
Genome-Wide Analysis of the UDP-Glycosyltransferase Family Reveals Its Roles in Coumarin Biosynthesis and Abiotic Stress in Melilotus albus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Genome-Wide Identification and Characteristics of the UGT Genes in M. albus
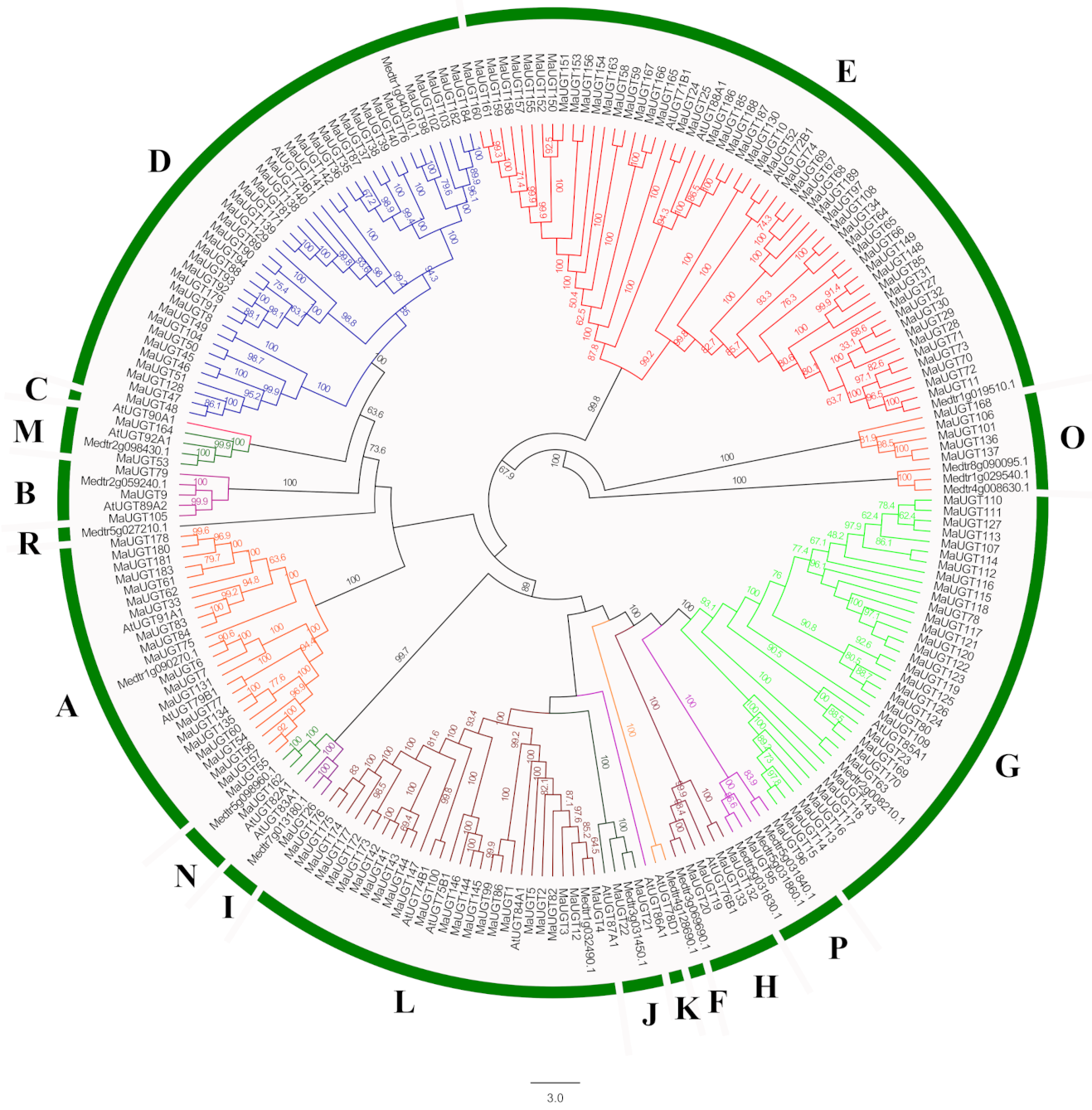
2.2. Phylogenetic Analysis of MaUGTs
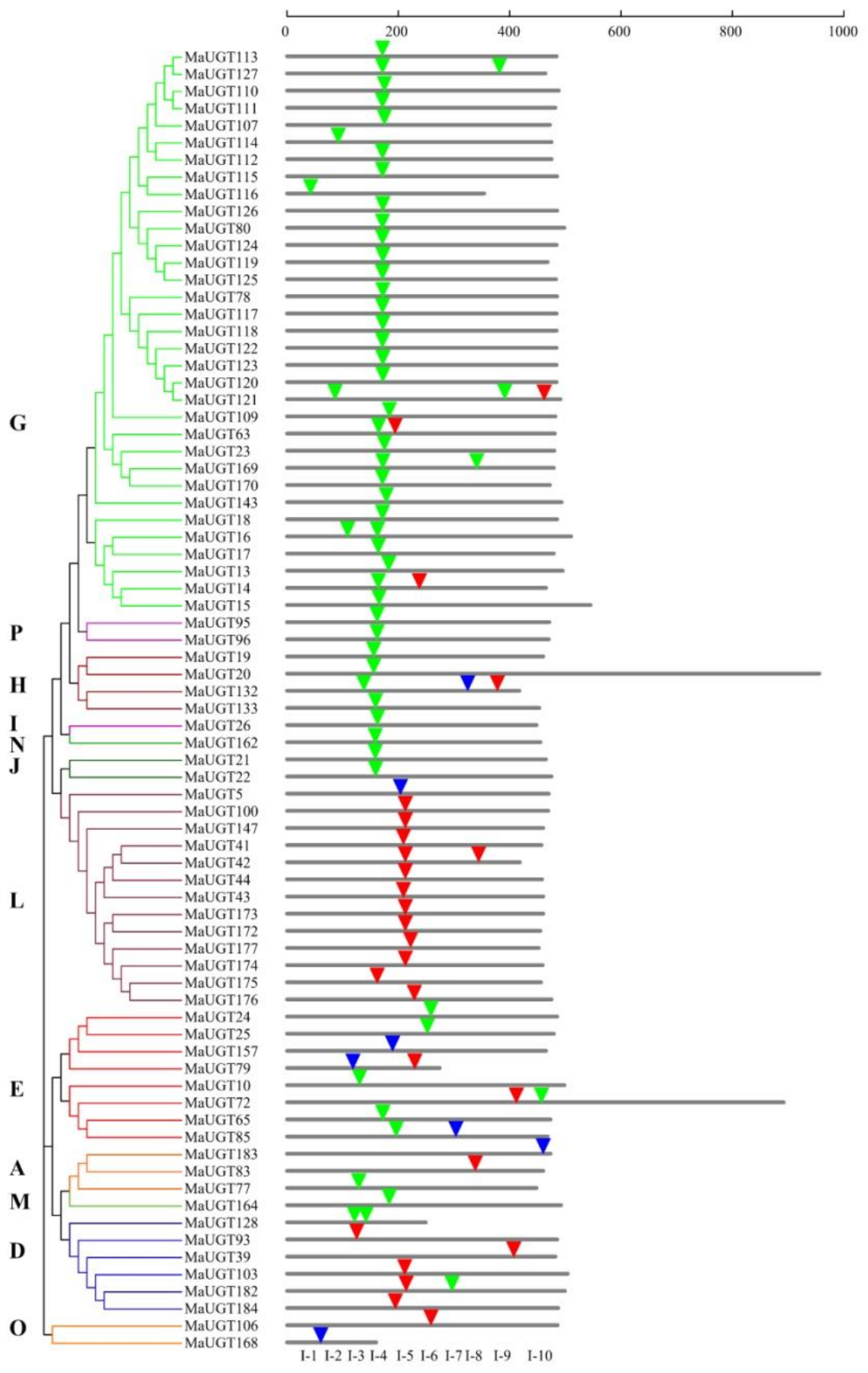
2.3. Structure, Motif Distribution Analysis, and Domain Architecture of the MaUGT Genes
2.4. Characterization of Cis-Acting Elements in the MaUGT Gene Promoters
2.5. Transcriptome Analysis of Tissue-Specific Expression
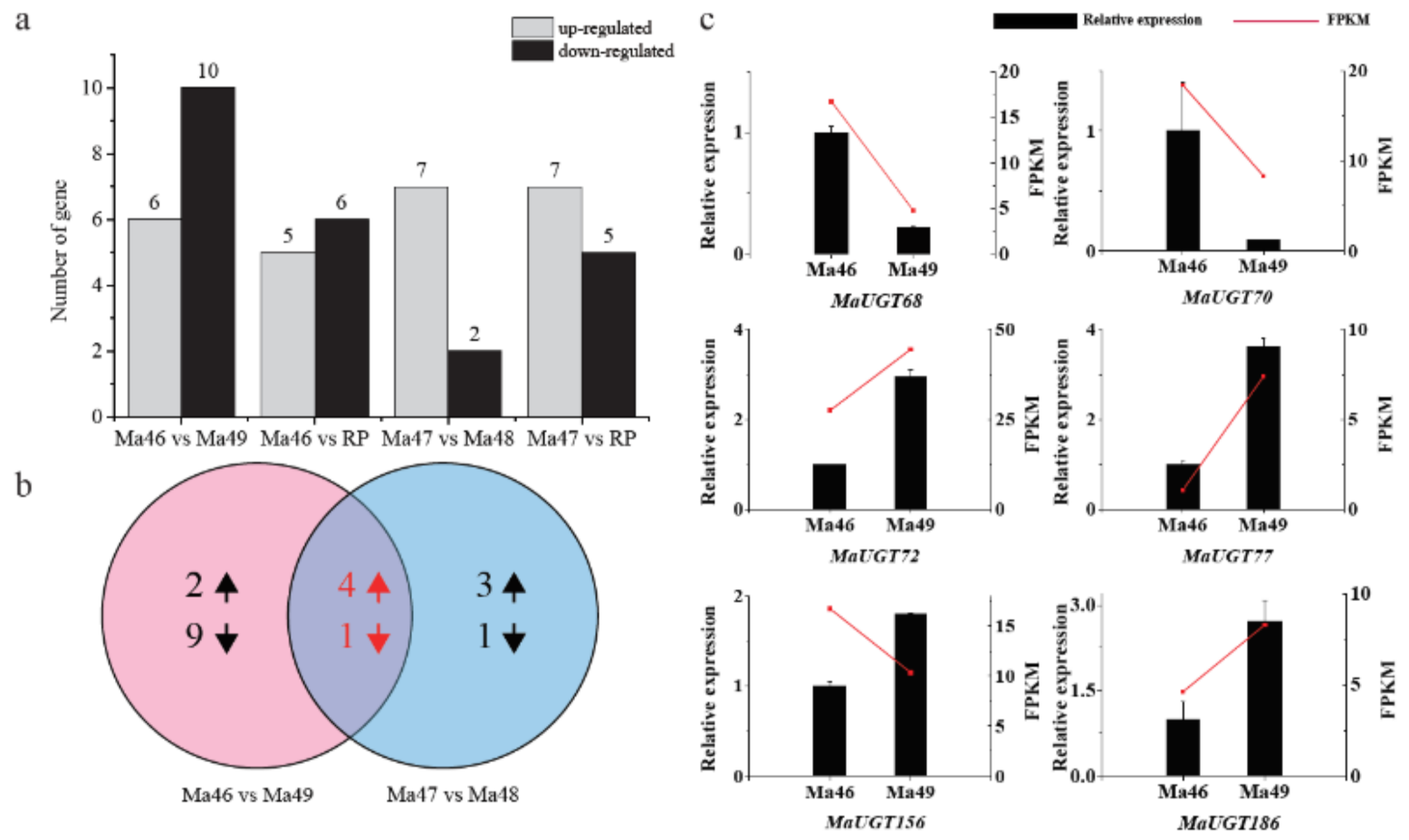
2.6. Expression Analysis of Coumarin Biosynthesis-Related MaUGT Genes
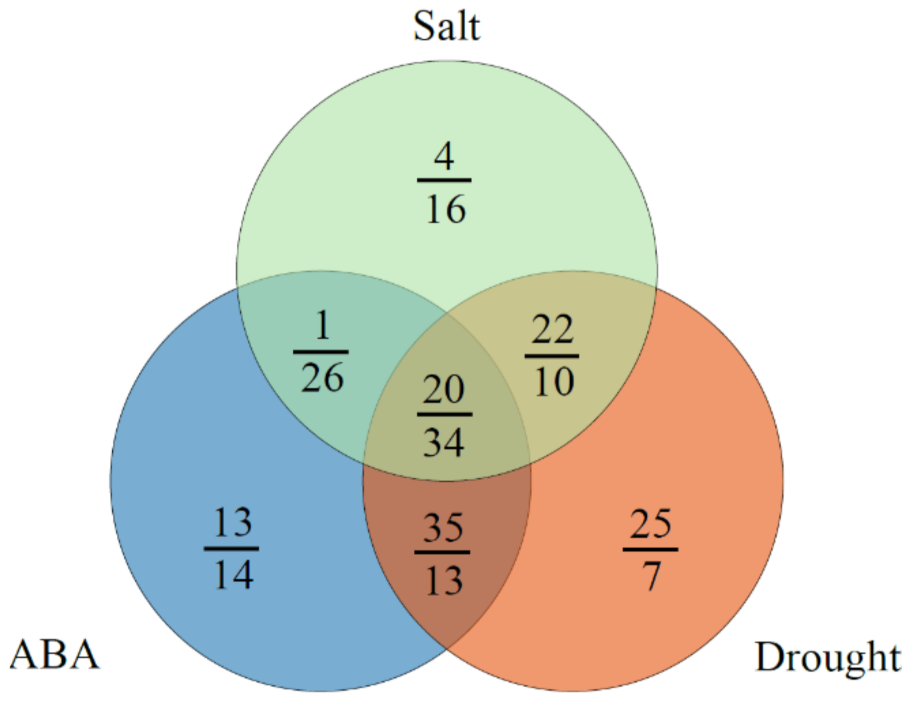
2.7. Expression Analysis of ABA and Abiotic Stress-Related MaUGT Genes
2.8. MaUGT68 and MaUGT186 Improve Drought Tolerance in Yeast
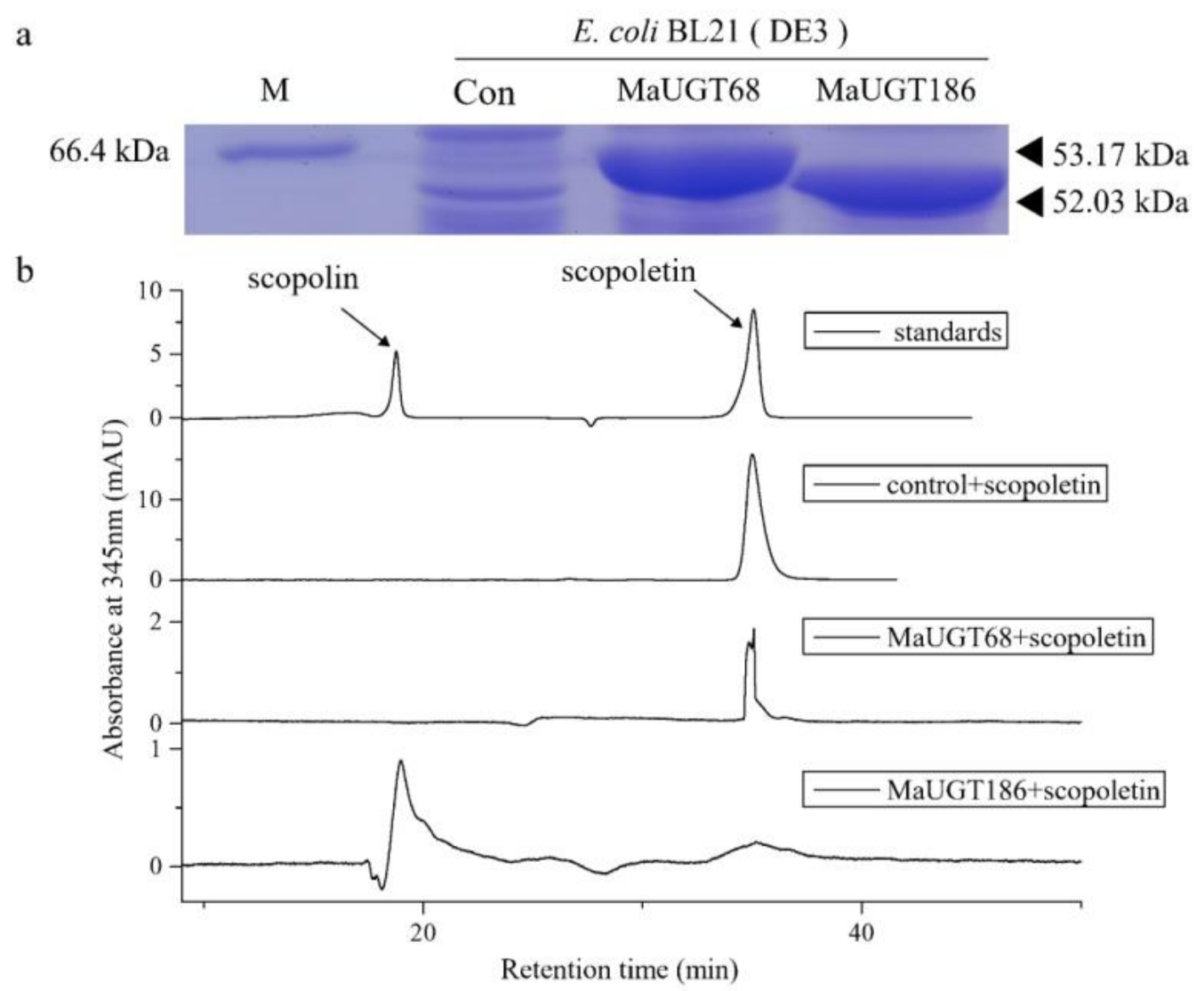
2.9. MaUGT186 Is Involved in Scopolin Biosynthesis
3. Discussion
4. Materials and Methods
4.1. Identification of the UGT Genes in M. albus
4.2. Phylogenetic and Classification Analysis
4.3. Analysis of Conserved Motifs, Gene Structure, and Exon/Intron Organization of the MaUGTs
4.4. Analysis of Promoter Cis-Acting Elements
4.5. Transcriptome Analysis for Tissue-Specific Expression
4.6. Stress Treatment and Transcriptomic Data Analysis
4.7. Quantitative Real-Time PCR Analysis
4.8. Validation of Heterologous Expression in Yeast
4.9. Heterologous Expression in E. coli and Glucosyltransferase Activity Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institution Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lončarić, M.; Gašo-Sokač, D.; Jokić, S.; Molnar, M. Recent Advances in the Synthesis of Coumarin Derivatives from Different Starting Materials. Biomolecules 2020, 10, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringlis, I.A.; de Jonge, R.; Pieterse, C.M.J. The Age of Coumarins in Plant-Microbe Interactions. Plant Cell Physiol. 2019, 60, 1405–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stassen, M.J.J.; Hsu, S.-H.; Pieterse, C.M.J.; Stringlis, I.A. Coumarin Communication Along the Microbiome-Root-Shoot Axis. Trends Plant Sci. 2021, 26, 169–183. [Google Scholar] [CrossRef]
