
RIF1 Links Replication Timing with Fork Reactivation and DNA Double-Strand Break Repair


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
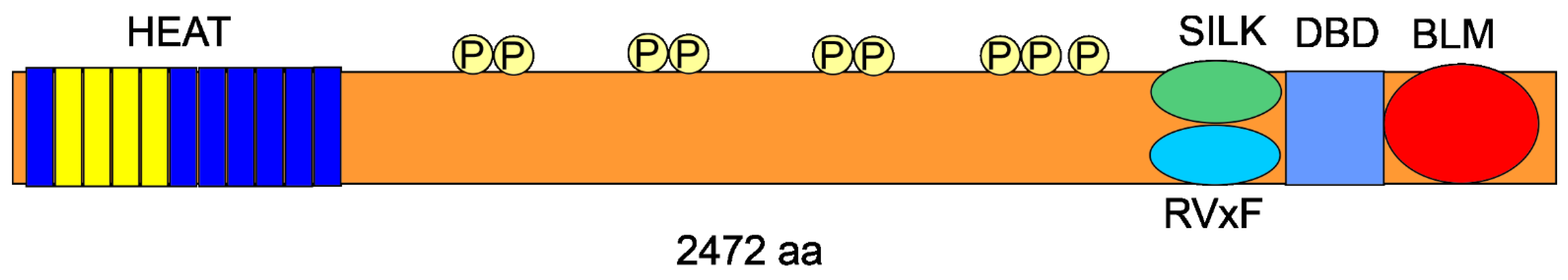
2. RIF1—The Gene and the Protein
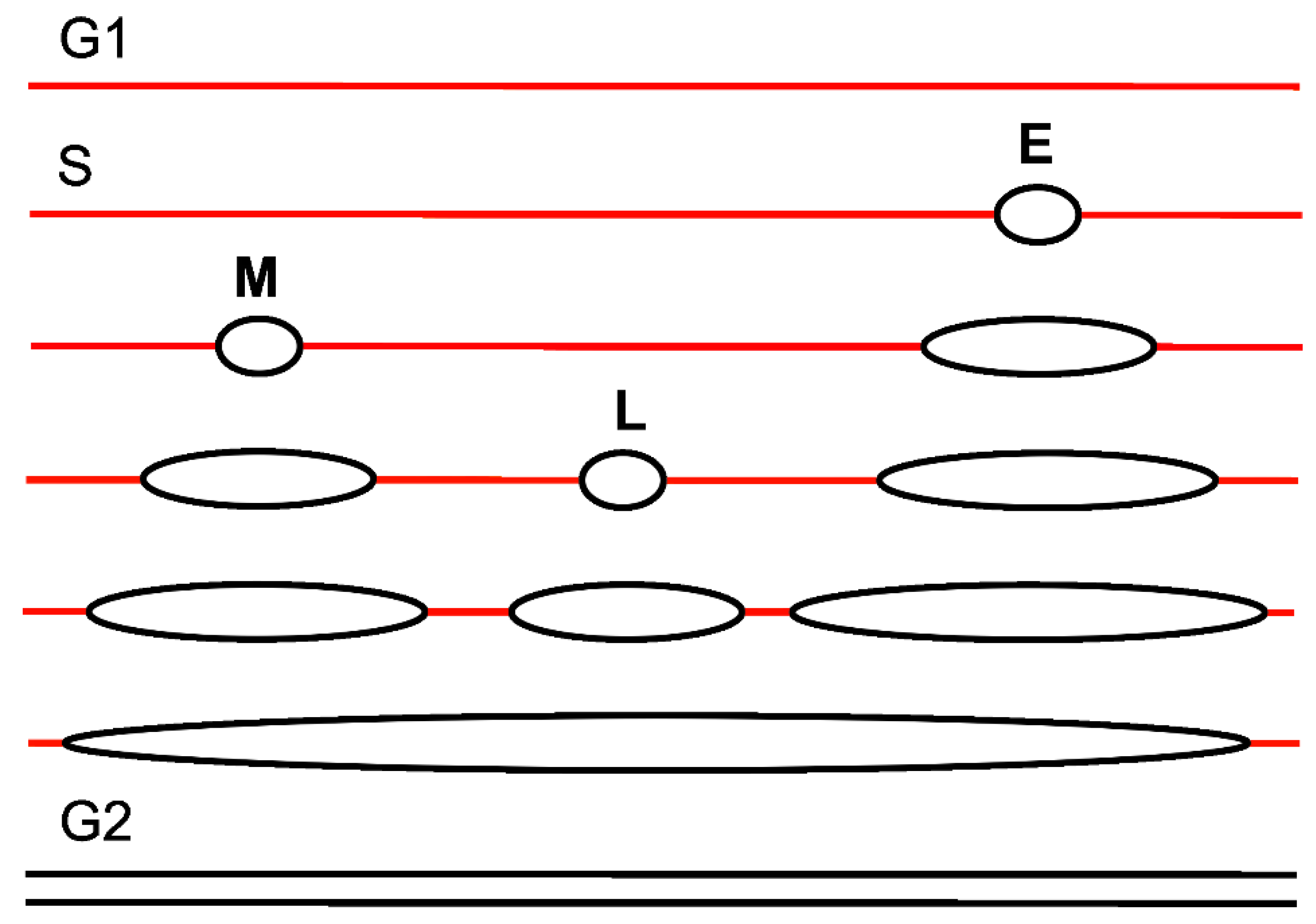
3. Replication Timing
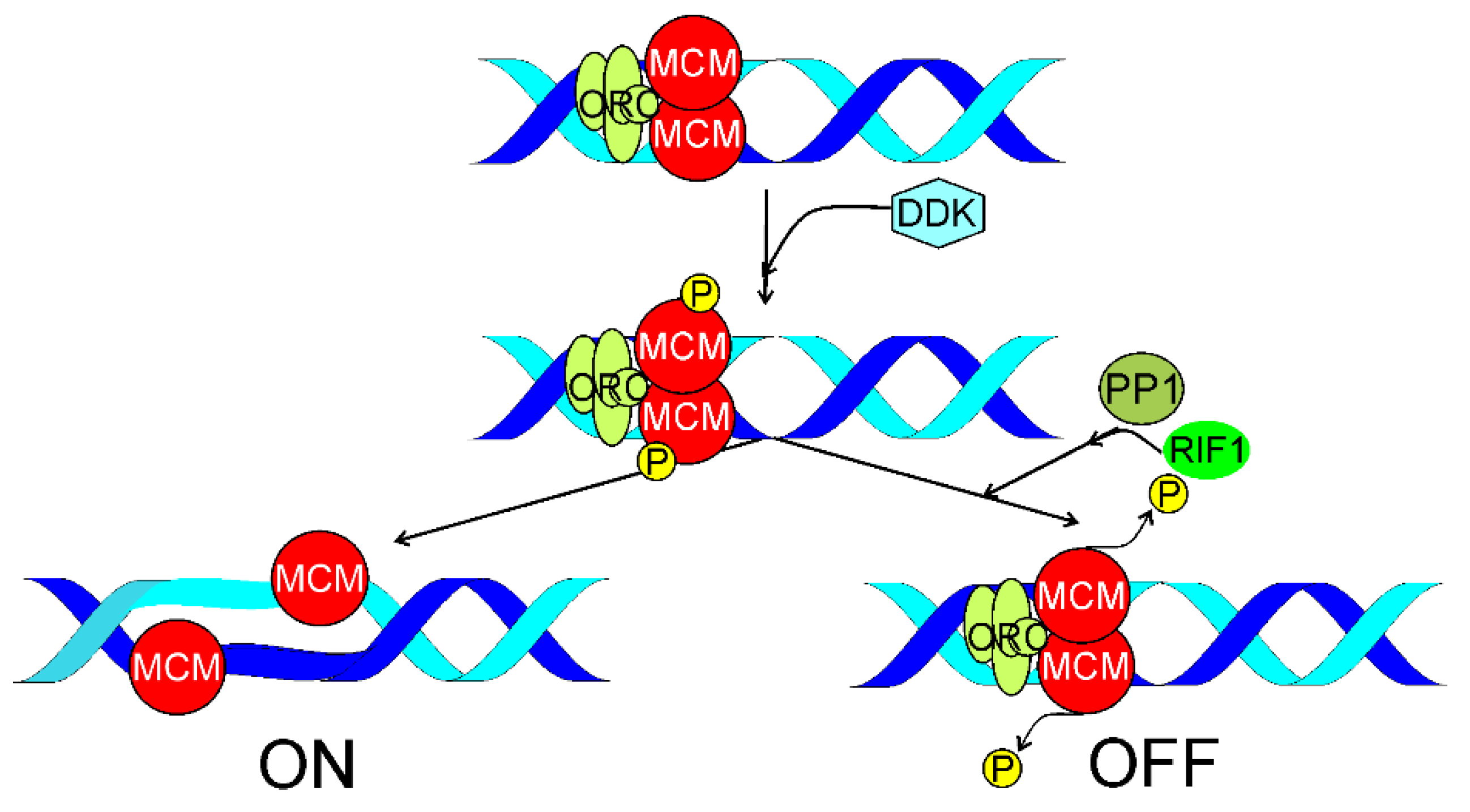
4. RIF1 in Replication Timing
4.1. Organizing the 3D Structure of Chromatin
4.2. Interaction with G4-Quadruplexes
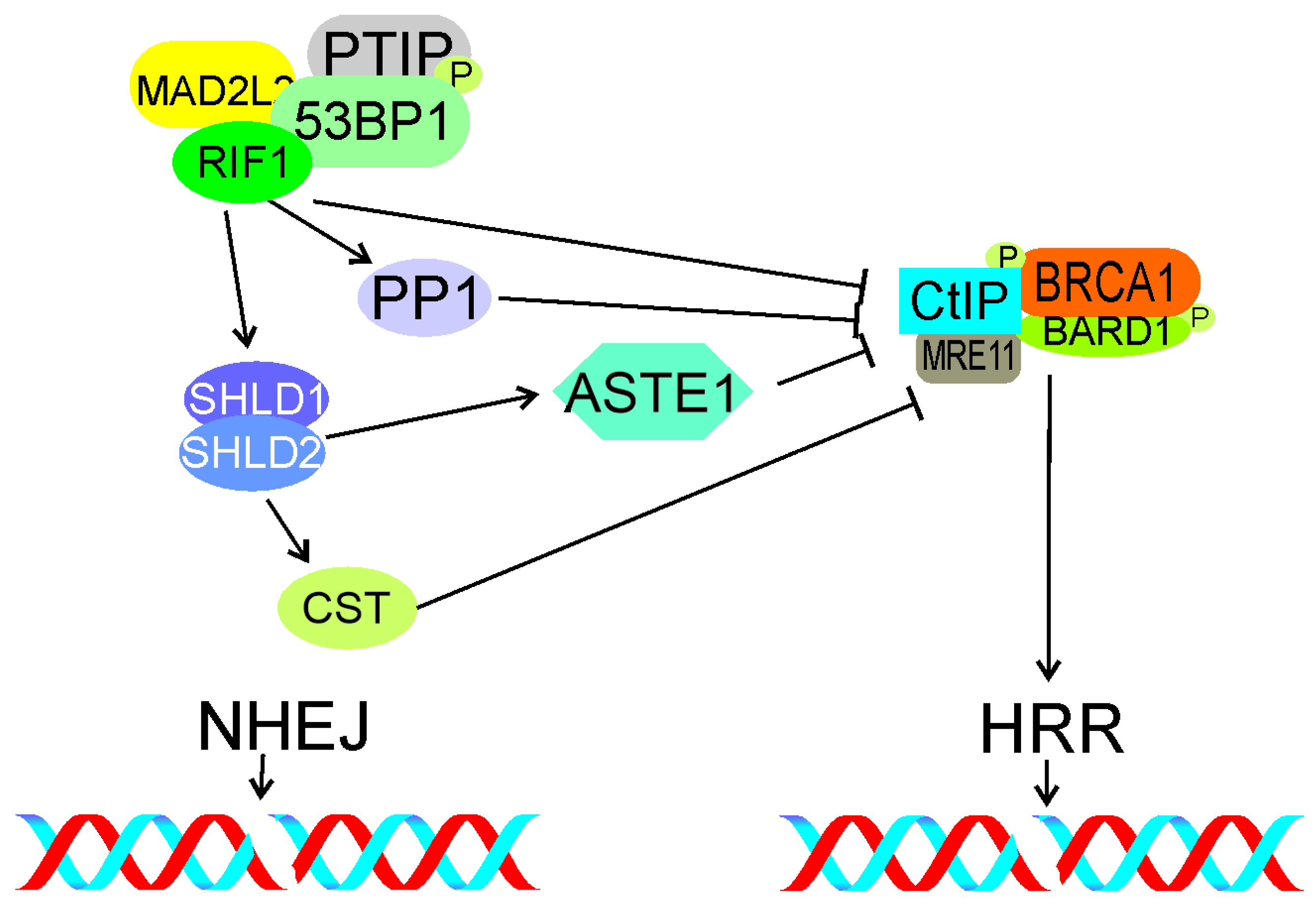
5. RIF1 in Double-Strand Break Processing
5.1. Essential Role of TP53BP1
5.2. DNA End Resection
5.3. Interaction with Shieldin
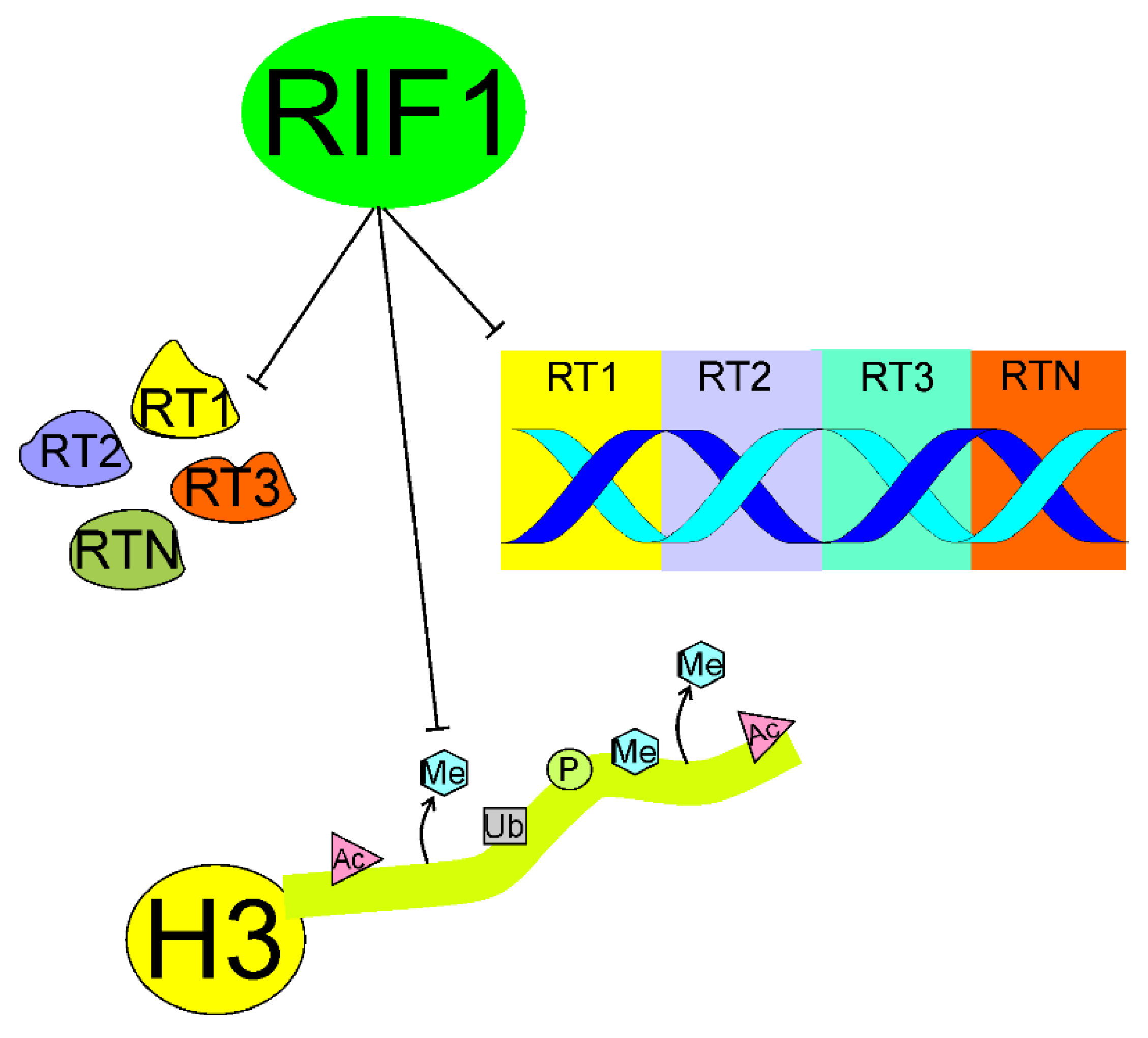
5.4. Epigenetics
5.5. Local 3D Chromatin Changes
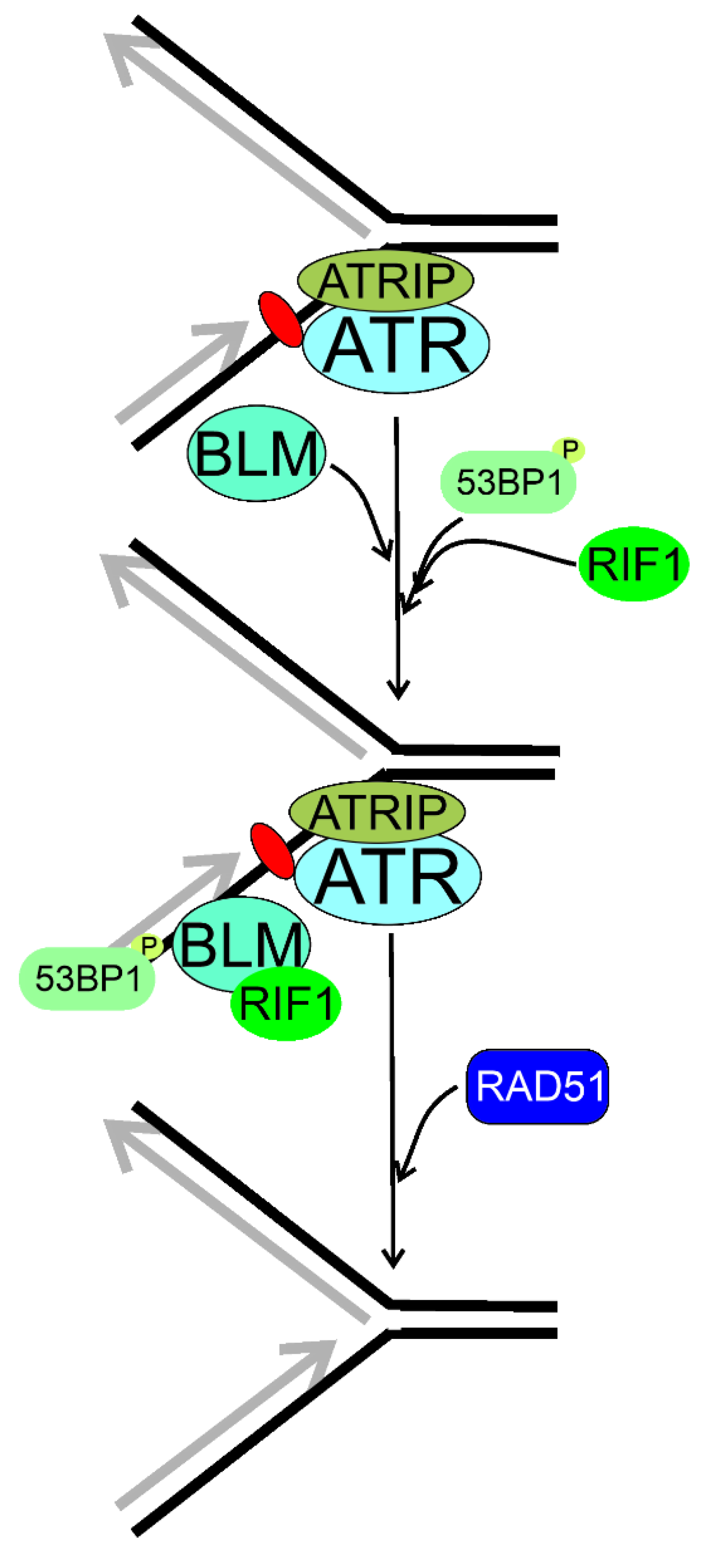
6. RIF1 in Reactivation of Impaired Replication Fork
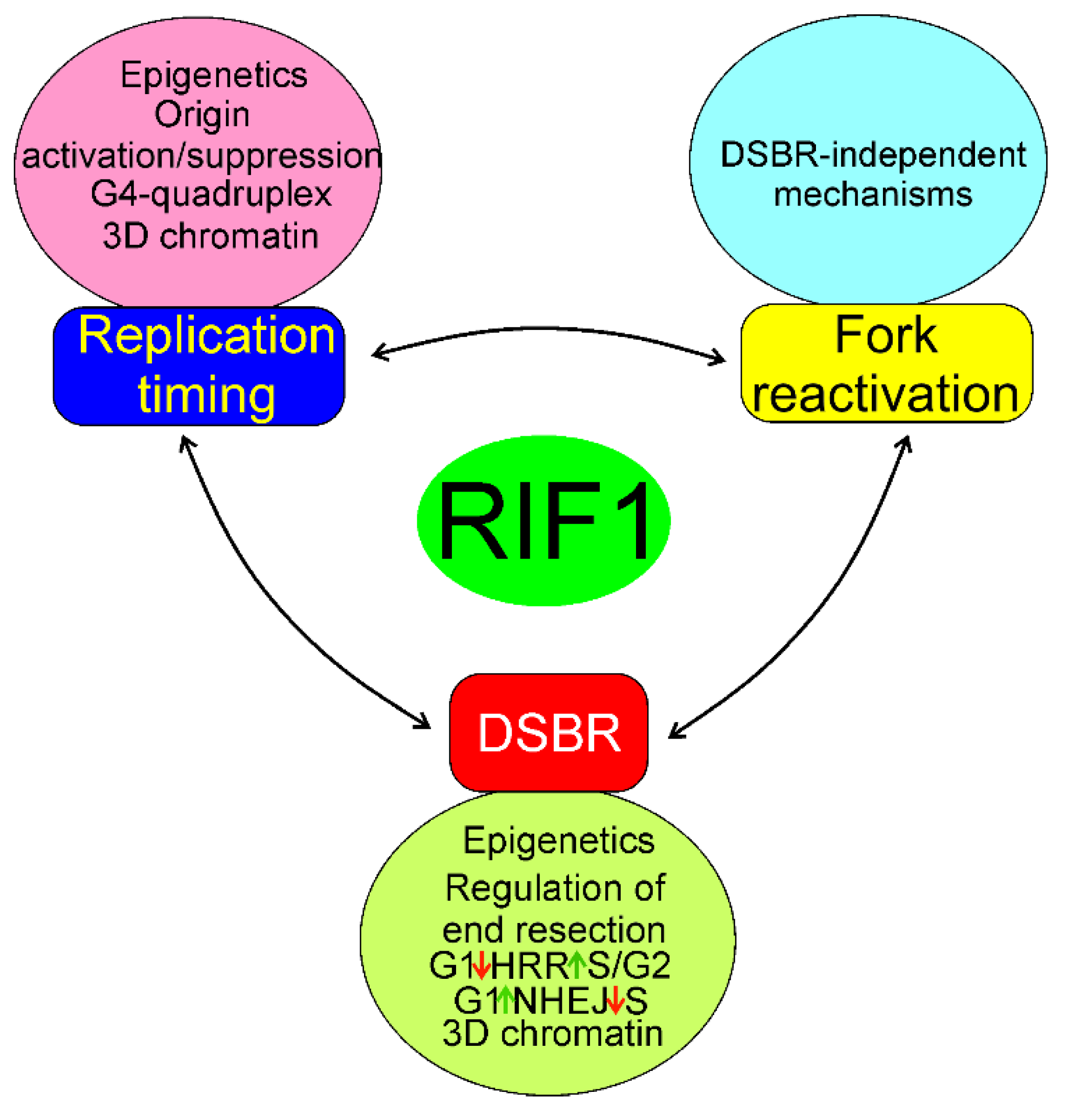
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dimitrova, D.S.; Gilbert, D.M. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol. Cell 1999, 4, 983–993. [Google Scholar] [CrossRef]
