
Heterogeneity of Response to Iron-Based Metallodrugs in Glioblastoma Is Associated with Differences in Chemical Structures and Driven by FAS Expression Dynamics and Transcriptomic Subtypes

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Ferrocifens Induce Diverse Impacts on PDCL Viability According to the Genetic Context
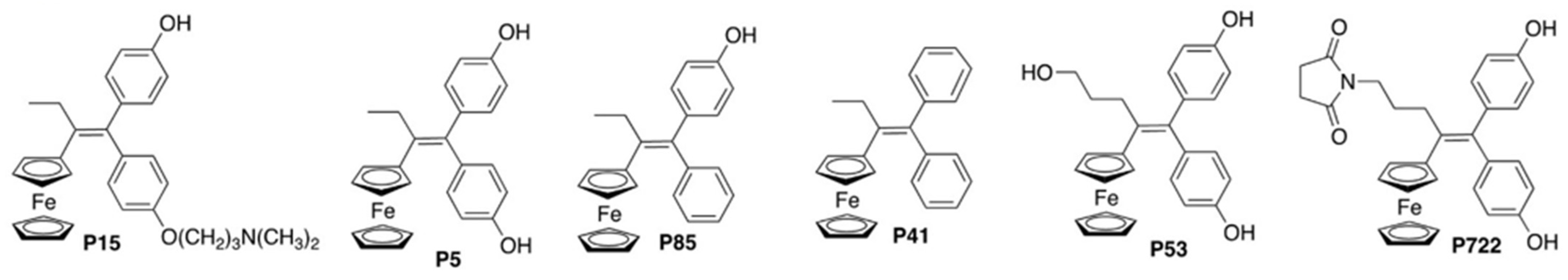
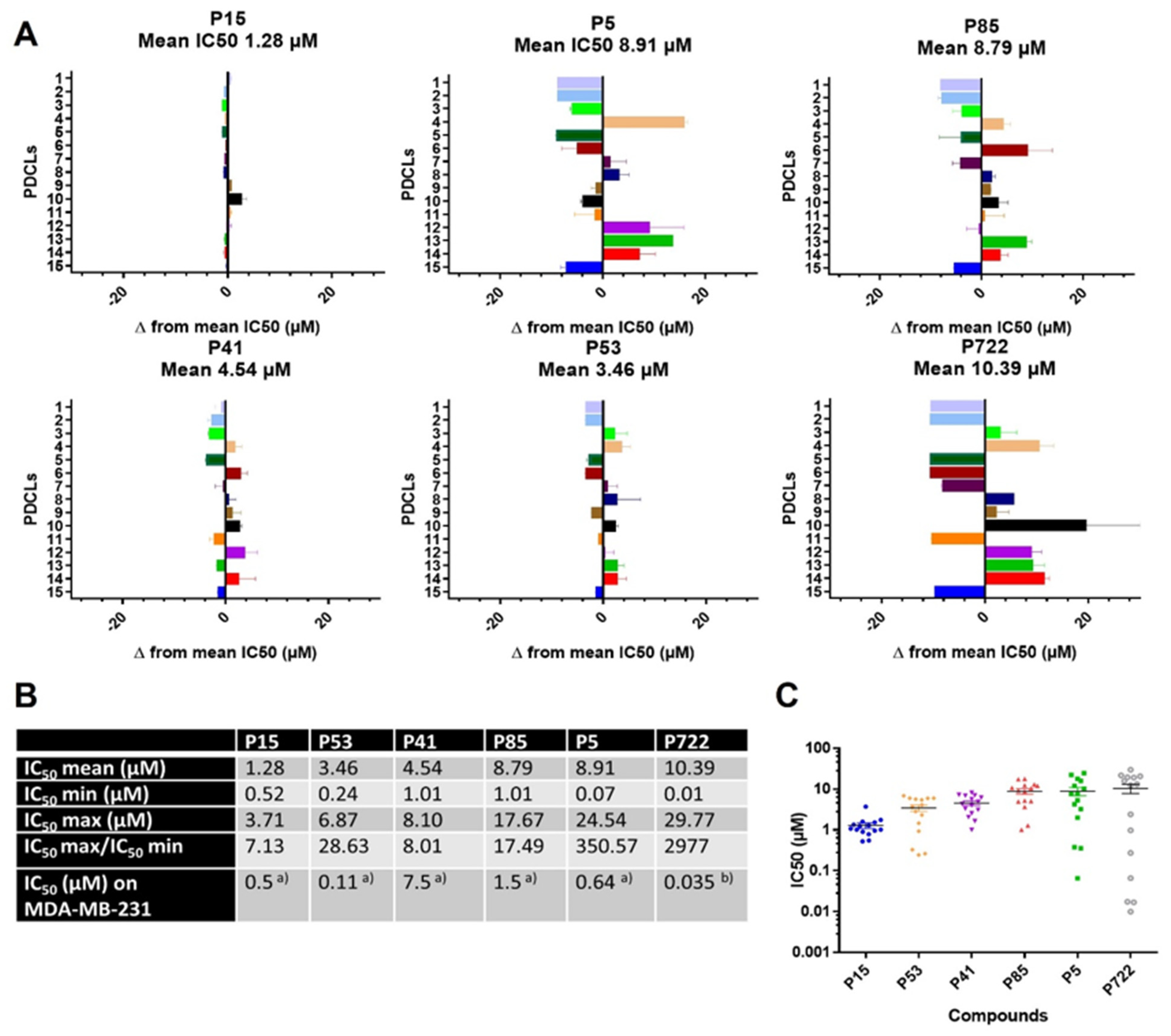
2.1.1. IC50 Values of Ferrocifens on the Panel of PDCLs
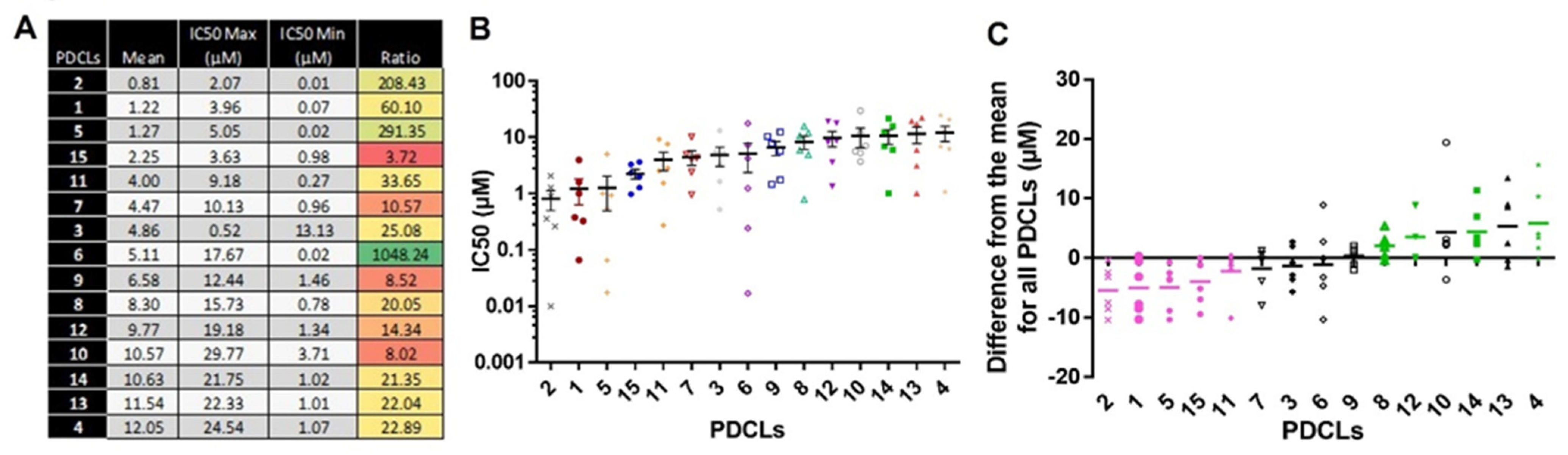
2.1.2. IC50 Values According to the PDCLs
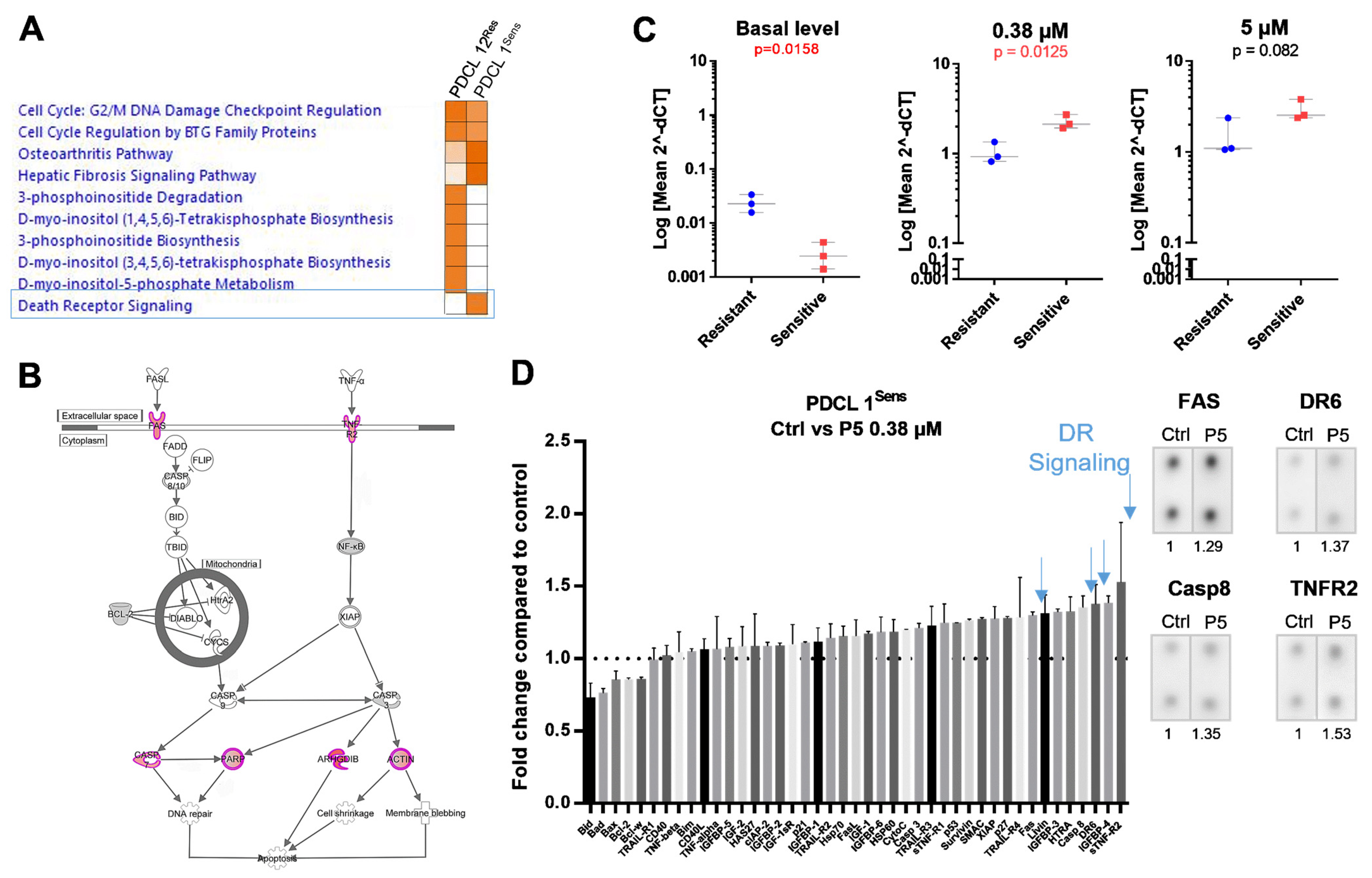
2.2. Death Receptor Signaling and FAS Expression Dynamics Predict the Response to P5
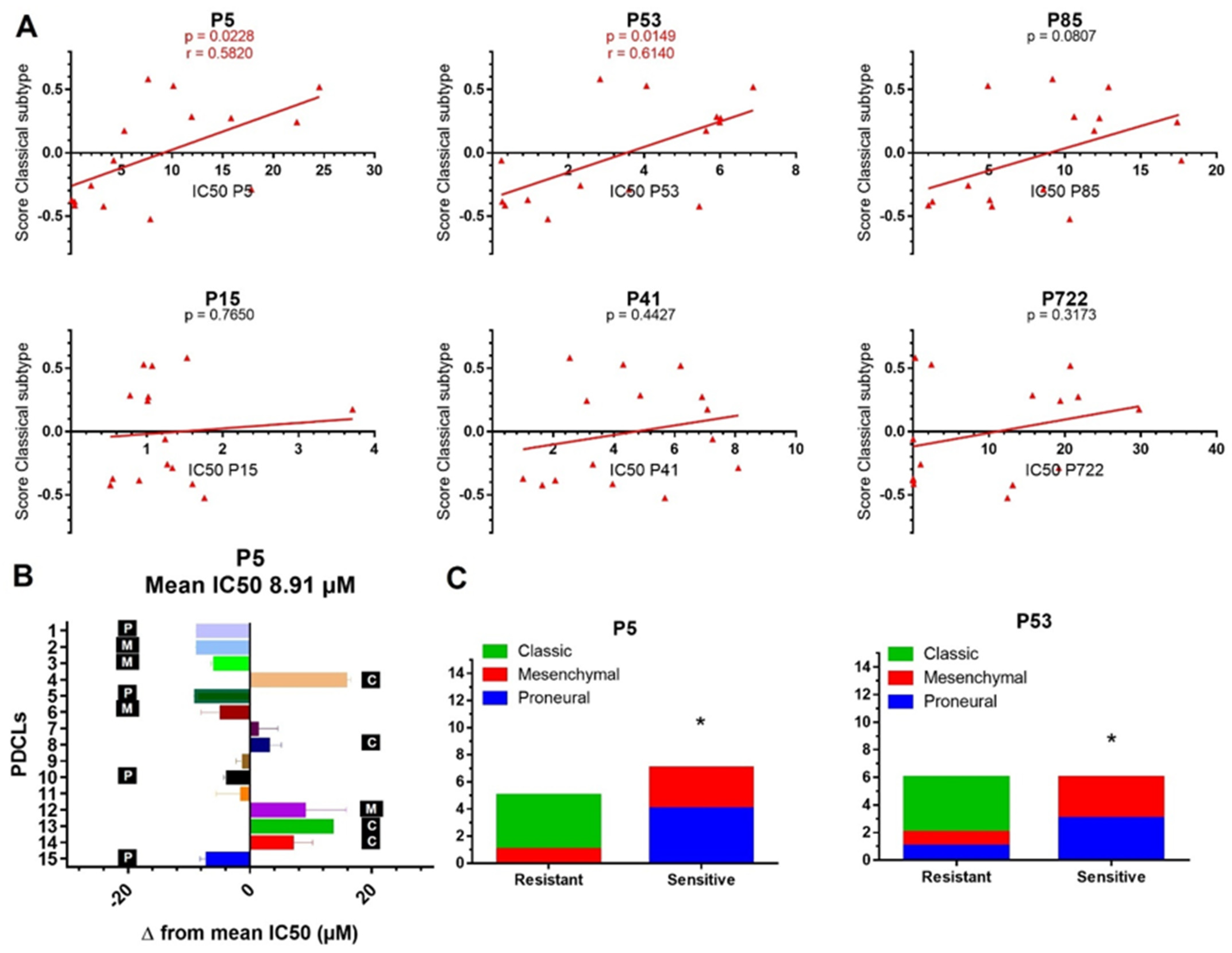
2.3. Transcriptomic Subtypes Predict the Response to P5 and P53
3. Discussion
4. Materials and Methods
4.1. Synthesis of the Compounds
4.2. Cell Lines
4.3. Cell Viability Assay
4.4. RNA Sequencing and Analysis and TP53 Mutation Status
4.5. RT-qPCR
4.6. Protein Array
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hess, K.R.; Broglio, K.R.; Bondy, M.L. Adult Glioma Incidence Trends in the United States, 1977–2000. Cancer 2004, 101, 2293–2299. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauchet, L.; Rigau, V.; Mathieu-Daude, H.; Figarella-Branger, D.; Hugues, D.; Palusseau, L.R.; Bauchet, F.; Fabbro, M.; Campello, C.; Capelle, L.; et al. French Brain Tumor Data Bank: Methodology and First Results on 10,000 Cases. J. Neuro-Oncol. 2007, 84, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Keime-Guibert, F.; Chinot, O.; Taillandier, L.; Cartalat-Carel, S.; Frenay, M.; Kantor, G.; Guillamo, J.S.; Jadaud, E.; Colin, P.; Bondiau, P.Y.; et al. Radiotherapy for Glioblastoma in the Elderly. N. Engl. J. Med. 2007, 356, 1527–1535. [Google Scholar] [CrossRef]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E.; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J.; et al. Bevacizumab plus Irinotecan in Recurrent Glioblastoma Multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar] [CrossRef] [Green Version]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma Heterogeneity and the Tumour Microenvironment: Implications for Preclinical Research and Development of New Treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Kyritsis, A.P.; Bondy, M.L.; Hess, K.R.; Cunningham, J.E.; Zhu, D.; Amos, C.J.; Yung, W.K.A.; Levin, V.A.; Bruner, J.M. Prognostic Significance of P53 Immunoreactivity in Patients with Glioma. Clin. Cancer Res. 1995, 1, 1617–1622. [Google Scholar]
