
PRC1 Stabilizes Cardiac Contraction by Regulating Cardiac Sarcomere Assembly and Cardiac Conduction System Construction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Loss of Rnf2 Caused Embryonic Lethality with Severe Cardiac Defects
2.2. The Cardiac Contraction Was Disrupted in the Rnf2-Null Embryos
2.3. The Mesoderm Formed Normally in the Rnf2-Null Zebrafish Embryos
2.4. The Heart Tube Structure Appeared Normal in the rnf2−/− Embryos
2.5. Rnf2 Deficiency Disorganized the Sarcomere Assembly in Zebrafish Hearts
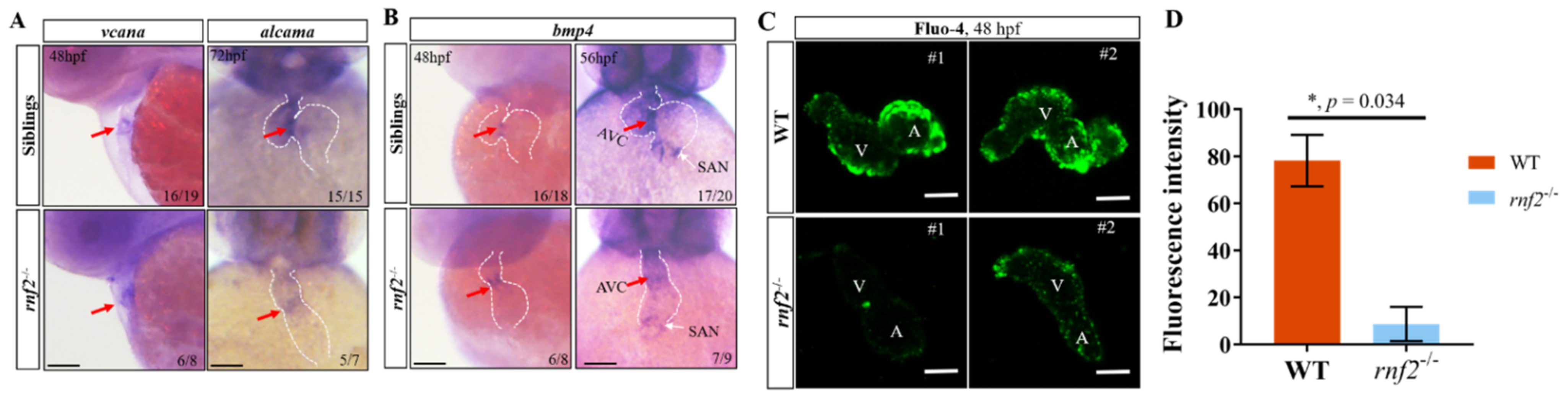
2.6. Rnf2 Deficiency Caused Defects in the Cardiac Conduction System
3. Discussion
- PRC1 maintains regular cardiac sarcomere assembly by orchestrating the sarcomere gene transcription program.
- 2.
- PRC1 represses sarcomere genes indirectly.
- 3.
- PRC1 is involved in the construction of the zebrafish cardiac conduction system.
4. Materials and Methods
4.1. Zebrafish Lines and Maintenance
4.2. Generation of Rnf2 Mutants
4.3. Western Blotting Experiments
4.4. Whole Mount In Situ Hybridization (WISH)
4.5. Histological Analysis
4.6. Quantitative Real-Time PCR
4.7. Calcium Signaling Detection Using Fluo-4 AM
4.8. Imaging, Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Bakkers, J.; Verhoeven, M.C.; Abdelilah-Seyfried, S. Shaping the zebrafish heart: From left-right axis specification to epithelial tissue morphogenesis. Dev. Biol. 2009, 330, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Houk, A.R.; Yelon, D. Strategies for analyzing cardiac phenotypes in the zebrafish embryo. In Zebrafish: Cellular and Developmental Biology, Pt B: Developmental Biology; Detrich, H.W., Westerfield, M., Zon, L.I., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 335–368. [Google Scholar]
- Miura, G.I.; Yelon, D. A guide to analysis of cardiac phenotypes in the zebrafish embryo. Methods Cell Biol. 2011, 101, 161–180. [Google Scholar] [PubMed] [Green Version]
- Peterson, R.T.; Mably, J.D.; Chen, J.-N.; Fishman, M.C. Convergence of distinct pathways to heart patterning revealed by the small molecule concentramide and the mutation heart-and-soul. Curr. Biol. 2001, 11, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Yelon, D.; Horne, S.A.; Stainier, D.Y.R. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev. Biol. 1999, 214, 23–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Shih, Y.-H.; Xu, X. Understanding cardiac sarcomere assembly with Zebrafish genetics. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2014, 297, 1681–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorio, C.C.; Antin, P.B. To the heart of myofibril assembly. Trends Cell Biol. 2000, 10, 355–362. [Google Scholar] [CrossRef]
- Squire, J.M. Architecture and function in the muscle sarcomere. Curr. Opin. Struct. Biol. 1997, 7, 247–257. [Google Scholar] [CrossRef]
- Wilczewski, C.M.; Hepperla, A.J.; Shimbo, T.; Wasson, L.; Robbe, Z.; Davis, I.; Wade, P.; Conlon, F.L. CHD4 and the NuRD complex directly control cardiac sarcomere formation. Proc. Natl. Acad. Sci. USA 2018, 115, 6727–6732. [Google Scholar] [CrossRef] [Green Version]
- Van Weerd, J.H.; Christoffels, V.M. The formation and function of the cardiac conduction system. Development 2016, 143, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Peal, D.S.; Lynch, S.N.; Milan, D.J. Patterning and development of the atrioventricular canal in zebrafish. J. Cardiovasc. Transl. Res. 2011, 4, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Walsh, E.C.; Stainier, D.Y.R. UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Science 2001, 293, 1670. [Google Scholar] [CrossRef]
