression. Stroke 2007, 38, 770–773. [Google Scholar] [CrossRef]
- Becker, K.J.; Kalil, A.J.; Tanzi, P.; Zierath, D.K.; Savos, A.V.; Gee, J.M.; Hadwin, J.; Carter, K.T.; Shibata, D.; Cain, K.C. Autoimmune responses to the brain after stroke are associated with worse outcome. Stroke 2011, 42, 2763–2769. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.J.; Kindrick, D.L.; Lester, M.P.; Shea, C.; Ye, Z.-C. Sensitization to brain antigens after stroke is augmented by lipopolysaccharide. J. Cereb. Blood Flow Metab. 2005, 25, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Zierath, D.; Thullbery, M.; Hadwin, J.; Gee, J.M.; Savos, A.; Kalil, A.; Becker, K.J. CNS immune responses following experimental stroke. Neurocrit. Care 2010, 12, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Gee, J.M.; Zierath, D.; Hadwin, J.; Savos, A.; Kalil, A.; Thullbery, M.; Becker, K.J. Long term immunologic consequences of experimental stroke and mucosal tolerance. Exp. Transl. Stroke Med. 2009, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.-J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Chastain, E.M.; Terry, R.L.; Miller, S.D. Virus infection, antiviral immunity, and autoimmunity. Immunol. Rev. 2013, 255, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berer, K.; Krishnamoorthy, G. Microbial view of central nervous system autoimmunity. FEBS Lett. 2014, 588, 4207–4213. [Google Scholar] [CrossRef]
- Haeusler, K.G.; Schmidt, W.U.; Föhring, F.; Meisel, C.; Helms, T.; Jungehulsing, G.J.; Nolte, C.H.; Schmolke, K.; Wegner, B.; Meisel, A. Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc. Dis. 2008, 25, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Klehmet, J.; Harms, H.; Richter, M.; Prass, K.; Volk, H.; Dirnagl, U.; Meisel, A.; Meisel, C. Stroke-induced immunodepression and post-stroke infections: Lessons from the preventive antibacterial therapy in stroke trial. Neuroscience 2009, 158, 1184–1193. [Google Scholar] [CrossRef]
- Urra, X.; Cervera, Á.; Villamor, N.; Planas, A.; Chamorro, A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience 2009, 158, 1174–1183. [Google Scholar] [CrossRef]
- Trakhtenberg, E.F.; Goldberg, J.L. Neuroimmune communication. Science 2011, 334, 47–48. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Zhang, B.; Kosaka, Y.; Burrows, G.G.; Grafe, M.R.; Vandenbark, A.A.; Hurn, P.D.; Offner, H. Recombinant T cell receptor ligand treats experimental stroke. Stroke 2009, 40, 2539–2545. [Google Scholar] [CrossRef] [PubMed]
- Ortega, S.B.; Noorbhai, I.; Poinsatte, K.; Kong, X.; Anderson, A.; Monson, N.L.; Stowe, A.M. Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov. Med. 2015, 19, 381–392. [Google Scholar]