- Wang, X.; Chen, J.; Liu, J.; You, C.; Liu, Y.; Mao, Q. Gain of Function of Mutant TP53 in Glioblastoma: Prognosis and Response to Temozolomide. Ann. Surg. Oncol. 2014, 21, 1337–1344. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen Type Anti Cancer Drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Top, S.; Vessieres, A.; Leclercq, G.; Quivy, J.; Tang, J.; Vaissermann, J.; Huche, M.; Jaouen, G. Synthesis, Biochemical Properties and Molecular Modelling Studies of Organometallic Specific Estrogen Receptor Modulators (SERMs), the Ferrocifens and Hydroxyferrocifens: Evidence for an Antiproliferative Effect of Hydroxyferrocifens on Both Hormone-Dependent and Hormone-Independent Breast Cancer Cell Lines. Chem. Eur. J. 2003, 9, 5223–5236. [Google Scholar] [CrossRef] [PubMed]
- Görmen, M.; Pigeon, P.; Top, S.; Hillard, E.A.; Huché, M.; Hartinger, C.G.; de Montigny, F.; Plamont, M.-A.; Vessières, A.; Jaouen, G. Synthesis, Cytotoxicity, and COMPARE Analysis of Ferrocene and [3]Ferrocenophane Tetrasubstituted Olefin Derivatives against Human Cancer Cells. ChemMedChem 2010, 5, 2039–2050. [Google Scholar] [CrossRef] [Green Version]
- Lainé, A.-L.; Adriaenssens, E.; Vessières, A.; Jaouen, G.; Corbet, C.; Desruelles, E.; Pigeon, P.; Toillon, R.-A.; Passirani, C. The in Vivo Performance of Ferrocenyl Tamoxifen Lipid Nanocapsules in Xenografted Triple Negative Breast Cancer. Biomaterials 2013, 34, 6949–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, E.; Passirani, C.; Garcion, E.; Pigeon, P.; Vessieres, A.; Jaouen, G.; Benoit, J.P. Lipid Nanocapsules Loaded with an Organometallic Tamoxifen Derivative as a Novel Drug-Carrier System for Experimental Malignant Gliomas. J. Control. Release 2008, 130, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Huynh, N.T.; Morille, M.; Bejaud, J.; Legras, P.; Vessieres, A.; Jaouen, G.; Benoit, J.P.; Passirani, C. Treatment of 9L Gliosarcoma in Rats by Ferrociphenol-Loaded Lipid Nanocapsules Based on a Passive Targeting Strategy via the EPR Effect. Pharm. Res. 2011, 28, 3189–3198. [Google Scholar] [CrossRef] [Green Version]