- Robe, K.; Izquierdo, E.; Vignols, F.; Rouached, H.; Dubos, C. The Coumarins: Secondary Metabolites Playing a Primary Role in Plant Nutrition and Health. Trends Plant Sci. 2021, 26, 248–259. [Google Scholar] [CrossRef]
- Chen, B.-X.; Peng, Y.-X.; Gao, J.-D.; Zhang, Q.; Liu, Q.-J.; Fu, H.; Liu, J. Coumarin-induced delay of rice seed germination is mediated by suppression of abscisic acid catabolism and reactive oxygen species production. Front. plant sci. 2019, 10, 828. [Google Scholar] [CrossRef] [Green Version]
- Stringlis, I.A.; Yu, K.; Feussner, K.; de Jonge, R.; van Bentum, S.; Van Verk, M.C.; Berendsen, R.L.; Bakker, P.A.H.M.; Feussner, I.; Pieterse, C.M.J. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA 2018, 115, E5213–E5222. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Baltz, R.; Schmitt, C.; Beffa, R.; Fritig, B.; Saindrenan, P. Downregulation of a pathogen-responsive tobacco UDP-Glc: Phenylpropanoid glucosyltransferase reduces scopoletin glucoside accumulation, enhances oxidative stress, and weakens virus resistance. Plant Cell 2002, 14, 1093–1107. [Google Scholar] [CrossRef] [Green Version]
- Lupini, A.; Araniti, F.; Mauceri, A.; Princi, M.P.; Sorgona, A.; Sunseri, F.; Varanini, Z.; Abenavoli, M.R. Coumarin enhances nitrate uptake in maize roots through modulation of plasma membrane H+-ATPase activity. Plant Biol. 2018, 20, 390–398. [Google Scholar] [CrossRef]
- Santhanam, R.; Menezes, R.C.; Grabe, V.; Li, D.; Baldwin, I.T.; Groten, K. A suite of complementary biocontrol traits allows a native consortium of root-associated bacteria to protect their host plant from a fungal sudden-wilt disease. Mol. Ecol. 2019, 28, 1154–1169. [Google Scholar] [CrossRef]