- Mracsko, E.; Javidi, E.; Na, S.-Y.; Kahn, A.; Liesz, A.; Veltkamp, R. Leukocyte invasion of the brain after experimental intracerebral hemorrhage in mice. Stroke 2014, 45, 2107–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubbe, T.; Ebner, F.; Richter, D.; Engel, O.R.; Klehmet, J.; Royl, G.; Meisel, A.; Nitsch, R.; Meisel, C.; Brandt, C. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J. Cereb. Blood Flow Metab. 2013, 33, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Schwab, J.; Nguyen, T.; Meyermann, R.; Schluesener, H. Human focal cerebral infarctions induce differential lesional interleukin-16 (IL-16) expression confined to infiltrating granulocytes, CD8+ T-lymphocytes and activated microglia/macrophages. J. Neuroimmunol. 2001, 114, 232–241. [Google Scholar] [CrossRef]
- Hogquist, K.A.; Baldwin, T.A.; Jameson, S.C. Central tolerance: Learning self-control in the thymus. Nat. Rev. Immunol. 2005, 5, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Camara-Lemarroy, C.R.; Ibarra-Yruegas, B.E.; Gongora-Rivera, F. Gastrointestinal complications after ischemic stroke. J. Neurol. Sci. 2014, 346, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Houlden, A.; Goldrick, M.; Brough, D.; Vizi, E.S.; Lénárt, N.; Martinecz, B.; Roberts, I.S.; Denes, A. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav. Immun. 2016, 57, 10–20. [Google Scholar] [CrossRef]
- Singh, V.; Roth, S.; Llovera, G.; Sadler, R.; Garzetti, D.; Stecher, B.; Dichgans, M.; Liesz, A. Microbiota Dysbiosis Controls the Neuroinflammatory Response after Stroke. J. Neurosci. 2016, 36, 7428–7440. [Google Scholar] [CrossRef]
- Liu, Q.; Johnson, E.M.; Lam, R.K.; Wang, Q.; Bo Ye, H.; Wilson, E.N.; Minhas, P.S.; Liu, L.; Swarovski, M.S.; Tran, S.; et al. Peripheral TREM1 responses to brain and intestinal immunogens amplify stroke severity. Nat. Immunol. 2019, 20, 1023–1034. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, L.Y.; Orban, L.; Cuylear, D.; Thompson, J.; Simon, B.; Yang, Y. Non-invasive vagus nerve stimulation reduces blood-brain barrier disruption in a rat model of ischemic stroke. Brain Stimul. 2018, 11, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, O.; Akyüz, L.; da Costa Goncalves, A.C.; Winek, K.; Dames, C.; Thielke, M.; Herold, S.; Böttcher, C.; Priller, J.; Volk, H.D.; et al. Cholinergic Pathway Suppresses Pulmonary Innate Immunity Facilitating Pneumonia After Stroke. Stroke 2015, 46, 3232–3240. [Google Scholar] [CrossRef]
- Benakis, C.; Brea, D.; Caballero, S.; Faraco, G.; Moore, J.; Murphy, M.; Sita, G.; Racchumi, G.; Ling, L.; Pamer, E.G.; et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med. 2016, 22, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Thornton, P.; Rothwell, N.; Allan, S. Inflammation and brain injury: Acute cerebral ischaemia, peripheral and central inflammation. Brain Behav. Immun. 2010, 24, 708–723. [Google Scholar] [CrossRef]
- Liesz, A.; Zhou, W.; Mracsko, E.; Karcher, S.; Bauer, H.; Schwarting, S.; Sun, L.; Bruder, D.; Stegemann, S.; Cerwenka, A. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain 2011, 134, 704–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkind, M.S.V.; Veltkamp, R.; Montaner, J.; Johnston, S.C.; Singhal, A.B.; Becker, K.; Lansberg, M.G.; Tang, W.; Kasliwal, R.; Elkins, J. Natalizumab in acute ischemic stroke (ACTION II). Neurology 2020, 95, e1091. [Google Scholar] [CrossRef]