- Vouzas, A.E.; Gilbert, D.M. Mammalian DNA replication timing. Cold Spring Harb. Perspect. Biol. 2021, 13, a040162. [Google Scholar] [CrossRef]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA double-strand break repair: An ever-growing toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Fontana, G.A.; Rass, U. Compartmentalized DNA repair: Rif1 S-acylation links DNA double-strand break repair to the nuclear membrane. Mol. Cell. Oncol. 2019, 6, e1648025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnan, S.; Flyamer, I.M.; Klein, K.N.; Castelli, E.; Rapp, A.; Maiser, A.; Chen, N.; Weber, P.; Enervald, E.; Cardoso, M.C.; et al. Nuclear organisation and replication timing are coupled through RIF1-PP1 interaction. Nat. Commun. 2021, 12, 2910. [Google Scholar] [CrossRef] [PubMed]
- Nathanailidou, P.; Taraviras, S.; Lygerou, Z. Chromatin and nuclear architecture: Shaping DNA replication in 3D. Trends Genet. 2020, 36, 967–980. [Google Scholar] [CrossRef]
- Smogorzewska, A.; de Lange, T. Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem. 2004, 73, 177–208. [Google Scholar] [CrossRef] [Green Version]
- Toteva, T.; Mason, B.; Kanoh, Y.; Brøgger, P.; Green, D.; Verhein-Hansen, J.; Masai, H.; Thon, G. Establishment of expression-state boundaries by Rif1 and Taz1 in fission yeast. Proc. Natl. Acad. Sci. USA 2017, 114, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Wang, L.; Ge, F.; Gong, P.; Wang, H.; Wang, F.; Chen, L.; Liu, L. Pold3 is required for genomic stability and telomere integrity in embryonic stem cells and meiosis. Nucleic Acids Res. 2018, 46, 3468–3486. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Sung, P. RIF1 in DNA break repair pathway choice. Mol. Cell 2013, 49, 840–841. [Google Scholar] [CrossRef] [Green Version]
- Hayano, M.; Kanoh, Y.; Matsumoto, S.; Renard-Guillet, C.; Shirahige, K.; Masai, H. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 2012, 26, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Kanoh, Y.; Matsumoto, S.; Fukatsu, R.; Kakusho, N.; Kono, N.; Renard-Guillet, C.; Masuda, K.; Iida, K.; Nagasawa, K.; Shirahige, K.; et al. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015, 22, 889–897. [Google Scholar] [CrossRef]