- Kai, K.; Shimizu, B.; Mizutani, M.; Watanabe, K.; Sakata, K. Accumulation of coumarins in Arabidopsis thaliana. Phytochemistry 2006, 67, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, J.; Huss, B.; Creach, A.; Hawkins, S.; Neutelings, G. Glycosylation is a major regulator of phenylpropanoid availability and biological activity in plants. Front. Plant Sci. 2016, 7, 735. [Google Scholar] [CrossRef] [Green Version]
- Wang, X. Structure, mechanism and engineering of plant natural product glycosyltransferases. Febs Lett. 2009, 583, 3303–3309. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Shen, G.; Di, S.; Fan, C.; Chang, Z.; Pang, Y. Genome-wide identification and functional characterization of UDP-glucosyltransferase genes involved in flavonoid biosynthesis in Glycine max. Plant Cell Physiol. 2017, 58, 1558–1572. [Google Scholar] [CrossRef]
- Xiao, X.; Lu, Q.; Liu, R.; Gong, J.; Gong, W.; Liu, A.; Ge, Q.; Li, J.; Shang, H.; Li, P.; et al. Genome-wide characterization of the UDP-glycosyltransferase gene family in upland cotton. 3 Biotech 2019, 9, 453. [Google Scholar] [CrossRef]
- Song, Z.; Niu, L.; Yang, Q.; Dong, B.; Wang, L.; Dong, M.; Fan, X.; Jian, Y.; Meng, D.; Fu, Y. Genome-wide identification and characterization of UGT family in pigeonpea (Cajanus cajan) and expression analysis in abiotic stress. Trees-Struct. Funct. 2019, 33, 987–1002. [Google Scholar] [CrossRef]
- Wu, B.; Liu, X.; Xu, K.; Zhang, B. Genome-wide characterization, evolution and expression profiling of UDP-glycosyltransferase family in pomelo (Citrus grandis) fruit. BMC Plant Biol. 2020, 20, 459. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Su, Y.; Chen, N.; Shen, S. Genome-wide analysis of the UGT gene family and identification of flavonoids in Broussonetia papyrifera. Molecules 2021, 26, 3449. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Baldauf, S.; Lim, E.K.; Bowles, D.J. Phylogenetic analysis of the UDP-glycosyltransferase multigene family of Arabidopsis thaliana. J. Biol. Chem. 2001, 276, 4338–4343. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-S.; Huang, X.-X.; Li, Q.; Cao, Y.; Bao, Y.; Meng, X.-F.; Li, Y.-J.; Fu, C.; Hou, B.-K. UDP-glycosyltransferase 72B1 catalyzes the glucose conjugation of monolignols and is essential for the normal cell wall lignification in Arabidopsis thaliana. Plant J. 2016, 88, 26–42. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Strebl, M.; Merz, M.; Blamberg, R.; Huang, F.-C.; McGraphery, K.; Hoffmann, T.; Schwab, W. Glucosylation of the phytoalexin N-feruloyl tyramine modulates the levels of pathogen-responsive metabolites in Nicotiana benthamiana. Plant J. 2019, 100, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, B.; Lacchini, E.; Bicalho, K.U.; Mertens, J.; Arendt, P.; Vanden Bossche, R.; Calegario, G.; Gryffroy, L.; Ceulemans, E.; Buitink, J.; et al. A seed-specific regulator of triterpene saponin biosynthesis in Medicago truncatula. Plant Cell 2020, 32, 2020–2042. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, X.-X.; Zhao, S.-M.; Xiao, D.-W.; Xiao, L.-T.; Tong, J.-H.; Wang, W.-S.; Li, Y.-J.; Ding, Z.; Hou, B.-K. IPyA glucosylation mediates light and temperature signaling to regulate auxin-dependent hypocotyl elongation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2020, 117, 6910–6917. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-X.; Wang, Y.; Lin, J.-S.; Chen, L.; Li, Y.-J.; Liu, Q.; Wang, G.-F.; Xu, F.; Liu, L.; Hou, B.-K. The novel pathogen-responsive glycosyltransferase UGT73C7 mediates the redirection of phenylpropanoid metabolism and promotes SNC1-dependent Arabidopsis immunity. Plant J. 2021, 107, 149–165. [Google Scholar] [CrossRef]
- Wu, F.; Luo, K.; Yan, Z.; Zhang, D.; Yan, Q.; Zhang, Y.; Yi, X.; Zhang, J. Analysis of miRNAs and their target genes in five Melilotus albus NILs with different coumarin content. Sci. Rep. 2018, 8, 14138. [Google Scholar] [CrossRef] [PubMed]