- Elkins, J.; Veltkamp, R.; Montaner, J.; Johnston, S.C.; Singhal, A.B.; Becker, K.; Lansberg, M.G.; Tang, W.; Chang, I.; Muralidharan, K.; et al. Safety and efficacy of natalizumab in patients with acute ischaemic stroke (ACTION): A randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2017, 16, 217–226. [Google Scholar] [CrossRef]
- Llovera, G.; Hofmann, K.; Roth, S.; Salas-Pérdomo, A.; Ferrer-Ferrer, M.; Perego, C.; Zanier, E.R.; Mamrak, U.; Rex, A.; Party, H.; et al. Results of a preclinical randomized controlled multicenter trial (pRCT): Anti-CD49d treatment for acute brain ischemia. Sci. Transl. Med. 2015, 7, 299ra121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.H.; Okada, T.; Matloubian, M.; Lo, C.G.; Cyster, J.G. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity 2008, 28, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Rolland, W.B., 2nd; Manaenko, A.; Lekic, T.; Hasegawa, Y.; Ostrowski, R.; Tang, J.; Zhang, J.H. FTY720 is neuroprotective and improves functional outcomes after intracerebral hemorrhage in mice. Acta Neurochir. Suppl. 2011, 111, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Hao, J.; Zhang, N.; Ren, L.; Sun, N.; Li, Y.J.; Yan, Y.; Huang, D.; Yu, C.; Shi, F.D. Fingolimod for the treatment of intracerebral hemorrhage: A 2-arm proof-of-concept study. JAMA Neurol. 2014, 71, 1092–1101. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, Y.; Suzuki, H.; Sozen, T.; Rolland, W.; Zhang, J.H. Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke 2010, 41, 368–374. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Yemisci, M.; Kim, H.H.; Yung, L.M.; Shin, H.K.; Hwang, S.K.; Guo, S.; Qin, T.; Alsharif, N.; Brinkmann, V.; et al. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann. Neurol. 2011, 69, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Kraft, P.; Göb, E.; Schuhmann, M.K.; Göbel, K.; Deppermann, C.; Thielmann, I.; Herrmann, A.M.; Lorenz, K.; Brede, M.; Stoll, G.; et al. FTY720 ameliorates acute ischemic stroke in mice by reducing thrombo-inflammation but not by direct neuroprotection. Stroke 2013, 44, 3202–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesz, A.; Sun, L.; Zhou, W.; Schwarting, S.; Mracsko, E.; Zorn, M.; Bauer, H.; Sommer, C.; Veltkamp, R. FTY720 reduces post-ischemic brain lymphocyte influx but does not improve outcome in permanent murine cerebral ischemia. PLoS ONE 2011, 6, e21312. [Google Scholar] [CrossRef] [Green Version]
- Kwak, B.; Mulhaupt, F.; Myit, S.; Mach, F. Statins as a newly recognized type of immunomodulator. Nat. Med. 2000, 6, 1399–1402. [Google Scholar] [CrossRef]
- Yilmaz, A.; Reiss, C.; Tantawi, O.; Weng, A.; Stumpf, C.; Raaz, D.; Ludwig, J.; Berger, T.; Steinkasserer, A.; Daniel, W.G.; et al. HMG-CoA reductase inhibitors suppress maturation of human dendritic cells: New implications for atherosclerosis. Atherosclerosis 2004, 172, 85–93. [Google Scholar] [CrossRef]
