
Dexmedetomidine Promotes Lipopolysaccharide-Induced Differentiation of Cardiac Fibroblasts and Collagen I/III Synthesis through α2A Adrenoreceptor-Mediated Activation of the PKC-p38-Smad2/3 Signaling Pathway in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. DEX Promoted LPS-Induced Differentiation of CFs to Myofibroblasts In Vitro
2.2. DEX Enhanced LPS-Induced Collagen I/III Synthesis and Phosphorylation of PKC, p38 and Smad2/3 in Mouse CFs
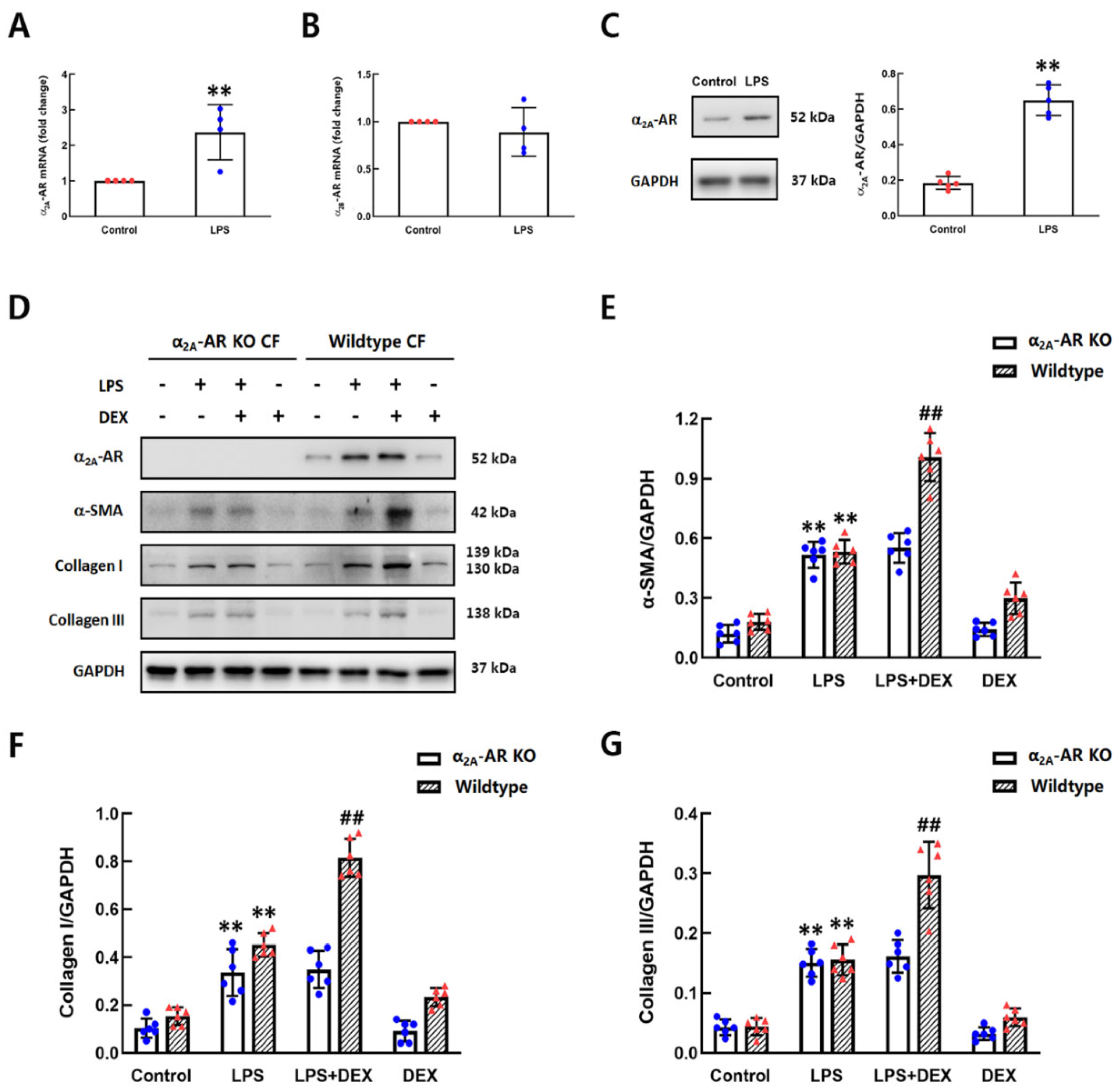
2.3. The Role of DEX in LPS-Induced Differentiation of CFs Was Dependent on α2A-AR
2.4. The Enhancing Effect of DEX on CF Differentiation Was TNF-α and IL-6 Independent
2.5. DEX Accelerated LPS-Induced Differentiation of CFs to Cardiac Myofibroblasts by Activating the PKC-p38-Smad2/3-Mediated Signaling Pathway
2.6. DEX Aggravated LPS-Induced Cardiac Fibrosis in Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Adult Mouse Cardiac Fibroblast Culture and Treatment
4.3. Animals and Treatment Protocols
4.4. Immunofluorescence Staining
4.5. Western Blotting Analysis
4.6. Quantitative Real-Time Polymerase Chain Reaction (qPCR) Analysis
4.7. Assays for Concentrations of TNF-α, IL-6 and IL-10
4.8. Masson’s Trichrome Stain Analysis
4.9. Migration Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Van Den Borne, S.W.M.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2009, 7, 30–37. [Google Scholar] [CrossRef]
- Ni, S.Y.; Zhong, X.L.; Li, Z.H.; Huang, D.J.; Xu, W.T.; Zhou, Y.; Ou, C.W.; Chen, M.S. Puerarin Alleviates Lipopolysaccharide-Induced Myocardial Fibrosis by Inhibiting PARP-1 to Prevent HMGB1-Mediated TLR4-NF-κB Signaling Pathway. Cardiovasc. Toxicol. 2020, 20, 482–491. [Google Scholar] [CrossRef]
- Hu, H.; Fu, Y.; Li, M.; Xia, H.; Liu, Y.; Sun, X.; Hu, Y.; Song, F.; Cheng, X.; Li, P.; et al. Interleukin-35 pretreatment attenuates lipopolysaccharide-induced heart injury by inhibition of inflammation, apoptosis and fibrotic reactions. Int. Immunopharmacol. 2020, 86, 106725. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Takashina, M.; Mizuno, N.; Sakata, K.; Hattori, K.; Imura, J.; Ohashi, W.; Hattori, Y. Cardiac fibroblasts: Contributory role in septic cardiac dysfunction. J. Surg. Res. 2015, 193, 874–887. [Google Scholar] [CrossRef]
- Asgharzadeh, F.; Bargi, R.; Hosseini, M.; Farzadnia, M.; Khazaei, M. Cardiac and renal fibrosis and oxidative stress balance in lipopolysaccharide-induced inflammation in male rats. ARYA Atheroscler. 2018, 14, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Cormack, J.; Orme, R.; Costello, T. The role of α2-agonists in neurosurgery. J. Clin. Neurosci. 2005, 12, 375–378. [Google Scholar] [CrossRef]
