
Difluoromethylornithine Induces Apoptosis through Regulation of AP-1 Signaling via JNK Phosphorylation in Epithelial Ovarian Cancer

 , , and
, , and 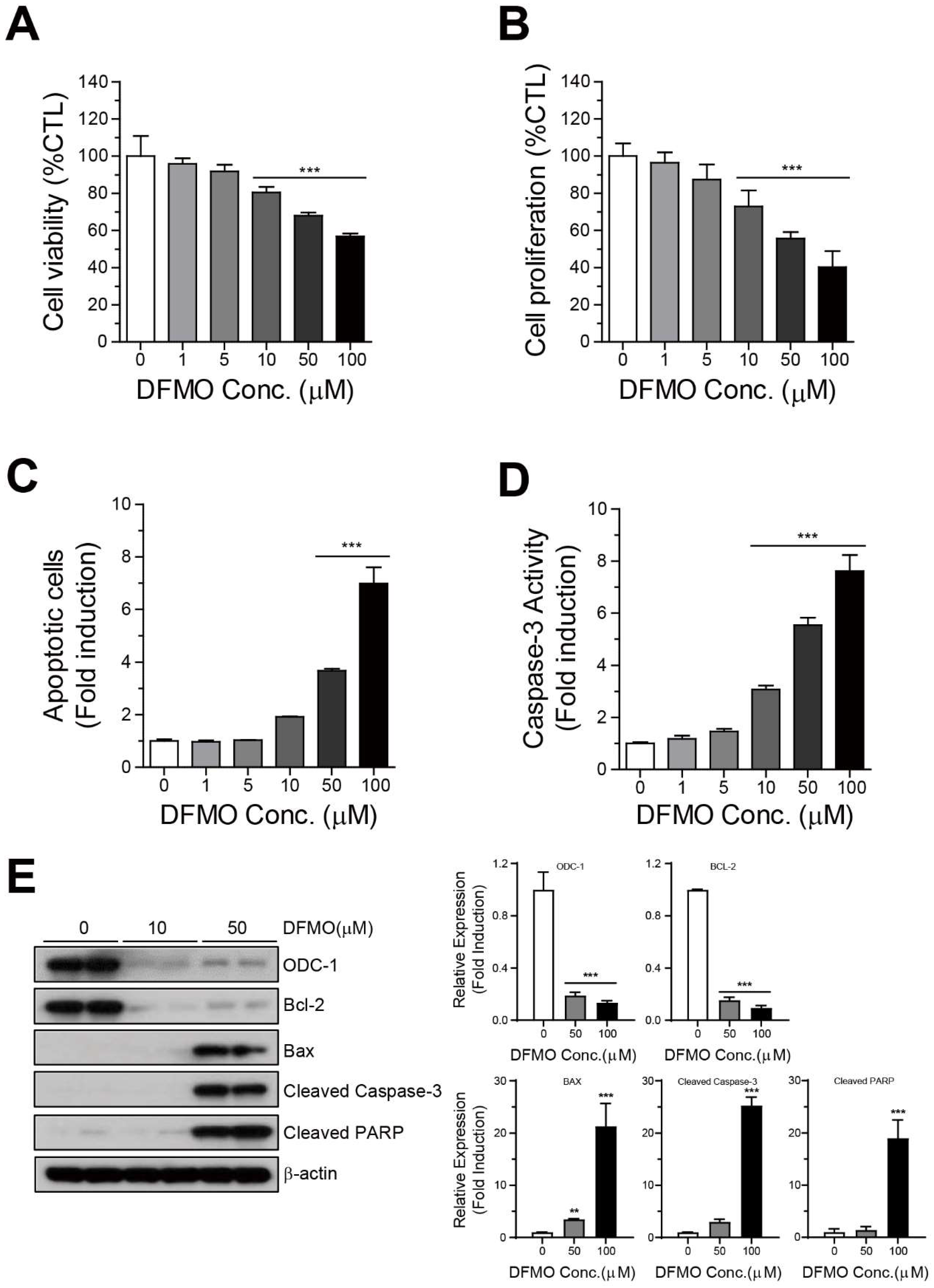{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. DFMO Suppresses Cancer Survival and Induces Apoptotic Cell Death in Ovarian Cancer Cells
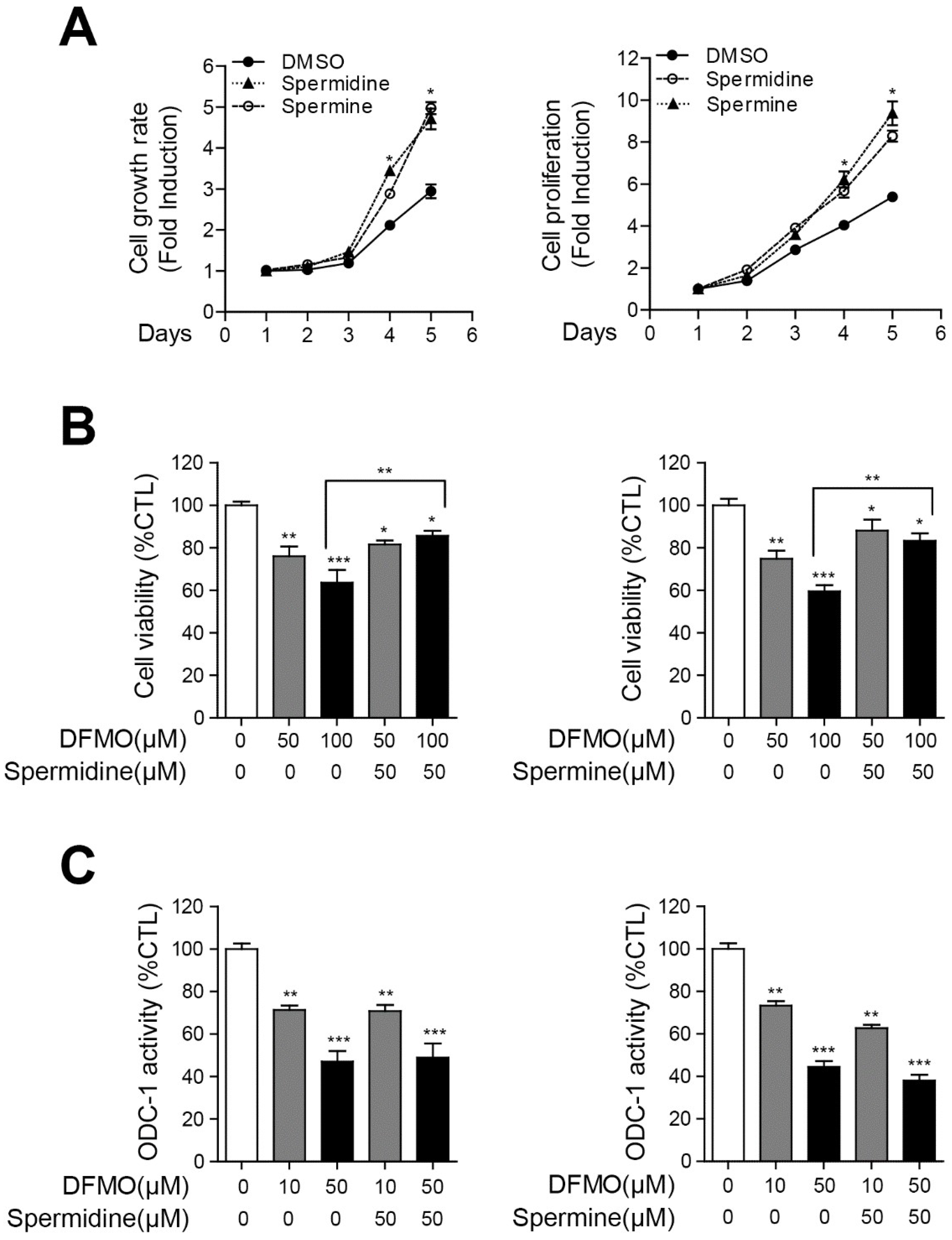
2.2. Polyamines Contribute to the Survival of Cancer Cells and Inhibit DFMO-Induced Cell Death in Ovarian Cancer Cells
2.3. Combination of DFMO and Cisplatin Therapy Reduces Cancer Cell Survival and Promotes Apoptotic Cell Death in Ovarian Cancer Cells
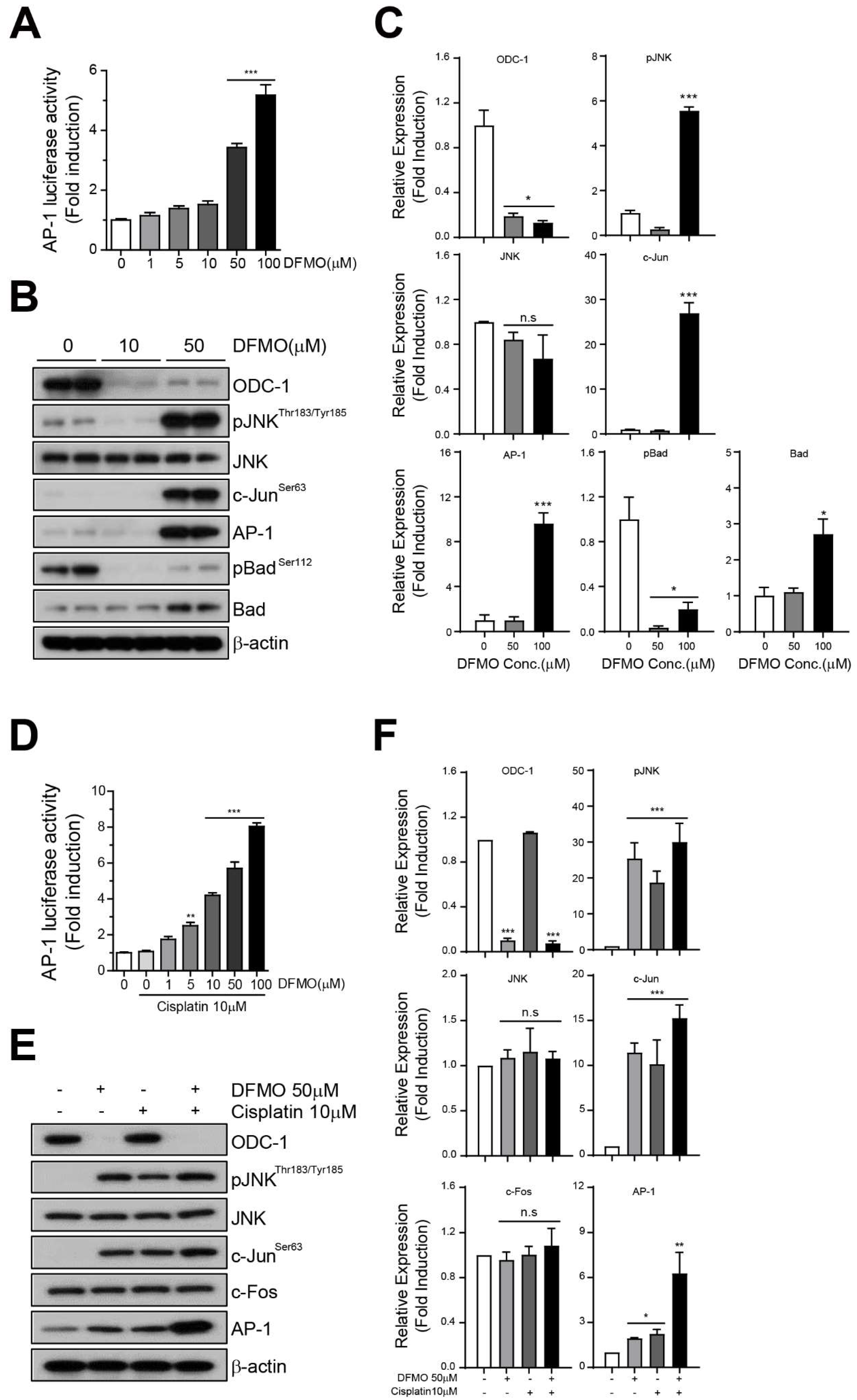
2.4. DFMO Induces Phosphorylation of JNK and Activates the AP-1 Signaling Pathway