- Chi, N.C.; Shaw, R.; De Val, S.; Kang, G.; Jan, L.; Black, B.; Stainier, D.Y. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 2008, 22, 734–739. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, M.C.; Haase, C.; Christoffels, V.M.; Weidinger, G.; Bakkers, J. Wnt signaling regulates atrioventricular canal formation upstream of BMP and Tbx2. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 435–440. [Google Scholar] [CrossRef]
- Puskaric, S.; Schmitteckert, S.; Mori, A.D.; Glaser, A.; Schneider, K.U.; Bruneau, B.; Blaschke, R.J.; Steinbeisser, H.; Rappold, G. Shox2 mediates Tbx5 activity by regulating Bmp4 in the pacemaker region of the developing heart. Hum. Mol. Genet. 2010, 19, 4625–4633. [Google Scholar] [CrossRef] [Green Version]
- Hashem, S.I.; Lam, M.L.; Mihardja, S.S.; White, S.M.; Lee, R.J.; Claycomb, W.C. Shox2 regulates the pacemaker gene program in embryoid bodies. Stem Cells Dev. 2013, 22, 2915–2926. [Google Scholar] [CrossRef]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef]
- Hamish, W.K.; Nadezda, A.F.; Neil, P.B.; Robert, J.K. PRC1 shapes the nucleosome landscape but not accessibility at target genes. Genome Res. 2018, 2018, 14. [Google Scholar]
- Brockdorff, N. Polycomb complexes in X chromosome inactivation. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20170021. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Pintacuda, G.; Masui, O.; Koseki, Y.; Gdula, M.; Cerase, A.; Brown, D.; Mould, A.; Innocent, C.; Nakayama, M.; et al. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 2017, 356, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Tong, H.; Huang, Y.; Yan, Y.; Teng, H.; Xia, Y.; Jiang, Q.; Qin, J. Essential role for Polycomb group protein Pcgf6 in Embryonic stem cell maintenance and a noncanonical Polycomb Repressive Complex 1 (PRC1) integrity. J. Biol. Chem. 2017, 292, 2773–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupret, B.; Völkel, P.; Le Bourhis, X.; Angrand, P.-O. The Polycomb group protein Pcgf1 is dispensable in Zebrafish but involved in early growth and aging. PLoS ONE 2016, 11, e0158700. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.L.; Beckedorff, F.; Zhang, Y.; Garcia-Huidobro, J.; Jiang, H.; Colaprico, A.; Bilbao, D.; Figueroa, M.E.; LaCava, J.; Shiekhattar, R.; et al. Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun. 2018, 9, 3377. [Google Scholar] [CrossRef] [Green Version]
- Banito, A.; Li, X.; Laporte, A.N.; Roe, J.-S.; Sanchez-Vega, F.; Huang, C.-H.; Dancsok, A.R.; Hatzi, K.; Chen, C.-C.; Tschaharganeh, D.F.; et al. The SS18-SSX oncoprotein hijacks KDM2B-PRC1.1 to drive synovial sarcoma. Cancer Cell 2018, 33, 527–541.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuettengruber, B.; Bourbon, H.-M.; Di Croce, L.; Cavalli, G. Genome regulation by Polycomb and trithorax: 70 years and counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Napoles, M.; Mermoud, J.E.; Wakao, R.; Tang, Y.A.; Endoh, M.; Appanah, R.; Brockdorff, N.; Silva, J.; Otte, A.P.; Vidal, M.; et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 2004, 7, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Tavares, L.; Dimitrova, E.; Oxley, D.; Webster, J.; Poot, R.; Demmers, J.; Bezstarosti, K.; Taylor, S.; Ura, H.; Koide, H.; et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 2012, 148, 664–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356.e8. [Google Scholar] [CrossRef] [Green Version]
- Van der Velden, Y.U.; Wang, L.; van Lohuizen, M.; Haramis, A.-P.G. The Polycomb group protein Ring1b is essential for pectoral fin development. Development 2012, 139, 2210–2220. [Google Scholar] [CrossRef] [Green Version]