- Devlin, J.W.; Skrobik, Y.; Gélinas, C.; Needham, D.M.; Slooter, A.J.C.; Pandharipande, P.; Watson, P.L.; Weinhouse, G.L.; Nunnally, M.E.; Rochwerg, B.; et al. Clinical Practice Guidelines for the Prevention and Management of Pain, Agitation/Sedation, Delirium, Immobility, and Sleep Disruption in Adult Patients in the ICU. Crit. Care Med. 2018, 46, e825–e873. [Google Scholar] [CrossRef] [Green Version]
- Shehabi, Y.; Howe, B.D.; Bellomo, R.; Arabi, Y.M.; Bailey, M.; Bass, F.E.; Kadiman, S.B.; McArthur, C.J.; Murray, L.; Reade, M.C.; et al. Early Sedation with Dexmedetomidine in Critically Ill Patients. N. Engl. J. Med. 2019, 380, 2506–2517. [Google Scholar] [CrossRef] [PubMed]
- Shehabi, Y.; Bellomo, R.; Kadiman, S.; Ti, L.K.; Howe, B.; Reade, M.C.; Khoo, T.M.; Alias, A.; Wong, Y.-L.; Mukhopadhyay, A.; et al. Sedation Intensity in the First 48 hour of Mechanical Ventilation and 180-Day Mortality: A Multinational Prospective Longitudinal Cohort Study. Crit. Care Med. 2018, 46, 850–859. [Google Scholar] [CrossRef]
- Schwinghammer, U.A.; Melkonyan, M.M.; Hunanyan, L.; Tremmel, R.; Weiskirchen, R.; Borkham-Kamphorst, E.; Schaeffeler, E.; Seferyan, T.; Mikulits, W.; Yenkoyan, K.; et al. α2-Adrenergic Receptor in Liver Fibrosis: Implications for the Adrenoblocker Mesedin. Cells 2020, 9, 456. [Google Scholar] [CrossRef] [Green Version]
- Nakamori, H.; Yoshida, S.-I.; Ishiguro, H.; Suzuki, S.; Yasuzaki, H.; Hashimoto, T.; Ishigami, T.; Hirawa, N.; Toya, Y.; Umemura, S.; et al. Arterial wall hypertrophy is ameliorated by α2-adrenergic receptor antagonist or aliskiren in kidneys of angiotensinogen-knockout mice. Clin. Exp. Nephrol. 2017, 22, 773–781. [Google Scholar] [CrossRef]
- Kakkar, R.; Lee, R.T. Intramyocardial Fibroblast Myocyte Communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore-Morris, T.; Guimarães-Camboa, N.; Yutzey, K.; Puceat, M.; Evans, S.M. Cardiac fibroblasts: From development to heart failure. J. Mol. Med. 2015, 93, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, K.T. Cardiac interstitium in health and disease: The fibrillar collagen network. J. Am. Coll. Cardiol. 1989, 13, 1637–1652. [Google Scholar] [CrossRef] [Green Version]
- Medugorac, I.; Jacob, R. Characterisation of left ventricular collagen in the rat. Cardiovasc. Res. 1983, 17, 15–21. [Google Scholar] [CrossRef]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C. The Cardiac Fibroblast: Therapeutic Target in Myocardial Remodeling and Failure. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxidative Med. Cell. Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef] [PubMed]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Fail. Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef]