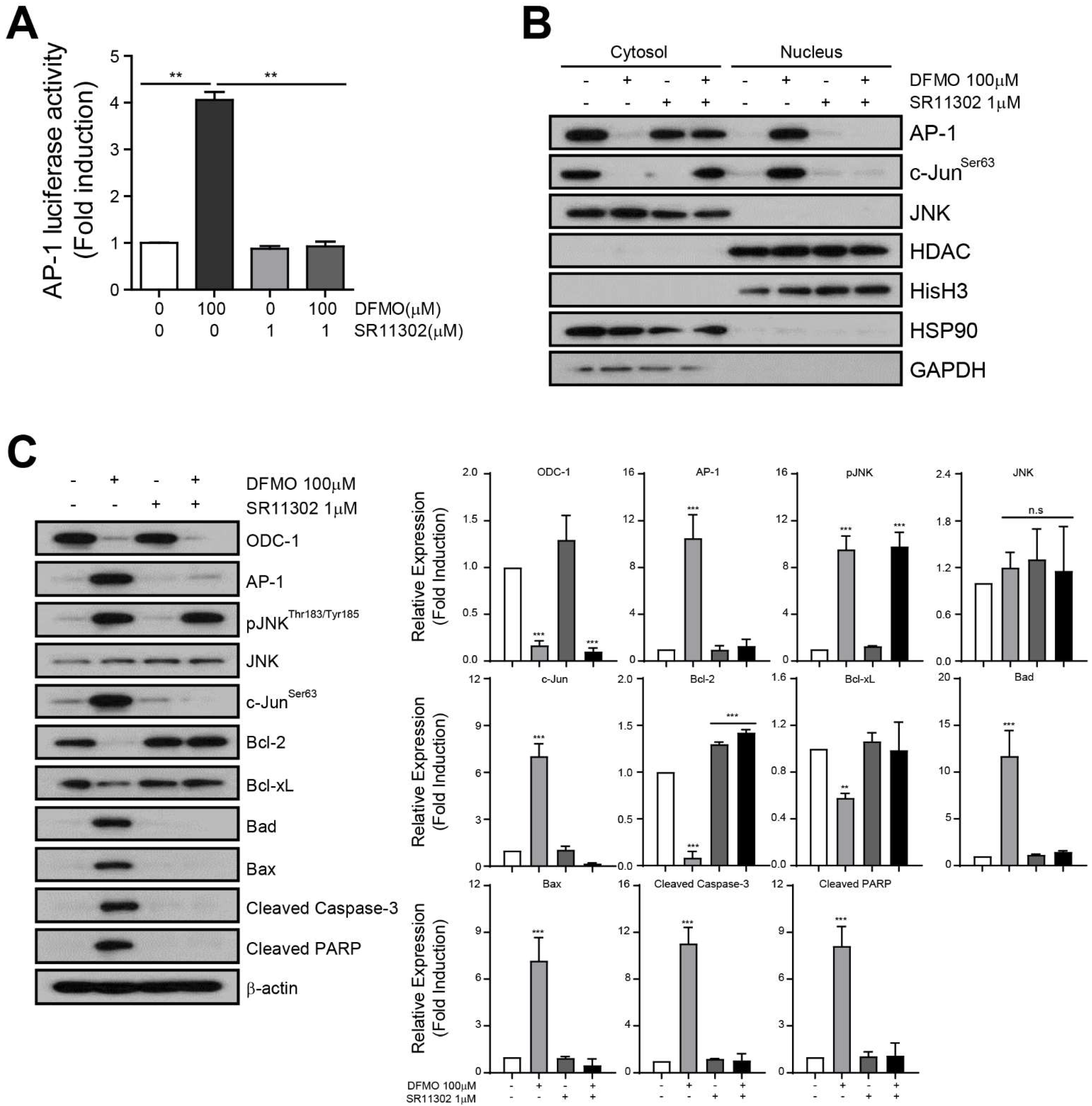
2.5. AP-1 Mediates DFMO-Induced Apoptotic Cell Death in Ovarian Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Drugs
4.2. Cell Viability
4.3. Cell Proliferation
4.4. Caspase 3/7 Activity
4.5. Ornithine Decarboxylase-1 (ODC-1) Activity
4.6. AP-1 Luciferase Activity
4.7. Annexin V-FITC/PI Assay via Flow Cytometry
4.8. Subcellular Fractionation
4.9. Western Blot
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel Molecular Subtypes of Serous and Endometrioid Ovarian Cancer Linked to Clinical Outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexiou, G.A.; Lianos, G.D.; Ragos, V.; Galani, V.; Kyritsis, A.P. Difluoromethylornithine in cancer: New advances. Futur. Oncol. 