- Li, X.L.; Liu, Y.; Cao, L.L.; Li, H.; Yue, L.T.; Wang, S.; Zhang, M.; Li, X.H.; Dou, Y.C.; Duan, R.S. Atorvastatin-modified dendritic cells in vitro ameliorate experimental autoimmune myasthenia gravis by up-regulated Treg cells and shifted Th1/Th17 to Th2 cytokines. Mol. Cell. Neurosci. 2013, 56, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Palinski, W.; Miller, E.; Witztum, J.L. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 821–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, R.; Sukhova, G.; Faria, A.M.; Hoffmann, E.; Mach, F.; Libby, P.; Weiner, H.L. Mucosal administration of heat shock protein-65 decreases atherosclerosis and inflammation in aortic arch of low-density lipoprotein receptor-deficient mice. Circulation 2002, 106, 1708–1715. [Google Scholar] [CrossRef] [Green Version]
- Binder, C.J.; Hörkkö, S.; Dewan, A.; Chang, M.K.; Kieu, E.P.; Goodyear, C.S.; Shaw, P.X.; Palinski, W.; Witztum, J.L.; Silverman, G.J. Pneumococcal vaccination decreases atherosclerotic lesion formation: Molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat. Med. 2003, 9, 736–743. [Google Scholar] [CrossRef]
- Xu, K.; Lee, J.-Y.; Kaneko, Y.; Tuazon, J.P.; Vale, F.; van Loveren, H.; Borlongan, C.V. Human stem cells transplanted into the rat stroke brain migrate to the spleen via lymphatic and inflammation pathways. Haematologica 2019, 104, 1062–1073. [Google Scholar] [CrossRef]
- Xie, Y.; Bagby, T.R.; Cohen, M.S.; Forrest, M.L. Drug delivery to the lymphatic system: Importance in future cancer diagnosis and therapies. Expert Opin. Drug Deliv. 2009, 6, 785–792. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Seyfried, D.; Meng, Y.; Yang, D.; Schultz, L.; Chopp, M.; Seyfried, D. Multipotent mesenchymal stromal cell-derived exosomes improve functional recovery after experimental intracerebral hemorrhage in the rat. J. Neurosurg. 2018, 131, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Smith, A.J.; Phuan, P.-W.; Tradtrantip, L.; Anderson, M.O. The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin. Ther. Targets 2017, 21, 1161–1170. [Google Scholar] [CrossRef]
- Abir-Awan, M.; Kitchen, P.; Salman, M.M.; Conner, M.T.; Conner, A.C.; Bill, R.M. Inhibitors of Mammalian Aquaporin Water Channels. Int. J. Mol. Sci. 2019, 20, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Brown, J.E.; Bill, R.M.; Conner, A.C.; Conner, M.T. Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)- and calmodulin-mediated mechanism. Eur. J. Neurosci. 2017, 46, 2542–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [Green Version]
- Eura, N.; Matsui, T.K.; Luginbühl, J.; Matsubayashi, M.; Nanaura, H.; Shiota, T.; Kinugawa, K.; Iguchi, N.; Kiriyama, T.; Zheng, C.; et al. Brainstem Organoids From Human Pluripotent Stem Cells. Front. Neurosci. 2020, 14, 538. [Google Scholar] [CrossRef]
- Pellegrini, L.; Bonfio, C.; Chadwick, J.; Begum, F.; Skehel, M.; Lancaster, M.A. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 2020, 369, eaaz5626. [Google Scholar] [CrossRef]
- Iwasa, N.; Matsui, T.K.; Iguchi, N.; Kinugawa, K.; Morikawa, N.; Sakaguchi, Y.M.; Shiota, T.; Kobashigawa, S.; Nakanishi, M.; Matsubayashi, M.; et al. Gene Expression Profiles of Human Cerebral Organoids Identify PPAR Pathway and PKM2 as Key Markers for Oxygen-Glucose Deprivation and Reoxygenation. Front. Cell. Neurosci. 2021, 15, 210. [Google Scholar] [CrossRef]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef]
- Matsui, T.K.; Tsuru, Y.; Hasegawa, K.; Kuwako, K.-I. Vascularization of human brain organoids. Stem Cells 2021, 39, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-N.; Wang, Z.; Xu, T.-Y.; Cheng, M.-H.; Li, W.-L.; Miao, C.-Y. Cerebral Organoids Repair Ischemic Stroke Brain Injury. Transl. Stroke Res. 2020, 11, 983–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, L.; Zhu, Y.; Qin, J. Human brain organoid-on-a-chip to model prenatal nicotine exposure. Lab Chip 2018, 18, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 1077. [Google Scholar] [CrossRef]
- Song, G.; Zhao, M.; Chen, H.; Zhou, X.; Lenahan, C.; Ou, Y.; He, Y. The Application of Brain Organoid Technology in Stroke Research: Challenges and Prospects. Front. Cell. Neurosci. 2021, 15, 203. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Thompson, J.F.; Carter, M.B.; Edwards, B.S.; Sklar, L.A.; Rosenberg, G.A. Identification of Isoxsuprine Hydrochloride as a Neuroprotectant in Ischemic Stroke through Cell-Based High-Throughput Screening. PLoS ONE 2014, 9, e96761. [Google Scholar] [CrossRef] [Green Version]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef]




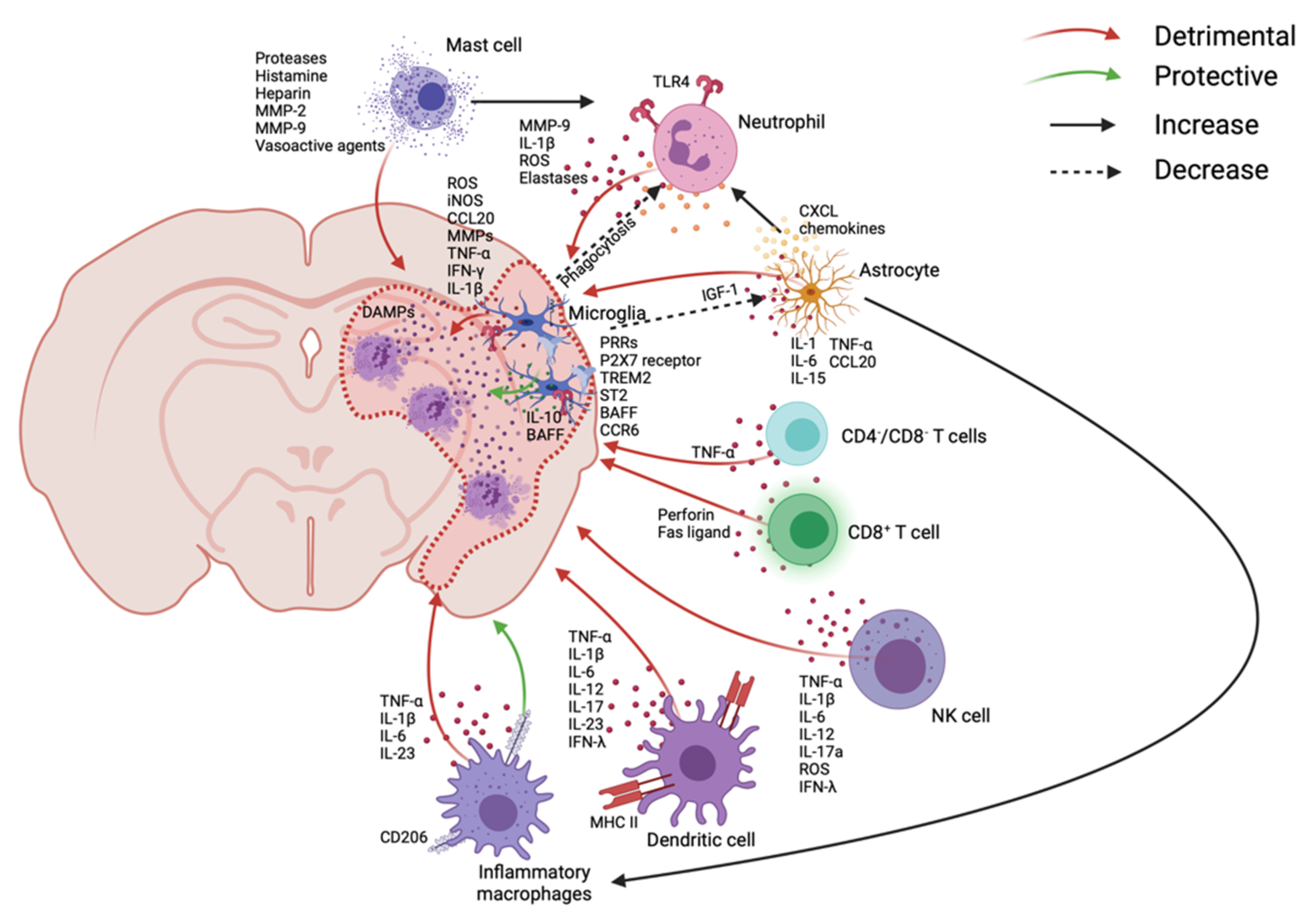{kind=link}
{kind=link}
{kind=link}
| Immune Cells | Temporal Trend | Produced Cytokines/Chemokines | Pro- or Anti-Inflammatory | Action(s) |
|---|---|---|---|---|
| Microglia | Peak at 48–72 h and last for weeks | iNOS | Pro | BBB destruction, leukocyte recruitment, increase adhesion molecules |
| MMPs | Pro | |||
| TNF-α | Pro | |||
| IL-1β | Pro | |||
| ROS | Pro | |||
| RNS | Pro | |||
| BAFF | Anti | IFN-γ and IL-10 production | ||
| IGF-1 | Anti | Suppress astrocyte activities | ||
| Astrocytes | Shown within 3–5 days | CCL20 | Pro | Recruit macrophages |
| CXCL chemokines | Pro | Recruit neutrophils | ||
| Adhesion molecules | Pro | Leukocyte recruitment | ||
| IL-15 | Pro | Recruit CD8+ T-cells and NK cells | ||
| TNF-α, IL-1, 6 | Pro | Augment immune responses | ||
| TGF-β | Anti | Develop blood vessels, maintain BBB integrity and endothelial progenitor cells | ||
| GDNF | Anti | |||
| Angiopoietin 1, 2 | Anti | |||
| VEGF | Anti | |||
| bFGF | Anti | |||
| IL-17a | Anti | Brain tissue repair and recovery in later period | ||
| Neutrophils | Accumulate after 3 h, peak at day 1–3, and dissipate over 7 days | Elastases | Pro | Cerebral edema, BBB destruction, and neuronal death |
| MMP-9 | Pro | |||
| IL-1β | Pro | |||
| ROS | Pro | |||
| MMP-9 | Anti | Degradation of DAMP signaling and vascular remodeling | ||
| VEGF | Anti | Cerebral angiogenesis | ||
| Annexin-1 | Anti | Microglia migration toward the infarct core after 1 day | ||