- Zabala, J.M.; Marinoni, L.; Giavedoni, J.A.; Schrauf, G.E. Breeding strategies in Melilotus albus Desr., a salt-tolerant forage legume. Euphytica 2018, 214, 22. [Google Scholar] [CrossRef]
- Zhang, J.; Di, H.; Luo, K.; Jahufer, Z.; Wu, F.; Duan, Z.; Stewart, A.; Yan, Z.; Wang, Y. Coumarin content, morphological variation, and molecular phylogenetics of Melilotus. Molecules 2018, 23, 810. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Ahmad, D.; Zhang, X.; Zhang, Y.; Wu, L.; Jiang, P.; Ma, H. Genome-wide analysis of family-1 UDP glycosyltransferases (UGT) and identification of UGT genes for FHB resistance in wheat (Triticum aestivum L.). BMC Plant Biol. 2018, 18, 67. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Tsukamoto, C.; Ishimoto, M. Reconstruction of the evolutionary histories of UGT gene superfamily in Legumes Clarifies the functional divergence of duplicates in specialized metabolism. Int. J. Mol. Sci. 2020, 21, 1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K.; Wu, F.; Zhang, D.; Dong, R.; Fan, Z.; Zhang, R.; Yan, Z.; Wang, Y.; Zhang, J. Transcriptomic profiling of Melilotus albus near-isogenic lines contrasting for coumarin content. Sci. Rep. 2017, 7, 4577. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, H.; Cao, X.; Zhang, B.; Chen, K. Genome-wide identification, expression patterns, and functional analysis of UDP glycosyltransferase family in Peach (Prunus persica L. Batsch). Front. plant sci. 2017, 8, 389. [Google Scholar]
- Li, Y.; Li, P.; Wang, Y.; Dong, R.; Yu, H.; Hou, B. Genome-wide identification and phylogenetic analysis of Family-1 UDP glycosyltransferases in maize (Zea mays). Planta 2014, 239, 1265–1279. [Google Scholar] [CrossRef] [PubMed]