- Van der Velden, Y.U.; Wang, L.; Cano, L.Q.; Haramis, A.-P.G. The Polycomb group protein Ring1b/Rnf2 is specifically required for craniofacial development. PLoS ONE 2013, 8, e73997. [Google Scholar] [CrossRef] [Green Version]
- Chrispijn, N.D.; Elurbe, D.M.; Mickoleit, M.; Aben, M.; De Bakker, D.E.; Andralojc, K.M.; Huisken, J.; Bakkers, J.; Kamminga, L.M. Loss of the Polycomb group protein Rnf2 results in derepression of tbx-transcription factors and defects in embryonic and cardiac development. Sci. Rep. 2019, 9, 4327. [Google Scholar] [CrossRef] [Green Version]
- Bakkers, J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Keegan, B.R.; Meyer, D.; Yelon, D. Organization of cardiac chamber progenitors in the zebrafish blastula. Development 2004, 131, 3081–3091. [Google Scholar] [CrossRef] [Green Version]
- Stainier, D.Y. Zebrafish genetics and vertebrate heart formation. Nat. Rev. Genet. 2001, 2, 39. [Google Scholar] [CrossRef] [PubMed]
- Puceat, M. Embryological origin of the endocardium and derived valve progenitor cells: From developmental biology to stem cell-based valve repair. Biochim. Et Biophys. Acta Mol. Cell Res. 2013, 1833, 917–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, A.; Ma, Q.; Cao, J.; Von Gise, A.; Zhou, P.; Xie, H.; Zhang, B.; Hsing, M.; Christodoulou, D.C.; Cahan, P.; et al. Polycomb Repressive Complex 2 regulates normal development of the mouse heart. Circ. Res. 2012, 110, 406–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San, B.; Chrispijn, N.; Wittkopp, N.; van Heeringen, S.; Lagendijk, A.K.; Aben, M.; Bakkers, J.; Ketting, R.; Kamminga, L.M. Normal formation of a vertebrate body plan and loss of tissue maintenance in the absence of ezh2. Sci. Rep. 2016, 6, 24658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirai, M.; Takihara, Y.; Morisaki, T. Pcgf5 contributes to PRC1 (Polycomb Repressive Complex 1) in developing cardiac cells. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Ed.; Springer: Tokyo, Japan, 2016; pp. 305–312. [Google Scholar]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef]
- Piunti, A.; Shilatifard, A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat. Rev. Mol. Cell Biol. 2021, 22, 326–345. [Google Scholar] [CrossRef]
- Osugi, T.; Shirai, M.; Koga, H.; Nishiguchi, S.; Yamauchi-Takihara, K.; Takihara, Y. Rae28, a member of the mammalian Polycomb group of genes, is required for maintenance of NKX2.5 expression in cardiac development. Circulation 2000, 102, 99. [Google Scholar]
- Voncken, J.W.; Roelen, B.A.; Roefs, M.; de Vries, S.; Verhoeven, E.; Marino, S.; Deschamps, J.; van Lohuizen, M. Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc. Natl. Acad. Sci. USA 2003, 100, 2468–2473. [Google Scholar] [CrossRef] [Green Version]
- Buchwald, G.; Van Der Stoop, P.; Weichenrieder, O.; Perrakis, A.; Van Lohuizen, M.; Sixma, T.K. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. 2006, 25, 2465–2474. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Moffat, M.; Mann, A.R.; Read, D.; Hunter, C.J.; Pradeepa, M.M.; Adams, I.R.; Bickmore, W.A. The E3 ubiquitin ligase activity of RING1B is not essential for early mouse development. Genes Dev. 2015, 29, 1897–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskeland, R.; Leeb, M.; Grimes, G.R.; Kress, C.; Boyle, S.; Sproul, D.; Gilbert, N.; Fan, Y.; Skoultchi, A.I.; Wutz, A.; et al. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell 2010, 38, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, S.; Flyamer, I.M.; Williamson, I.; Sengupta, D.; Bickmore, W.A.; Illingworth, R.S. A central role for canonical PRC1 in shaping the 3D nuclear landscape. Genes Dev. 2020, 34, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.A.; McElhinny, A.S.; Beckerle, M.C.; Gregorio, C.C. Striated muscle cytoarchitecture: An intricate web of form and function. Annu. Rev. Cell Dev. Biol. 2002, 18, 637–706. [Google Scholar] [CrossRef] [Green Version]