- Hillard, E.; Vessieres, A.; Thouin, L.; Jaouen, G.; Amatore, C. Ferrocene-Mediated Proton-Coupled Electron Transfer in a Series of Ferrocifen-Type Breast-Cancer Drug Candidates. Angew. Chem. Int. Edit. 2006, 45, 285–290. [Google Scholar] [CrossRef]
- Messina, P.; Labbé, E.; Buriez, O.; Hillard, E.A.; Vessières, A.; Hamels, D.; Top, S.; Jaouen, G.; Frapart, Y.M.; Mansuy, D.; et al. Deciphering the Activation Sequence of Ferrociphenol Anticancer Drug Candidates. Chem. Eur. J. 2012, 18, 6581–6587. [Google Scholar] [CrossRef] [PubMed]
- Vessières, A.; Wang, Y.; McGlinchey, M.J.; Jaouen, G. Multifaceted Chemical Behaviour of Metallocene (M = Fe, Os) Quinone Methides. Their Contribution to Biology. Coord. Chem. Rev. 2021, 430, 213658. [Google Scholar] [CrossRef]
- Hamels, D.; Dansette, P.M.; Hillard, E.A.; Top, S.; Vessieres, A.; Herson, P.; Jaouen, G.; Mansuy, D. Ferrocenyl Quinone Methides as Strong Antiproliferative Agents: Formation by Metabolic and Chemical Oxidation of Ferrocenyl Phenols. Angew. Chem. Int. Ed. 2009, 48, 9124–9126. [Google Scholar] [CrossRef]
- Lu, C.; Heldt, J.M.; Guille-Collignon, M.; Lemaitre, F.; Jaouen, G.; Vessieres, A.; Amatore, C. Quantitative Analyses of ROS and RNS Production in Breast Cancer Cell Lines Incubated with Ferrocifens. ChemMedChem 2014, 9, 1286–1293. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Kim, M.K.; Osada, T.; Barry, W.T.; Yang, X.Y.; Freedman, J.A.; Tsamis, K.A.; Datto, M.; Clary, B.M.; Clay, T.; Morse, M.A.; et al. Characterization of an Oxaliplatin Sensitivity Predictor in a Preclinical Murine Model of Colorectal Cancer. Mol. Cancer Ther. 2012, 11, 1500–1509. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor Stem Cells Derived from Glioblastomas Cultured in BFGF and EGF More Closely Mirror the Phenotype and Genotype of Primary Tumors than Do Serum-Cultured Cell Lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.; Verreault, M.; Schmitt, C.; Guegan, J.; Guehennec, J.; Levasseur, C.; Marie, Y.; Bielle, F.; Mokhtari, K.; Hoang-Xuan, K.; et al. Multi-Omics Analysis of Primary Glioblastoma Cell Lines Shows Recapitulation of Pivotal Molecular Features of Parental Tumors. Neuro Oncol. 