- Caputi, L.; Malnoy, M.; Goremykin, V.; Nikiforova, S.; Martens, S. A genome-wide phylogenetic reconstruction of family 1 UDP-glycosyltransferases revealed the expansion of the family during the adaptation of plants to life on land. Plant J. 2012, 69, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.K.; Doucet, C.J.; Li, Y.; Elias, L.; Worrall, D.; Spencer, S.P.; Ross, J.; Bowles, D.J. The activity of Arabidopsis glycosyltransferases toward salicylic acid, 4-hydroxybenzoic acid, and other benzoates. J. Biol. Chem. 2002, 277, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.K.; Ashford, D.A.; Hou, B.K.; Jackson, R.G.; Bowles, D.J. Arabidopsis glycosyltransferases as biocatalysts in fermentation for regioselective synthesis of diverse quercetin glucosides. Biotechnol. Bioeng. 2004, 87, 623–631. [Google Scholar] [CrossRef]
- Poppenberger, B.; Fujioka, S.; Soeno, K.; George, G.L.; Vaistij, F.E.; Hiranuma, S.; Seto, H.; Takatsuto, S.; Adam, G.; Yoshida, S.; et al. The UGT73C5 of Arabidopsis thaliana glucosylates brassinosteroids. Proc. Natl. Acad. Sci. USA 2005, 102, 15253–15258. [Google Scholar] [CrossRef] [Green Version]
- Caputi, L.; Lim, E.-K.; Bowles, D.J. Discovery of new biocatalysts for the glycosylation of terpenoid scaffolds. Chem. Eur. J. 2008, 14, 6656–6662. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.W.; Gilbert, W. Rates of intron loss and gain: Implications for early eukaryotic evolution. Proc. Natl. Acad. Sci. USA 2005, 102, 5773–5778. [Google Scholar] [CrossRef] [Green Version]
- Kubo, A.; Arai, Y.; Nagashima, S.; Yoshikawa, T. Alteration of sugar donor specificities of plant glycosyltransferases by a single point mutation. Arch. Biochem. Biophys. 2004, 429, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, K.; Khurshid, M.; Li, J.; He, M.; Georgiev, M.I.; Zhang, X.; Zhou, M. MYB transcription repressors regulate plant secondary metabolism. Crit. Rev. Plant Sci. 2019, 38, 159–170. [Google Scholar] [CrossRef]