- Kumar, R.; Cheok, C.F. RIF1: A novel regulatory factor for DNA replication and DNA damage response signaling. DNA Repair 2014, 15, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Mattarocci, S.; Hafner, L.; Lezaja, A.; Shyian, M.; Shore, D. Rif1: A conserved regulator of DNA replication and repair hijacked by telomeres in yeasts. Front. Genet. 2016, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, C.F.; Sussel, L.; Shore, D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992, 6, 801–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.; Takai, H.; Buonomo, S.B.; Eisenhaber, F.; de Lange, T. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 2004, 18, 2108–2119. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Blackburn, E.H. Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J. Cell Biol. 2004, 167, 819–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Muniandy, P.; Leo, E.; Yin, J.; Thangavel, S.; Shen, X.; Ii, M.; Agama, K.; Guo, R.; Fox, D., 3rd; et al. Rif1 provides a new DNA-binding interface for the Bloom syndrome complex to maintain normal replication. EMBO J. 2010, 29, 3140–3155. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Fukatsu, R.; Kanoh, Y.; Kakusho, N.; Matsumoto, S.; Chaen, S.; Masai, H. Both a unique motif at the C terminus and an N-terminal heat repeat contribute to G-quadruplex binding and origin regulation by the Rif1 protein. Mol. Cell. Biol. 2019, 39, e00364-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, K.; Lai, M.S.; Masai, H. Interaction of Rif1 protein with G-quadruplex in control of chromosome transactions. Adv. Exp. Med. Biol. 2017, 1042, 287–310. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, H.; Al Shibar, M.; Willard, B.; Ray, A.; Runge, K.W. Rif1 phosphorylation site analysis in telomere length regulation and the response to damaged telomeres. DNA Repair 2018, 65, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Sukackaite, R.; Jensen, M.R.; Mas, P.J.; Blackledge, M.; Buonomo, S.B.; Hart, D.J. Structural and biophysical characterization of murine rif1 C terminus reveals high specificity for DNA cruciform structures. J. Biol. Chem. 2014, 289, 13903–13911. [Google Scholar] [CrossRef] [Green Version]
- Sreesankar, E.; Senthilkumar, R.; Bharathi, V.; Mishra, R.K.; Mishra, K. Functional diversification of yeast telomere associated protein, Rif1, in higher eukaryotes. BMC Genom. 2012, 13, 255. [Google Scholar] [CrossRef] [Green Version]
- Masai, H.; Fukatsu, R.; Kakusho, N.; Kanoh, Y.; Moriyama, K.; Ma, Y.; Iida, K.; Nagasawa, K. Rif1 promotes association of G-quadruplex (G4) by its specific G4 binding and oligomerization activities. Sci. Rep. 2019, 9, 8618. [Google Scholar] [CrossRef] [Green Version]
- Masai, H.; Kanoh, Y.; Moriyama, K.; Yamazaki, S.; Yoshizawa, N.; Matsumoto, S. Telomere-binding factors in the regulation of DNA replication. Genes Genet. Syst. 2018, 92, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Alavi, S.; Ghadiri, H.; Dabirmanesh, B.; Moriyama, K.; Khajeh, K.; Masai, H. G-quadruplex binding protein Rif1, a key regulator of replication timing. J. Biochem. 