2017, 19, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pigeon, P.; Wang, Y.; Top, S.; Najlaoui, F.; Alvarez, M.C.G.; Bignon, J.; McGlinchey, M.J.; Jaouen, G. A New Series of Succinimido-Ferrociphenols and Related Heterocyclic Species Induce Strong Antiproliferative Effects, Especially against Ovarian Cancer Cells Resistant to Cisplatin. J. Med. Chem. 2017, 60, 8358–8368. [Google Scholar] [CrossRef] [Green Version]
- Folda, A.; Citta, A.; Scalcon, V.; Cali, T.; Zonta, F.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Mitochondrial Thioredoxin System as a Modulator of Cyclophilin D Redox State. Sci. Rep. 2016, 6, 23071. [Google Scholar] [CrossRef] [Green Version]
- Scalcon, V.; Salmain, M.; Folda, A.; Top, S.; Pigeon, P.; Shirley Lee, H.Z.; Jaouen, G.; Bindoli, A.; Vessières, A.; Rigobello, M.P. Tamoxifen-like Metallocifens Target the Thioredoxin System Determining Mitochondrial Impairment Leading to Apoptosis in Jurkat Cells. Metallomics 2017, 9, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michard, Q.; Jaouen, G.; Vessieres, A.; Bernard, B.A. Evaluation of Cytotoxic Properties of Organometallic Ferrocifens on Melanocytes, Primary and Metastatic Melanoma Cell Lines. J. Inorg. Chem. 2008, 102, 1980–1985. [Google Scholar] [CrossRef]
- Citta, A.; Folda, A.; Bindoli, A.; Pigeon, P.; Top, S.; Vessieres, A.; Salmain, M.; Jaouen, G.; Rigobello, M.P. Evidence for Targeting Thioredoxin Reductases with Ferrocenyl Quinone Methides. A Possible Molecular Basis for the Antiproliferative Effect of Hydroxyferrocifens on Cancer Cells. J. Med. Chem. 2014, 57, 8849–8859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonolo, F.; Salmain, M.; Scalcon, V.; Top, S.; Pigeon, P.; Folda, A.; Caron, B.; McGlinchey, M.J.; Toillon, R.-A.; Bindoli, A.; et al. Small Structural Differences between Two Ferrocenyl Diphenols Determine Large Discrepancies of Reactivity and Biological Effects. ChemMedChem 2019, 14, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pigeon, P.; Top, S.; Garcia, J.S.; Troufflard, C.; Ciofini, I.; McGlinchey, M.J.; Jaouen, G. Atypical Lone Pair-Pi Interaction with Quinone Methides in a Series of Imido-Ferrociphenol Anticancer Drug Candidates. Angew. Chem. Int. Ed. 2019, 58, 8421–8425. [Google Scholar] [CrossRef]