- Tsai, H.H.; Schmidt, W. Mobilization of iron by plant-borne coumarins. Trends Plant Sci. 2017, 22, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yu, H.-M.; Meng, X.-F.; Lin, J.-S.; Li, Y.-J.; Hou, B.-K. Ectopic expression of glycosyltransferase UGT76E11 increases flavonoid accumulation and enhances abiotic stress tolerance in Arabidopsis. Plant Biology 2018, 20, 10–19. [Google Scholar] [CrossRef]
- Sun, Y.; Ji, K.; Liang, B.; Du, Y.; Jiang, L.; Wang, J.; Kai, W.; Zhang, Y.; Zhai, X.; Chen, P.; et al. Suppressing ABA uridine diphosphate glucosyltransferase (SlUGT75C1) alters fruit ripening and the stress response in tomato. Plant J. 2017, 91, 574–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Sun, Y.; Li, M.; Long, R. CrUGT87A1, a UDP-sugar glycosyltransferases (UGTs) gene from Carex rigescens, increases salt tolerance by accumulating flavonoids for antioxidation in Arabidopsis thaliana. Plant Physiol. Bioch. 2021, 159, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Parvin, K.; Hasanuzzaman, M.; Mohsin, S.M.; Nahar, K.; Fujita, M. Coumarin improves tomato plant tolerance to salinity by enhancing antioxidant defence, glyoxalase system and ion homeostasis. Plant Biology 2021, 23, 181–192. [Google Scholar] [CrossRef]
- Osmani, S.A.; Bak, S.; Moller, B.L. Substrate specificity of plant UDP-dependent glycosyltransferases predicted from crystal structures and homology modeling. Phytochemistry 2009, 70, 325–347. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic acids res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar]
- Jeanmougin, F.; Thompson, J.D.; Gouy, M.; Higgins, D.G.; Gibson, T.J. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 1998, 23, 403–405. [Google Scholar] [CrossRef]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular evolutionary genetics analysis software for microcomputers. Comput. Appl. Biosci. 1994, 10, 189–191. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Mikael, B.; Buske, F.A.; Martin, F.; Grant, C.E.; Luca, C. MEME suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a data-base of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Wang, S.; Han, Y.; Zhao, Q.; Xu, P.; Yan, Q.; Wu, F.; Zhang, J. Genome-wide profiling of the potential regulatory network of lncRNA and mRNA in Melilotus albus under salt stress. Environ. Exp. Bot. 2021, 189, 302–312. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Nan, Z. Relative and absolute quantification expression analysis of CsSAMDC gene as a case. China Biotechnol. 2009, 29, 86–91. [Google Scholar]
- Zhang, Z.; Jin, X.; Liu, Z.; Zhang, J.; Liu, W. Genome-wide identification of FAD gene family and functional analysis of MsFAD3.1 involved in the accumulation of alpha-linolenic acid in alfalfa. Crop Sci. 2021, 61, 566–579. [Google Scholar] [CrossRef]
- Kim, O.T.; Jin, M.L.; Lee, D.Y.; Jetter, R. Characterization of the asiatic acid glucosyltransferase, UGT73AH1, involved in asiaticoside biosynthesis in Centella asiatica (L.) Urban. Int. J. Mol. Sci. 2017, 18, 2630. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, Z.; Yan, Q.; Wu, F.; Wang, Y.; Wang, S.; Zong, X.; Zhou, P.; Zhang, J. Genome-Wide Analysis of the UDP-Glycosyltransferase Family Reveals Its Roles in Coumarin Biosynthesis and Abiotic Stress in Melilotus albus. Int. J. Mol. Sci. 2021, 22, 10826. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910826
Duan Z, Yan Q, Wu F, Wang Y, Wang S, Zong X, Zhou P, Zhang J. Genome-Wide Analysis of the UDP-Glycosyltransferase Family Reveals Its Roles in Coumarin Biosynthesis and Abiotic Stress in Melilotus albus. International Journal of Molecular Sciences. 2021; 22(19):10826. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910826
Chicago/Turabian StyleDuan, Zhen, Qi Yan, Fan Wu, Yimeng Wang, Shengsheng Wang, Xifang Zong, Pei Zhou, and Jiyu Zhang. 2021. "Genome-Wide Analysis of the UDP-Glycosyltransferase Family Reveals Its Roles in Coumarin Biosynthesis and Abiotic Stress in Melilotus albus" International Journal of Molecular Sciences 22, no. 19: 10826. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910826