2021, 169, 1–14. [Google Scholar] [CrossRef]
- Garzón, J.; Ursich, S.; Lopes, M.; Hiraga, S.I.; Donaldson, A.D. Human RIF1-protein phosphatase 1 prevents degradation and breakage of nascent DNA on replication stalling. Cell Rep. 2019, 27, 2558–2566.e2554. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; de Boer, H.R.; Demmers, J.; van Vugt, M.; Ray Chaudhuri, A. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef]
- Li, P.; Wang, L.; Bennett, B.D.; Wang, J.; Li, J.; Qin, Y.; Takaku, M.; Wade, P.A.; Wong, J.; Hu, G. Rif1 promotes a repressive chromatin state to safeguard against endogenous retrovirus activation. Nucleic Acids Res. 2017, 45, 12723–12738. [Google Scholar] [CrossRef]
- Buonomo, S.B.; Wu, Y.; Ferguson, D.; de Lange, T. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J. Cell Biol. 2009, 187, 385–398. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhao, A.; Chen, L.; Zhong, X.; Liao, J.; Gao, M.; Cai, M.; Lee, D.H.; Li, J.; Chowdhury, D.; et al. Human RIF1 encodes an anti-apoptotic factor required for DNA repair. Carcinogenesis 2009, 30, 1314–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.B.; Mei, Y.; Long, J.; Zhang, Y.; Hu, D.L.; Zhou, H.H. RIF1 promotes human epithelial ovarian cancer growth and progression via activating human telomerase reverse transcriptase expression. J. Exp. Clin. Cancer Res. 2018, 37, 182. [Google Scholar] [CrossRef]
- Mei, Y.; Liu, Y.B.; Cao, S.; Tian, Z.W.; Zhou, H.H. RIF1 promotes tumor growth and cancer stem cell-like traits in NSCLC by protein phosphatase 1-mediated activation of Wnt/β-catenin signaling. Cell Death Dis. 2018, 9, 942. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Liu, Y.B.; Cao, S.; Tian, Z.W.; Zhou, H.H. Correction: RIF1 promotes tumor growth and cancer stem cell-like traits in NSCLC by protein phosphatase 1-mediated activation of Wnt/β-catenin signaling. Cell Death Dis. 2021, 12, 812. [Google Scholar] [CrossRef]
- