
Identification of a Novel Mutation of β-Spectrin in Hereditary Spherocytosis Using Whole Exome Sequencing

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Case Presentation
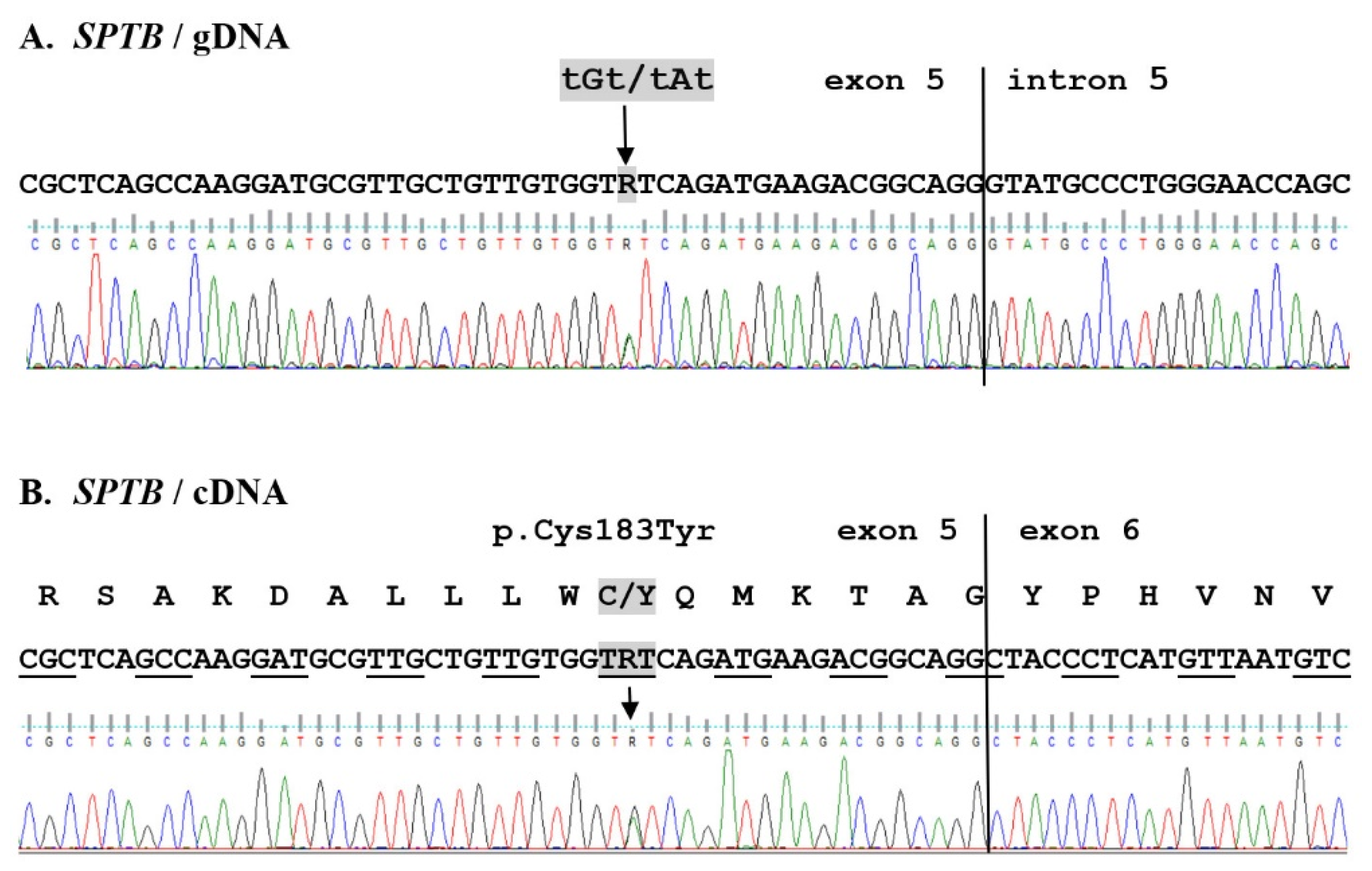
2.2. WES Results Analysis
3. Discussion
4. Materials and Methods
4.1. Hematological Parameters
4.2. DNA Isolation
4.3. Whole Exome Sequencing
4.4. Mutation Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Delaunay, J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Perrotta, S.; Gallagher, P.G.; Mohandas, N. Hereditary spherocytosis. Lancet 2008, 372, 1411–1426. [Google Scholar] [CrossRef]
- Bianchi, P.; Fermo, E.; Vercellati, C.; Marcello, A.P.; Porretti, L.; Cortelezzi, A.; Barcellini, W.; Zanella, A. Diagnostic power of laboratory tests for hereditary spherocytosis: A comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica 2012, 97, 516–523. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.B.; Langer, J.C.; Iolascon, A.; Tittensor, P.; King, M.-J. Guidelines for the diagnosis and management of hereditary spherocytosis—2011 update. Br. J. Haematol. 2012, 156, 37–49. [Google Scholar] [CrossRef]
- Narla, J.; Mohandas, N. Red cell membrane disorders. Int. J. Lab. Hematol. 2017, 39 (Suppl. 1), 47–52. [Google Scholar] [CrossRef]
- Pugi, J.; Drury, L.J.; Langer, J.C.; Butchart, S.; Fantauzzi, M.; Baker, J.; Blanchette, V.S.; Kirby-Allen, M.A.; Carcao, M. Genotype/Phenotype Correlations in 103 Children from 87 Families with Hereditary Spherocytosis. Blood 2016, 128, 2432. [Google Scholar] [CrossRef]
- Li, H.; Papageorgiou, D.; Chang, H.-Y.; Lu, L.; Yang, J.; Deng, Y. Synergistic Integration of Laboratory and Numerical Approaches in Studies of the Biomechanics of Diseased Red Blood Cells. Biosensors 2018, 8, 76. [Google Scholar] [CrossRef]
- Mohandas, N. Inherited hemolytic anemia: A possessive beginner’s guide. Hematology 2018, 2018, 377–381. [Google Scholar] [CrossRef]
- Liem, R.I.; Gallagher, P.G. Molecular mechanisms in the inherited red cell membrane disorders. Drug Discov. Today Dis. Mech. 2005, 2, 539–545. [Google Scholar] [CrossRef]
- Hassoun, H.; Vassiliadis, J.; Murray, J.; Yi, S.; Hanspal, M.; Johnson, C.; Palek, J. Hereditary spherocytosis with spectrin deficiency due to an unstable truncated beta spectrin. Blood 1996, 87, 2538–2545. [Google Scholar] [CrossRef]
- Jarolim, P.; Rubin, H.L.; Brabec, V.; Palek, J. A nonsense mutation 1669Glu→Ter within the regulatory domain of human erythroid ankyrin leads to a selective deficiency of the major ankyrin isoform (band 2.1) and a phenotype of autosomal dominant hereditary spherocytosis. J. Clin. Investig. 1995, 95, 941–947. [Google Scholar] [CrossRef]
- Fan, J.; Yao, L.; Lu, D.; Yao, Y.; Sun, Y.; Tian, Y.; Mou, L.; Chen, L.; Zhao, L.; Qiao, S.; et al. The updated beta-spectrin mutations in patients with hereditary spherocytosis by targeted next-generation sequencing. J. Hum. Genet. 2021, 1–6. [Google Scholar] [CrossRef]
- Bogusławska, D.M.; Heger, E.; Chorzalska, A.; Nierzwicka, M.; Hołojda, J.; Świderska, A.; Straburzyńska, A.; Paździor, G.; Langner, M.; Sikorski, A.F. Hereditary spherocytosis: Identification of several HS families with ankyrin and band 3 deficiency in a population of southwestern Poland. Ann. Hematol. 2004, 83, 28–33. [Google Scholar] [CrossRef]
- Paździor, G.; Langner, M.; Chmura, A.; Bogusławska, D.; Heger, E.; Chorzalska, A.; Sikorski, A.F. The kinetics of haemolysis of spherocytic erythrocytes. Cell. Mol. Biol. Lett. 2003, 8, 639–648. [Google Scholar]
- Bogusławska, D.M.; Heger, E.; Baldy-Chudzik, K.; Zagulski, M.; Maciejewska, M.; Likwiarz, A.; Sikorski, A.F. (AC)n microsatellite polymorphism and 14-nucleotide deletion in exon 42 ankyrin-1 gene in several families with hereditary spherocytosis in a population of South-Western Poland. Ann. Hematol. 2006, 85, 337–339. [Google Scholar] [CrossRef]
- Maciąg, M.; Płochocka, D.; Adamowicz-Salach, A.; Burzyńska, B. Novel beta-spectrin mutations in hereditary spherocytosis associated with decreased levels of mRNA. Br. J. Haematol. 2009, 146, 326–332. [Google Scholar] [CrossRef]
- Bogusławska, D.M.; Heger, E.; Machnicka, B.; Skulski, M.; Kuliczkowski, K.; Sikorski, A.F. A new frameshift mutation of the β-spectrin gene associated with hereditary spherocytosis. Ann. Hematol. 2017, 96, 163–165. [Google Scholar] [CrossRef]
- Bogusławska, D.M.; Heger, E.; Listowski, M.; Wasiński, D.; Kuliczkowski, K.; Machnicka, B.; Sikorski, A.F. A novel L1340P mutation in the ANK1 gene is associated with hereditary spherocytosis? Br. J. Haematol. 2014, 167, 269–271. [Google Scholar] [CrossRef]
- Chasis, J.A.; Agre, P.; Mohandas, N. Decreased membrane mechanical stability and in vivo loss of surface area reflect spectrin deficiencies in hereditary spherocytosis. J. Clin. Investig. 1988, 82, 617–623. [Google Scholar] [CrossRef]
- Reliene, R.; Mariani, M.; Zanella, A.; Reinhart, W.H.; Ribeiro, M.L.; del Giudice, E.M.; Perrotta, S.; Iolascon, A.; Eber, S.; Lutz, H.U. Splenectomy prolongs in vivo survival of erythrocytes differently in spectrin/ankyrin- and band 3–deficient hereditary spherocytosis. Blood 2002, 100, 2208–2215. [Google Scholar] [CrossRef]
- Manciu, S.; Matei, E.; Trandafir, B. Hereditary Spherocytosis—Diagnosis, Surgical Treatment and Outcomes. A Literature Review. Chirurgia 2017, 112, 110. [Google Scholar] [CrossRef]
- Andolfo, I.; Martone, S.; Rosato, B.E.; Marra, R.; Gambale, A.; Forni, G.L.; Pinto, V.; Göransson, M.; Papadopoulou, V.; Gavillet, M.; et al. Complex modes of inheritance in hereditary red blood cell disorders: A case series study of 155 patients. Genes 2021, 12, 958. [Google Scholar] [CrossRef]
- Iolascon, A.; Andolfo, I.; Russo, R. Advances in understanding the pathogenesis of red cell membrane disorders. Br. J. Haematol. 2019, 187, 13–24. [Google Scholar] [CrossRef]
- Tole, S.; Dhir, P.; Pugi, J.; Drury, L.J.; Butchart, S.; Fantauzzi, M.; Langer, J.C.; Baker, J.M.; Blanchette, V.S.; Kirby-Allen, M.; et al. Genotype–phenotype correlation in children with hereditary spherocytosis. Br. J. Haematol. 2020, 191, 486–496. [Google Scholar] [CrossRef]
- Cortesi, V.; Manzoni, F.; Raffaeli, G.; Cavallaro, G.; Fattizzo, B.; Amelio, G.S.; Gulden, S.; Amodeo, I.; Giannotta, J.A.; Mosca, F.; et al. Severe Presentation of Congenital Hemolytic Anemias in the Neonatal Age: Diagnostic and Therapeutic Issues. Diagnostics 2021, 11, 1549. [Google Scholar] [CrossRef]
- Farias, M.G. Advances in laboratory diagnosis of hereditary spherocytosis. Clin. Chem. Lab. Med. (CCLM) 2017, 55, 944–948. [Google Scholar] [CrossRef]
- Ciepiela, O. Old and new insights into the diagnosis of hereditary spherocytosis. Ann. Transl. Med. 2018, 6, 339. [Google Scholar] [CrossRef]
- King, M.-J.; Smythe, J.S.; Mushens, R. Eosin-5-maleimide binding to band 3 and Rh-related proteins forms the basis of a screening test for hereditary spherocytosis. Br. J. Haematol. 2004, 124, 106–113. [Google Scholar] [CrossRef]
- Russo, R.; Andolfo, I.; Manna, F.; Gambale, A.; Marra, R.; Rosato, B.E.; Caforio, P.; Pinto, V.; Pignataro, P.; Radhakrishnan, K.; et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am. J. Hematol. 2018, 93, 672–682. [Google Scholar] [CrossRef]
- Park, J.; Jeong, D.C.; Yoo, J.; Jang, W.; Chae, H.; Kim, J.; Kwon, A.; Choi, H.; Lee, J.W.; Chung, N.G.; et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin. Genet. 2016, 90, 69–78. [Google Scholar] [CrossRef]
- Jang, W.; Kim, J.; Chae, H.; Kim, M.; Koh, K.-N.; Park, C.-J.; Kim, Y. Hereditary spherocytosis caused by copy number variation in SPTB gene identified through targeted next-generation sequencing. Int. J. Hematol. 2019, 110, 250–254. [Google Scholar] [CrossRef]
- Shin, S.; Jang, W.; Kim, M.; Kim, Y.; Park, S.Y.; Park, J.; Yang, Y.J. Targeted next-generation sequencing identifies a novel nonsense mutation in SPTB for hereditary spherocytosis. Medicine 2018, 97, e9677. [Google Scholar] [CrossRef]
- Wang, R.; Yang, S.; Xu, M.; Huang, J.; Liu, H.; Gu, W.; Zhang, X. Exome sequencing confirms molecular diagnoses in 38 Chinese families with hereditary spherocytosis. Sci. China Life Sci. 2018, 61, 947–953. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Kim, M. Diagnostic approaches for inherited hemolytic anemia in the genetic era. Blood Res. 2017, 52, 84. [Google Scholar] [CrossRef]
- Paila, U.; Chapman, B.A.; Kirchner, R.; Quinlan, A.R. GEMINI: Integrative Exploration of Genetic Variation and Genome Annotations. PLoS Comput. Biology 2013, 9, e1003153. [Google Scholar] [CrossRef]
- Collin, M.; Dickinson, R.; Bigley, V. Haematopoietic and immune defects associated with GATA2 mutation. Br. J. Haematol. 2015, 169, 173–187. [Google Scholar] [CrossRef]
- Andolfo, I.; Alper, S.L.; De Franceschi, L.; Auriemma, C.; Russo, R.; De Falco, L.; Vallefuoco, F.; Esposito, M.R.; Vandorpe, D.H.; Shmukler, B.E.; et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood 2015, 121, 3925–3936. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H. Dehydrated hereditary stomatocytosis: Clinical perspectives. J. Blood Med. 2019, 10, 183–191. [Google Scholar] [CrossRef]
- Andolfo, I.; Russo, R.; Gambale, A.; Iolascon, A. Hereditary stomatocytosis: An underdiagnosed condition. Am. J. Hematol. 2018, 93, 107–121. [Google Scholar] [CrossRef]
- Orvain, C.; Da Costa, L.; Van Wijk, R.; Pissard, S.; Picard, V.; Mansour-Hendili, L.; Cunat, S.; Giansily-Blaizot, M.; Cartron, G.; Schved, J.-F.; et al. Inherited or acquired modifiers of iron status may dramatically affect the phenotype in dehydrated hereditary stomatocytosis. Eur. J. Haematol. 2018, 101, 566–569. [Google Scholar] [CrossRef]
- Becker, P.S.; Tse, W.T.; Lux, S.E.; Forget, B.G. Beta spectrin kissimmee: A spectrin variant associated with autosomal dominant hereditary spherocytosis and defective binding to protein 4.1. J. Clin. Investig. 1993, 92, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, H.; Vassiliadis, J.N.; Murray, J.; Njolstad, P.R.; Rogus, J.J.; Ballas, S.K.; Schaffer, F.; Jarolim, P.; Brabec, V.; Palek, J. Characterization of the underlying molecular defect in hereditary spherocytosis associated with spectrin deficiency. Blood 1997, 90, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Bassères, D.S.; Vicentim, D.L.; Costa, F.F.; Saad, S.T.O.; Hassoun, H. β-Spectrin Promissão: A Translation Initiation Codon Mutation of the β-Spectrin Gene (ATG → GTG) Associated With Hereditary Spherocytosis and Spectrin Deficiency in a Brazilian Family. Blood 1998, 91, 368–369. [Google Scholar] [CrossRef]
- Karinch, A.M.; Zimmer, W.E.; Goodman, S.R. The identification and sequence of the actin-binding domain of human red blood cell β-spectrin. J. Biol. Chem. 1990, 265, 11833–11840. [Google Scholar] [CrossRef]
- Djinovic-Carugo, K.; Gautel, M.; Ylänne, J.; Young, P. The spectrin repeat: A structural platform for cytoskeletal protein assemblies. FEBS Lett. 2002, 513, 119–123. [Google Scholar] [CrossRef]
- Salomao, M.; Zhang, X.; Yang, Y.; Lee, S.; Hartwig, J.H.; Chasis, J.A.; Mohandas, N.; An, X. Protein 4.1R-dependent multiprotein complex: New insights into the structural organization of the red blood cell membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 8026–8031. [Google Scholar] [CrossRef]
- Djinovic-Carugo, K.; Bañuelos, S.; Saraste, M. Crystal structure of a calponin homology domain. Nat. Struct. Biol. 1997, 4, 175–179. [Google Scholar] [CrossRef]