- Pierrat, O.A.; Paudyal, A.; Woodruff, J.; Koroleva, O.; Boateng, S.Y. The exon junction complex senses energetic stress and regulates contractility and cell architecture in cardiac myocytes. Biosci. Rep. 2017, 37, BSR20170707. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Olguín, P.; Huang, Y.; Li, X.; Christodoulou, D.; Seidman, C.E.; Seidman, J.G.; Tarakhovsky, A.; Bruneau, B.G. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat. Genet. 2012, 44, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Min, J.R.; Zhang, Y.; Xu, R.M. Structural basis for specific binding of polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003, 17, 1823–1828. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugishita, H.; Kondo, T.; Ito, S.; Nakayama, M.; Yakushiji-Kaminatsui, N.; Kawakami, E.; Koseki, Y.; Ohinata, Y.; Sharif, J.; Harachi, M.; et al. Variant PCGF1-PRC1 links PRC2 recruitment with differentiation-associated transcriptional inactivation at target genes. Nat. Commun. 2021, 12, 5341. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.M.; Berul, C.I. Inherited conduction system abnormalities—One group of diseases, many genes. J. Cardiovasc. Electrophysiol. 2006, 17, 446–455. [Google Scholar] [CrossRef]
- Jensen, B.; Boukens, B.J.; Postma, A.; Gunst, Q.D.; Hoff, M.J.B.V.D.; Moorman, A.F.M.; Wang, T.; Christoffels, V.M. Identifying the evolutionary building blocks of the cardiac conduction system. PLoS ONE 2012, 7, e44231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, M.L.; Boukens, B.J.; Mommersteeg, M.T.; Brons, J.F.; Wakker, V.; Moorman, A.F.; Christoffels, V.M. Transcription factor Tbx3 is required for the specification of the atrioventricular conduction system. Circ. Res. 2008, 102, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Chi, N.C.; Shaw, R.; Jungblut, B.; Huisken, J.; Ferrer, T.; Arnaout, R.; Scott, I.; Beis, D.; Xiao, T.; Baier, H.; et al. Genetic and physiologic dissection of the vertebrate cardiac conduction system. PLoS Biol. 2008, 6, e109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio); University of Oregon Press: Eugene, OR, USA, 1995. [Google Scholar]
- Sun, X.; Chen, J.; Zhang, Y.; Munisha, M.; Dougan, S.; Sun, Y. Mga modulates bmpr1a activity by antagonizing Bs69 in Zebrafish. Front. Cell Dev. Biol. 2018, 6, 126. [Google Scholar] [CrossRef]
- Thisse, C.; Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008, 3, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Jou, C.J.; Spitzer, K.W.; Tristani-Firouzi, M. Blebbistatin effectively uncouples the excitation-contraction process in Zebrafish embryonic heart. Cell. Physiol. Biochem. 2010, 25, 419–424. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, X.; Feng, G.; Zhang, Y.; Sun, Y. PRC1 Stabilizes Cardiac Contraction by Regulating Cardiac Sarcomere Assembly and Cardiac Conduction System Construction. Int. J. Mol. Sci. 2021, 22, 11368. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111368
Peng X, Feng G, Zhang Y, Sun Y. PRC1 Stabilizes Cardiac Contraction by Regulating Cardiac Sarcomere Assembly and Cardiac Conduction System Construction. International Journal of Molecular Sciences. 2021; 22(21):11368. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111368
Chicago/Turabian StylePeng, Xixia, Gang Feng, Yanyong Zhang, and Yuhua Sun. 2021. "PRC1 Stabilizes Cardiac Contraction by Regulating Cardiac Sarcomere Assembly and Cardiac Conduction System Construction" International Journal of Molecular Sciences 22, no. 21: 11368. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111368