- Barki-Harrington, L.; Perrino, C.; Rockman, H.A. Network integration of the adrenergic system in cardiac hypertrophy. Cardiovasc. Res. 2004, 63, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Faber, J.E.; Yang, N.; Xin, X. Expression of α-Adrenoceptor Subtypes by Smooth Muscle Cells and Adventitial Fibroblasts in Rat Aorta and in Cell Culture. J. Pharm. Exp. Ther. 2001, 298, 441–452. [Google Scholar]
- Driesen, R.B.; Nagaraju, C.K.; Abi-Char, J.; Coenen, T.; Lijnen, P.J.; Fagard, R.H.; Sipido, K.R.; Petrov, V.V. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc. Res. 2013, 101, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, F.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yoshida, K.; Sugano, Y.; Yamagata, H.; Matsushita, M.; Seki, T.; Inagaki, Y.; et al. p38 MAPK mediates fibrogenic signal through smad3 phosphorylation in rat myofibroblasts. Hepatology 2003, 38, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.Y.; Lee, S.W.; Kim, T.S. Stimulation of interleukin-12 production in mouse macrophages via activation of p38 mitogen-activated protein kinase by α2-adrenoceptor agonists. Eur. J. Pharmacol. 2003, 467, 223–231. [Google Scholar] [CrossRef]
- Humeres, C.; Vivar, R.; Boza, P.; Muñoz, C.; Bolivar, S.; Anfossi, R.; Osorio, J.M.; Olivares-Silva, F.; García, L.; Díaz-Aray, G. Cardiac fibroblast cytokine profiles induced by proinflammatory or profibrotic stimuli promote monocyte recruitment and modulate macrophage M1/M2 balance in vitro. J. Mol. Cell. Cardiol. 2016, 101, 69–80. [Google Scholar] [CrossRef]
- Christia, P.; Bujak, M.; Gonzalez-Quesada, C.; Chen, W.; Dobaczewski, M.; Reddy, A.; Frangogiannis, N.G. Systematic Characterization of Myocardial Inflammation, Repair, and Remodeling in a Mouse Model of Reperfused Myocardial Infarction. J. Histochem. Cytochem. 2013, 61, 555–570. [Google Scholar] [CrossRef]
- Huang, L.; Zhu, J.; Zheng, M.; Zou, R.; Zhou, Y.; Zhu, M. Tanshinone IIA protects against subclinical lipopolysaccharide induced cardiac fibrosis in mice through inhibition of NADPH oxidase. Int. Immunopharmacol. 2018, 60, 59–63. [Google Scholar] [CrossRef]
- Lew, W.Y.W.; Bayna, E.; Molle, E.D.; Dalton, N.D.; Lai, N.C.; Bhargava, V.; Mendiola, V.L.; Clopton, P.; Tang, T. Recurrent Exposure to Subclinical Lipopolysaccharide Increases Mortality and Induces Cardiac Fibrosis in Mice. PLoS ONE 2013, 8, e61057. [Google Scholar] [CrossRef] [Green Version]
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of severe sepsis. Virulence 2013, 5, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyle, N.H.; Pena, O.M.; Boyd, J.H.; Hancock, R. Barriers to the effective treatment of sepsis: Antimicrobial agents, sepsis definitions, and host-directed therapies. Ann. N. Y. Acad. Sci. 2014, 1323, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Soong, J.; Soni, N. Sepsis: Recognition and treatment. Clin. Med. 2012, 12, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Li, H.M.; Wang, H.D.; Peng, X.M.; Wang, Y.P.; Lu, D.X.; Qi, R.B.; Hu, C.F.; Jiang, J.W. Pretreatment with berberine and yohimbine protects against LPS-induced myocardial dysfunction via inhibition of cardiac I-[kappa]B[alpha] phosphorylation and apoptosis in mice. Shock 2011, 35, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Y.; Yang, D.; Tang, X.; Li, H.; Lv, X.; Qi, R.; Hu, C.; Lu, D.; Lv, B.; et al. α2A-adrenergic blockade attenuates septic cardiomyopathy by increasing cardiac norepinephrine concentration and inhibiting cardiac endothelial activation. Sci. Rep. 2018, 8, 5478. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Iyer, R.P.; Jung, M.; Czubryt, M.; Lindsey, M.L. Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps. Trends Pharmacol. Sci. 2017, 38, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Van Putten, S.; Shafieyan, Y.; Hinz, B. Mechanical control of cardiac myofibroblasts. J. Mol. Cell. Cardiol. 2016, 93, 133–142. [Google Scholar] [CrossRef]
- Miragoli, M.; Salvarani, N.; Rohr, S. Myofibroblasts Induce Ectopic Activity in Cardiac Tissue. Circ. Res. 2007, 101, 755–758. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2012, 10, 15–26. [Google Scholar] [CrossRef]
- Ouzounian, M.; Lee, D.S.; Liu, P.P. Diastolic heart failure: Mechanisms and controversies. Nat. Clin. Pract. Neurol. 2008, 5, 375–386. [Google Scholar] [CrossRef]
- Gyires, K.; Zadori, Z.S.; Torok, T.; Mátyus, P. Alpha(2)-Adrenoceptor subtypes-mediated physiological, pharmacological actions. Neurochem. Int. 2009, 55, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.M.; Vetrivelan, R.; Mallick, H.N. Noradrenergic afferents and receptors in the medial preoptic area: Neuroanatomical and neurochemical links between the regulation of sleep and body temperature. Neurochem. Int. 2007, 50, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Panaro, M.; Gagliardi, N.; Saponaro, C.; Calvello, R.; Mitolo, V.; Cianciulli, A. Toll-like Receptor 4 Mediates LPS-Induced Release of Nitric Oxide and Tumor Necrosis Factor-α by Embryonal Cardiomyocytes: Biological Significance and Clinical Implications in Human Pathology. Curr. Pharm. Des. 2010, 16, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Sekiguchi, K.; Tanaka, T.; Tomaru, K.; Arai, M.; Suzuki, T.; Nagai, R. Angiotensin II and mechanical stretch induce production of tumor necrosis factor in cardiac fibroblasts. Am. J. Physiol. Content 1999, 276, H1968–H1976. [Google Scholar] [CrossRef]