2017, 13, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Soda, K. The mechanisms by which polyamines accelerate tumor spread. J. Exp. Clin. Cancer Res. 2011, 30, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray-Stewart, T.; Ferrari, E.; Xie, Y.; Yu, F.; Marton, L.J.; Oupicky, D.; Casero, R.A., Jr. Biochemical evaluation of the anticancer potential of the polyamine-based nanocarrier Nano11047. PLoS ONE 2017, 12, e0175917. [Google Scholar]
- Thomas, T.J.; John, S.; Hsu, H.-C.; Yang, P.; Keinänen, T.A.; Hyvönen, M.T.; Thomas, T. Tamoxifen metabolite endoxifen interferes with the polyamine pathway in breast cancer. Amino Acids 2016, 48, 2293–2302. [Google Scholar] [CrossRef]
- Nowotarski, S.L.; Woster, P.; Casero, R.A. Polyamines and cancer: Implications for chemotherapy and chemoprevention. Expert Rev. Mol. Med. 2013, 15, e3. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cameron, G.A.; Wallace, H.M. Decreased sensitivity to aspirin is associated with altered polyamine metabolism in human prostate cancer cells. Amino Acids 2015, 48, 1003–1012. [Google Scholar] [CrossRef]
- Pegg, A.E.; McCann, P.P. Polyamine metabolism and function. Am. J. Physiol. Physiol. 1982, 243, C212–C221. [Google Scholar] [CrossRef]
- Pegg, A.E. Polyamine metabolism and its importance in neoplastic growth and a target for chemotherapy. Cancer Res. 1988, 48, 759–774. [Google Scholar]
- Elitsur, Y.; Gesell, M.; Luk, G. ODC activity and polyamine levels in isolated human colonocytes. Life Sci. 1993, 53, 945–952. [Google Scholar] [CrossRef]
- Gerner, E.W.; Meyskens, F.L. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shantz, L.M.; Levin, V.A. Regulation of ornithine decarboxylase during oncogenic transformation: Mechanisms and therapeutic potential. Amino Acids 2007, 33, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Hayashi, H.; Taira, M.; Isono, K. Elevated expression of the ornithine decarboxylase gene in human esophageal cancer. Cancer Res. 1992, 52, 6671–6675. [Google Scholar]
- Miao, X.-P.; Li, J.-S.; Li, H.-Y.; Zeng, S.-P.; Zhao, Y.; Zeng, J.-Z. Expression of ornithine decarboxylase in precancerous and cancerous gastric lesions. World J. Gastroenterol. 2007, 13, 2867–2871. [Google Scholar] [CrossRef] [PubMed]
- Cancer Cell Line Encyclopedia. Available online: https://sites.broadinstitute.org/ccle (accessed on 5 September 2021).