| Resolvins | Anti | Decrease neutrophil migration and pro-inflammatory cytokine release | ||
| Protectins | Anti | |||
| Mast cells | Significant increase after 24 h | Histamine | Pro | Destruct BBB, increase vascular permeability, leukocyte recruitment, cerebral edema, destroy tight junctions, and disrupt hemostasis |
| Heparin | Pro | |||
| Vasoactive agents | Pro | |||
| Chymase | Pro | |||
| MMP-2, 9 | Pro | |||
| Monocyte/ Macrophage | Shown as early as 3 h, peak at day 3, and become anti-inflammatory at day 7 | TNF-α | Pro | Augment immune responses |
| IL-1β | Pro | |||
| IL-23 | Pro | IL-17a production from γδ T-cells | ||
| IL-10 | Anti | Tissue repair and wound healing | ||
| TGF-β | Anti | |||
| CD302, 163, 206 | Anti | |||
| PDGF | Anti | |||
| Fibronectin 1 | Anti | |||
| Arginase 1 | Anti | |||
| DCs | Detected as early as 1 h and stay elevated till day 7 | IL-23 | Pro | IL-17a production from γδ T-cells and neutrophil infiltration |
| IL-6, 12, 17, 1β | Pro | Augment immune responses | ||
| TNF-α | Pro | |||
| IFN-γ | Pro | |||
| IL-10 | Anti | Immunosuppression | ||
| NK cells | 3 h, peak at 12 h, and remain elevated at least 4 days | IFN-γ | Pro | Augment immune responses and development of cerebral infarction |
| IL-17a, 6, 12, 1β | Pro | |||
| TNF-α | Pro | |||
| ROS | Pro | |||
| CD4-/CD8- T-cells | 1–3 days | TNF-α | Pro | Augment immune responses |
| CD8+ T-cells | Detected as early as 3 h and stay for about 30 days | Perforin | Pro | Neurotoxicity and augment immune responses |
| Fas ligand | Pro | |||
| CD4+ T-cells (Th1 and Th17) | Shown at 24 h and stay for about 30 days | IL-2, 12, 17, 21, 22, 23 | Pro | Augment immune responses |
| TNF-α | Pro | |||
| IFN-γ | Pro | |||
| CD4+ T-cells (Th2) | IL-4, 5, 6, 10, 13 | Anti | Immunosuppression | |
| γδ T-cells | Within 3 days | IL-17a | Pro | Neutrophil infiltration |
| Tregs | Shown after several days and stays for about 30 days | IL-10 | Anti | Suppress astrogliosis, regulate astrocyte neurotoxicity, and functional recovery |
| IL-17 (in certain conditions) | Anti | Inhibit CD4+ T-cell proliferation | ||
| B-cells | Delayed appearance after weeks of onset | IL-10 | Anti | Neuroprotection |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, Y.H.; Laaker, C.; Hsu, M.; Cismaru, P.; Sandor, M.; Fabry, Z. Molecular Mechanisms of Neuroimmune Crosstalk in the Pathogenesis of Stroke. Int. J. Mol. Sci. 2021, 22, 9486. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179486
Choi YH, Laaker C, Hsu M, Cismaru P, Sandor M, Fabry Z. Molecular Mechanisms of Neuroimmune Crosstalk in the Pathogenesis of Stroke. International Journal of Molecular Sciences. 2021; 22(17):9486. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179486
Chicago/Turabian StyleChoi, Yun Hwa, Collin Laaker, Martin Hsu, Peter Cismaru, Matyas Sandor, and Zsuzsanna Fabry. 2021. "Molecular Mechanisms of Neuroimmune Crosstalk in the Pathogenesis of Stroke" International Journal of Molecular Sciences 22, no. 17: 9486. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179486