- Wang, Y.; Dansette, P.M.; Pigeon, P.; Top, S.; McGlinchey, M.J.; Mansuy, D.; Jaouen, G. A New Generation of Ferrociphenols Leads to a Great Diversity of Reactive Metabolites, and Exhibits Remarkable Antiproliferative Properties. Chem. Sci. 2018, 9, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vessieres, A.; Corbet, C.; Heldt, J.M.; Lories, N.; Jouy, N.; Laios, I.; Leclercq, G.; Jaouen, G.; Toillon, R.-A. A Ferrocenyl Derivative of Hydroxytamoxifen Elicits an Estrogen Receptor-Independent Mechanism of Action in Breast Cancer Cell Lines. J. Inorg. Biochem. 2010, 104, 503–511. [Google Scholar] [CrossRef]
- Osella, D.; Ferrali, M.; Zanello, P.; Laschi, F.; Fontani, M.; Nervi, C.; Cavigiolio, G. On the Mechanism of the Antitumor Activity of Ferrocenium Derivatives. Inorg. Chim. Acta 2000, 306, 42–48. [Google Scholar] [CrossRef]
- Hearn, J.M.; Hughes, G.M.; Romero-Canelon, I.; Munro, A.F.; Rubio-Ruiz, B.; Liu, Z.; Carragher, N.O.; Sadler, P.J. Pharmaco-Genomic Investigations of Organo-Iridium Anticancer Complexes Reveal Novel Mechanism of Action. Metallomics 2018, 10, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Hearn, J.M.; Romero-Canelon, I.; Munro, A.F.; Fu, Y.; Pizarro, A.M.; Garnett, M.J.; McDermott, U.; Carragher, N.O.; Sadler, P.J. Potent Organo-Osmium Compound Shifts Metabolism in Epithelial Ovarian Cancer Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E3800–E3805. [Google Scholar] [CrossRef] [Green Version]
- Kotamraju, S.; Chitambar, C.R.; Kalivendi, S.V.; Joseph, J.; Kalyanaraman, B. Transferrin Receptor-Dependent Iron Uptake Is Responsible for Doxorubicin-Mediated Apoptosis in Endothelial Cells—Role of Oxidant-Induced Iron Signaling in Apoptosis. J. Biol. Chem. 2002, 277, 17179–17187. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.-M. Extrinsic versus Intrinsic Apoptosis Pathways in Anticancer Chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Friesen, C.; Fulda, S.; Debatin, K.M. Cytotoxic Drugs and the CD95 Pathway. Leukemia 1999, 13, 1854–1858. [Google Scholar] [CrossRef] [Green Version]