- Marton, J.L.; Pegg, A.E. Polyamines as Targets for Therapeutic Intervention. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 55–91. [Google Scholar] [CrossRef]
- Casero, R.A.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef]
- Qu, N.; Ignatenko, N.A.; Yamauchi, P.; Stringer, D.E.; Levenson, C.; Shannon, P.; Perrin, S.; Gerner, E.W. Inhibition of human ornithine decarboxylase activity by enantiomers of difluoromethylornithine. Biochem. J. 2003, 375, 465–470. [Google Scholar] [CrossRef] [Green Version]
- Alhosin, M.; Razvi, S.S.I.; Sheikh, R.; Khan, J.A.; Zamzami, M.A.; Choudhry, H. Thymoquinone and Difluoromethylornithine (DFMO) Synergistically Induce Apoptosis of Human Acute T Lymphoblastic Leukemia Jurkat Cells through the Modulation of Epigenetic Pathways. Technol. Cancer Res. Treat. 2020, 19, 1533033820947489. [Google Scholar] [CrossRef]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs-Helber, S.M.; Wickrema, A.; Birrer, M.J.; Sawyer, S.T. AP1 Regulation of Proliferation and Initiation of Apoptosis in Erythropoietin-Dependent Erythroid Cells. Mol. Cell. Biol. 1998, 18, 3699–3707. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Wang, S.; Sun, Y.; Matt, Y.; Colburn, N.H.; Shu, Y.; Han, X. JNK/AP-1 pathway is involved in tumor necrosis factor-α induced expression of vascular endothelial growth factor in MCF7 cells. Biomed. Pharmacother. 2009, 63, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manetta, A.; Satyaswarcoop, P.G.; Podczaski, E.S.; Hamilton, T.; Ozols, R.F.; Mortel, R. Effect of alpha-difluoromethylornithine (DFMO) on the growth of human ovarian carcinoma. Eur. J. Gynaecol. Oncol. 1988, 9, 222–227. [Google Scholar] [PubMed]
- Ye, C.; Geng, Z.; Dominguez, D.; Chen, S.; Fan, J.; Qin, L.; Long, A.; Zhang, Y.; Kuzel, T.M.; Zhang, B. Targeting Ornithine Decarboxylase by α-Difluoromethylornithine Inhibits Tumor Growth by Impairing Myeloid-Derived Suppressor Cells. J. Immunol. 2015, 196, 915–923. [Google Scholar] [CrossRef]
- Hogarty, M.D.; Norris, M.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1 Is a Critical Determinant of MYCN Oncogenesis and a Therapeutic Target in Neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef] [Green Version]
- Wallick, C.J.; Gamper, I.; Thorne, M.; Feith, D.J.; Takasaki, K.Y.; Wilson, S.M.; Seki, J.A.; Pegg, A.E.; Byus, C.V.; Bachmann, A.S. Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine inhibitor-induced G1 cell cycle arrest in MYCN-amplified human neuroblastoma cells. Oncogene 2005, 24, 5606–5618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geck, R.C.; Foley, J.R.; Murray Stewart, T.; Asara, J.M.; Casero, R.A., Jr.; Toker, A. Inhibition of the polyamine synthesis enzyme ornithine decarboxylase sensitizes triple-negative breast cancer cells to cytotoxic chemotherapy. J. Biol. Chem. 2020, 295, 6263–6277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajani, J.A.; Ota, D.M.; Grossie, V.B.; Abbruzzese, J.L.; Faintuch, J.S.; Patt, Y.Z.; Jackson, D.E.; Levin, B.; Nishioka, K. Evaluation of continuous-infusion alpha-difluoromethylornithine therapy for colorectal carcinoma. Cancer Chemother. Pharmacol. 1990, 26, 223–226. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.A.; Demers, L.M.; Jones, S.E.; Arseneau, J.; Khandelwal, P.; George, T.; Gersh, R.; Mauger, D.; Manni, A. Alpha-difluoromethylornithine as treatment for metastatic breast cancer patients. Clin. Cancer Res. 1999, 5, 3438–3444. [Google Scholar]
- McCann, P.P.; Pegg, A.E. Ornithine decarboxylase as an enzyme target for therapy. Pharmacol. Ther. 1992, 54, 195–215. [Google Scholar] [CrossRef]