- Sato, H.; Watanabe, A.; Tanaka, T.; Koitabashi, N.; Arai, M.; Kurabayashi, M.; Yokoyama, T. Regulation of the human tumor necrosis factor-alpha promoter by angiotensin II and lipopolysaccharide in cardiac fibroblasts: Different cis-acting promoter sequences and transcriptional factors. J. Mol. Cell. Cardiol. 2003, 35, 1197–1205. [Google Scholar] [CrossRef]
- Zhang, F.; Wu, R.; Qiang, X.; Zhou, M.; Wang, P. Antagonism of α2A-adrenoceptor: A novel approach to inhibit inflammatory responses in sepsis. J. Mol. Med. 2009, 88, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-J.; Lin, Z.-H.; Lin, Y.-Z.; Rao, Z.-H.; Lin, J.-F.; Wu, L.-P.; Li, L. Dexmedetomidine Exerted Anti-arrhythmic Effects in Rat With Ischemic Cardiomyopathy via Upregulation of Connexin 43 and Reduction of Fibrosis and Inflammation. Front. Physiol. 2020, 11, 33. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Yang, D.; Yu, X.; Li, H.; Lv, X.; Lu, D.; Wang, H. β1-adrenoceptor stimulation promotes LPS-induced cardiomyocyte apoptosis through activating PKA and enhancing CaMKII and IκBα phosphorylation. Crit. Care 2015, 19, 76. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, J.; Li, K.; Su, X.; Chen, Y.; Wang, Y.; Tang, X.; Xing, Y.; Xu, Y.; Dai, X.; Teng, J.; et al. Dexmedetomidine Promotes Lipopolysaccharide-Induced Differentiation of Cardiac Fibroblasts and Collagen I/III Synthesis through α2A Adrenoreceptor-Mediated Activation of the PKC-p38-Smad2/3 Signaling Pathway in Mice. Int. J. Mol. Sci. 2021, 22, 12749. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312749
Liao J, Li K, Su X, Chen Y, Wang Y, Tang X, Xing Y, Xu Y, Dai X, Teng J, et al. Dexmedetomidine Promotes Lipopolysaccharide-Induced Differentiation of Cardiac Fibroblasts and Collagen I/III Synthesis through α2A Adrenoreceptor-Mediated Activation of the PKC-p38-Smad2/3 Signaling Pathway in Mice. International Journal of Molecular Sciences. 2021; 22(23):12749. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312749
Chicago/Turabian StyleLiao, Jia, Kaiying Li, Xingyu Su, Yihua Chen, Yingwei Wang, Xiangxu Tang, Yun Xing, Yaqian Xu, Xiaomeng Dai, Jiashuo Teng, and et al. 2021. "Dexmedetomidine Promotes Lipopolysaccharide-Induced Differentiation of Cardiac Fibroblasts and Collagen I/III Synthesis through α2A Adrenoreceptor-Mediated Activation of the PKC-p38-Smad2/3 Signaling Pathway in Mice" International Journal of Molecular Sciences 22, no. 23: 12749. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312749