- Machnicka, B.; Czogalla, A.; Hryniewicz-Jankowska, A.; Bogusławska, D.M.; Grochowalska, R.; Heger, E.; Sikorski, A.F. Spectrins: A structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 620–634. [Google Scholar] [CrossRef]
- An, X.; Debnath, G.; Guo, X.; Liu, S.; Lux, S.E.; Baines, A.; Gratzer, W.; Mohandas, N. Identification and functional characterization of protein 4.1R and actin-binding sites in erythrocyte β spectrin: Regulation of the interactions by phosphatidylinositol-4,5-bisphosphate. Biochemistry 2005, 44, 10681–10688. [Google Scholar] [CrossRef]
- Galkin, V.E.; Orlova, A.; Salmazo, A.; Djinovic-Carugo, K.; Egelman, E.H. Opening of tandem calponin homology domains regulates their affinity for F-actin. Nat. Struct. Mol. Biol. 2010, 17, 614–616. [Google Scholar] [CrossRef]


{kind=link}
| Study J Family Members | RBC (T/L) | HCT (L/L) | Hb (mmol/L) | Total Bilirubin (µmol/L) | Direct Bilirubin (µmol/L) | Indirect Bilirubin (µmol/L) | Ret (Fraction) | EMA Test (IF) * |
|---|---|---|---|---|---|---|---|---|
| References for female | 3.7–5.1 | 0.37–0.47 | 7.45–9.93 | 5.13–20.53 | <5.13 | 1.71–17.10 | 0.005–0.015 | 98.86–117.20 |
| J41 | 3.70 ± 0.10 | 0.35 ± 0.10 | 7.63 ± 0.19 | 11.55 ± 1.28 | 4.70 ± 0.43 | 6.84 ± 0.86 | 0.010 ± 0.001 | 90.68 ± 0.49 |
| J42 | 2.95 ± 0.15 | 0.27 ± 0.01 | 5.93 ± 0.16 | 45.50 ± 2.22 | 16.93 ± 0.68 | 28.56 ± 1.54 | 0.075 ± 0.03 | 89.51 ± 1.29 |
| Translation Impact of Variants | Total Genes | 71 Genes Involved in Known RBC Pathologies | ||
|---|---|---|---|---|
| Total | Not Reported | Total | Not Reported | |
| Missense mutation | 8657 | 45 | 58 | 2 |
| Frameshift | 248 | 7 | 2 | 0 |
| In-frame | 177 | 0 | 2 | 0 |
| Start lost | 14 | 0 | 0 | 0 |
| Stop lost | 29 | 0 | 0 | 0 |
| Stop gained | 91 | 2 | 0 | 0 |
| Total variants | 9216 | 284 | 398 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogusławska, D.M.; Skulski, M.; Machnicka, B.; Potoczek, S.; Kraszewski, S.; Kuliczkowski, K.; Sikorski, A.F. Identification of a Novel Mutation of β-Spectrin in Hereditary Spherocytosis Using Whole Exome Sequencing. Int. J. Mol. Sci. 2021, 22, 11007. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011007
Bogusławska DM, Skulski M, Machnicka B, Potoczek S, Kraszewski S, Kuliczkowski K, Sikorski AF. Identification of a Novel Mutation of β-Spectrin in Hereditary Spherocytosis Using Whole Exome Sequencing. International Journal of Molecular Sciences. 2021; 22(20):11007. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011007
Chicago/Turabian StyleBogusławska, Dżamila M., Michał Skulski, Beata Machnicka, Stanisław Potoczek, Sebastian Kraszewski, Kazimierz Kuliczkowski, and Aleksander F. Sikorski. 2021. "Identification of a Novel Mutation of β-Spectrin in Hereditary Spherocytosis Using Whole Exome Sequencing" International Journal of Molecular Sciences 22, no. 20: 11007. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011007