- Ma, H.; Li, Q.; Wang, J.; Pan, J.; Su, Z.; Liu, S. Dual Inhibition of Ornithine Decarboxylase and A1 Adenosine Receptor Efficiently Suppresses Breast Tumor Cells. Front. Oncol. 2021, 11, 636373. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Weeks, R.S.; Burns, M.R.; Boorman, D.W.; Klein-Szanto, A.; O’Brien, T.G. Combination therapy with 2-difluoromethylornithine and a polyamine transport inhibitor against murine squamous cell carcinoma. Int. J. Cancer 2005, 118, 2344–2349. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Tsamis, K.I.; Vartholomatos, E.; Peponi, E.; Tzima, E.; Tasiou, I.; Lykoudis, E.; Tsekeris, P.; Kyritsis, A.P. Combination treatment of TRAIL, DFMO and radiation for malignant glioma cells. J. Neuro-Oncol. 2015, 123, 217–224. [Google Scholar] [CrossRef]
- Alexiou, G.; Vartholomatos, E.; Tsamis, K.I.; Peponi, E.; Markopoulos, G.; A Papathanasopoulou, V.; Tasiou, I.; Ragos, V.; Tsekeris, P.; Kyritsis, A.; et al. Combination treatment for glioblastoma with temozolomide, DFMO and radiation. J. BUON 2019, 24, 397–404. [Google Scholar]
- Gerner, E.W.; Bruckheimer, E.; Cohen, A. Cancer pharmacoprevention: Targeting polyamine metabolism to manage risk factors for colon cancer. J. Biol. Chem. 2018, 293, 18770–18778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Lu, Q.-B. New combination chemotherapy of cisplatin with an electron-donating compound for treatment of multiple cancers. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Steverding, D. The development of drugs for treatment of sleeping sickness: A historical review. Parasites Vectors 2010, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Levin, V.A.; Hess, K.R.; Choucair, A.; Flynn, P.J.; Jaeckle, K.A.; Kyritsis, A.P.; Yung, W.K.A.; Prados, M.D.; Bruner, J.M.; Ictech, S.; et al. Phase III randomized study of postradiotherapy chemotherapy with combination alpha-difluoromethylornithine-PCV versus PCV for anaplastic gliomas. Clin. Cancer Res. 2003, 9, 981–990. [Google Scholar] [PubMed]
- Levin, V.A.; Uhm, J.H.; Jaeckle, K.A.; Choucair, A.; Flynn, P.J.; Wka, Y.; Prados, M.D.; Bruner, J.M.; Chang, S.M.; Kyritsis, A.P.; et al. Phase III randomized study of postradiotherapy chemotherapy with alpha-difluoromethylornithine-procarbazine, N-(2-chloroethyl)-N’-cyclohexyl-N-nitrosurea, vincristine (DFMO-PCV) versus PCV for glioblastoma multiforme. Clin. Cancer Res. 2000, 6, 3878–3884. [Google Scholar]
- Horn, Y.; Schechter, P.J.; Marton, L.J. Phase I–II clinical trial with alpha-difluoromethylornithine—An inhibitor of polyamine biosynthesis. Eur. J. Cancer Clin. Oncol. 1987, 23, 1103–1107. [Google Scholar] [CrossRef]
- Levin, V.A.; Jochec, J.L.; Shantz, L.M.; Koch, P.E.; Pegg, A.E. Tissue-based Assay for Ornithine Decarboxylase to Identify Patients Likely to Respond to Difluoromethylornithine. J. Histochem. Cytochem. 2004, 52, 1467–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Hager, E.R.; Phillips, D.L.; Dunn, V.R.; Hacker, A.; Frydman, B.; Kink, J.A.; Valasinas, A.L.; Reddy, V.K.; Marton, L.J.; et al. A novel polyamine analog inhibits growth and induces apoptosis in human breast cancer cells. Clin. Cancer Res. 2003, 9, 2769–2777. [Google Scholar] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, W.Y.; Park, W.H.; Suh, D.H.; Kim, K.; Kim, Y.B.; No, J.H. Difluoromethylornithine Induces Apoptosis through Regulation of AP-1 Signaling via JNK Phosphorylation in Epithelial Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 10255. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910255
Hwang WY, Park WH, Suh DH, Kim K, Kim YB, No JH. Difluoromethylornithine Induces Apoptosis through Regulation of AP-1 Signaling via JNK Phosphorylation in Epithelial Ovarian Cancer. International Journal of Molecular Sciences. 2021; 22(19):10255. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910255
Chicago/Turabian StyleHwang, Woo Yeon, Wook Ha Park, Dong Hoon Suh, Kidong Kim, Yong Beom Kim, and Jae Hong No. 2021. "Difluoromethylornithine Induces Apoptosis through Regulation of AP-1 Signaling via JNK Phosphorylation in Epithelial Ovarian Cancer" International Journal of Molecular Sciences 22, no. 19: 10255. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910255