- Jian, S.; Song, M.J.; Shin, E.C.; Lee, M.O.; Kim, S.J.; Park, J.H. Apoptosis in Human Hepatoma Cell Lines by Chemotherapeutic Drugs via Fas-Dependent and Fas-Independent Pathways. Hepatology 1999, 29, 101–110. [Google Scholar] [CrossRef]
- Friesen, C.; Fulda, S.; Debatin, K.M. Deficient Activation of the CD95 (APO-1/Fas) System in Drug-Resistant Cells. Leukemia 1997, 11, 1833–1841. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Tsujimoto, Y.; Nagata, S. Effect of Bcl-2 on Fas Antigen-Mediated Cell-Death. J. Immunol. 1993, 151, 621–627. [Google Scholar] [PubMed]
- Steinbach, J.P.; Supra, P.; Huang, H.J.S.; Cavenee, W.K.; Weller, M. CD95-Mediated Apoptosis of Human Glioma Cells: Modulation by Epidermal Growth Factor Receptor Activity. Brain Pathol. 2002, 12, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Cascino, I.; Papoff, G.; DeMaria, R.; Testi, R.; Ruberti, G. Fas/Apo-1 (CD95) Receptor Lacking the Intracytoplasmic Signaling Domain Protects Tumor Cells from Fas-Mediated Apoptosis. J. Immunol. 1996, 156, 13–17. [Google Scholar]
- Wang, X.; Chen, J.; Liu, Y.; You, C.; Mao, Q. Mutant TP53 Enhances the Resistance of Glioblastoma Cells to Temozolomide by Up-Regulating O-6-Methylguanine DNA-Methyltransferase. Neurol. Sci. 2013, 34, 1421–1428. [Google Scholar] [CrossRef]
- Lau, J.; Ilkhanizadeh, S.; Wang, S.; Miroshnikova, Y.A.; Salvatierra, N.A.; Wong, R.A.; Schmidt, C.; Weaver, V.M.; Weiss, W.A.; Persson, A.I. STAT3 Blockade Inhibits Radiation-Induced Malignant Progression in Glioma. Cancer Res. 2015, 75, 4302–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Stupp, R.; Hegi, M.; Wick, W. Individualized Targeted Therapy for Glioblastoma Fact or Fiction? Cancer J. 2012, 18, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Wagle, N.; Nguyen, M.; Carrillo, J.; Truong, J.; Dobrawa, L.; Kesari, S. Characterization of Molecular Pathways for Targeting Therapy in Glioblastoma. Chin. Clin. Oncol. 2020, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Lenin, S.; Ponthier, E.; Scheer, K.G.; Yeo, E.C.F.; Tea, M.N.; Ebert, L.M.; Oksdath Mansilla, M.; Poonnoose, S.; Baumgartner, U.; Day, B.W.; et al. A Drug Screening Pipeline Using 2D and 3D Patient-Derived In Vitro Models for Pre-Clinical Analysis of Therapy Response in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 4322. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma Targeted Therapy: Updated Approaches from Recent Biological Insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Zawlik, I.; Vaccarella, S.; Kita, D.; Mittelbronn, M.; Franceschi, S.; Ohgaki, H. Promoter Methylation and Polymorphisms of the MGMT Gene in Glioblastomas: A Population-Based Study. Neuroepidemiology 2009, 32, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Stupp, R. Withholding Temozolomide in Glioblastoma Patients with Unmethylated MGMT Promoter-Still a Dilemma? Neuro Oncol. 2015, 17, 1425–1427. [Google Scholar] [CrossRef] [Green Version]
- Perego, P.; Robert, J. Oxaliplatin in the Era of Personalized Medicine: From Mechanistic Studies to Clinical Efficacy. Cancer Chemother. Pharmacol. 2016, 77, 5–18. [Google Scholar] [CrossRef]
- Vessieres, A.; Top, S.; Pigeon, P.; Hillard, E.; Boubeker, L.; Spera, D.; Jaouen, G. Modification of the Estrogenic Properties of Diphenols by the Incorporation of Ferrocene. Generation of Antiproliferative Effects in Vitro. J. Med. Chem. 2005, 48, 3937–3940. [Google Scholar] [CrossRef] [Green Version]
- Top, S.; Vessieres, A.; Cabestaing, C.; Laios, I.; Leclercq, G.; Provot, C.; Jaouen, G. Studies on Organometallic Selective Receptor Modulators (SERMs). Dual Activity in the Hydroxy Ferrocifen Series. J. Organomet. Chem. 2001, 637, 500–506. [Google Scholar] [CrossRef]
- Hillard, E.A.; Pigeon, P.; Vessières, A.; Amatore, C.; Jaouen, G. The Influence of Phenolic Hydroxy Substitution on the Electron Transfer and Anti-Cancer Properties of Compounds Based on the 2-Ferrocenyl-1-Phenyl-but-1-Ene Motif. Dalton Trans. 2007, 43, 5073–5081. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Pigeon, P.; Top, S.; McGlinchey, M.J.; Jaouen, G. Organometallic Antitumor Compounds: Ferrocifens as Precursors to Quinone Methides. Angew. Chem. Int. Ed. 2015, 54, 10230–10233. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IPA. 2016. Available online: www.Giagen.Com/Ingenuity (accessed on 1 June 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDCL ID | PDCL Name | TP53 Status | Molecular Subtype | PDCL ID | PDCL Name | TP53 Status | Molecular Subtype |
|---|---|---|---|---|---|---|---|
| 1 | N13-1300 | Pro177Ser/Lys132Asn | Proneural | 9 | GBM4339 | Ser241Phe | Mesenchymal |
| 2 | N15-0460 | Tyr220Cys | Mesenchymal | 10 | N15-0516 | WT | Proneural |
| 3 | GBM7097 | WT | Mesenchymal | 11 | N14-0522 | WT | Classical |
| 4 | N14-1208 | WT | Classical | 12 | N15-0385 | Met237Lys | Mesenchymal |
| 5 | N14-0072 | Tyr220Cys | Proneural | 13 | GBM6240 | WT | Classical |
| 6 | N16-0535 | Arg273His | Mesenchymal | 14 | GBM4371 | Arg175His | Classical |
| 7 | N15-0661 | WT | Classical | 15 | N13-1520 | Arg248Gln | Proneural |
| 8 | N14-1525 | WT | Classical |
| Gene | Forward Primer | Reverse Primer | UPL |
|---|---|---|---|
| FAS | GTGGACCCGCTCAGTACG | TCTAGCAACAGACGTAAGAACCA | #60 |
| TNFR2 | CTCCTTCCTGCTCCCAATG | CACACCCACAATCAGTCCAA | #23 |
| PPIA | ATGCTGGACCCAACACAAAT | TCTTTCACTTTGCCAAACACC | #48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vessières, A.; Quissac, E.; Lemaire, N.; Alentorn, A.; Domeracka, P.; Pigeon, P.; Sanson, M.; Idbaih, A.; Verreault, M. Heterogeneity of Response to Iron-Based Metallodrugs in Glioblastoma Is Associated with Differences in Chemical Structures and Driven by FAS Expression Dynamics and Transcriptomic Subtypes. Int. J. Mol. Sci. 2021, 22, 10404. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910404
Vessières A, Quissac E, Lemaire N, Alentorn A, Domeracka P, Pigeon P, Sanson M, Idbaih A, Verreault M. Heterogeneity of Response to Iron-Based Metallodrugs in Glioblastoma Is Associated with Differences in Chemical Structures and Driven by FAS Expression Dynamics and Transcriptomic Subtypes. International Journal of Molecular Sciences. 2021; 22(19):10404. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910404
Chicago/Turabian StyleVessières, Anne, Emie Quissac, Nolwenn Lemaire, Agusti Alentorn, Patrycja Domeracka, Pascal Pigeon, Marc Sanson, Ahmed Idbaih, and Maïté Verreault. 2021. "Heterogeneity of Response to Iron-Based Metallodrugs in Glioblastoma Is Associated with Differences in Chemical Structures and Driven by FAS Expression Dynamics and Transcriptomic Subtypes" International Journal of Molecular Sciences 22, no. 19: 10404. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910404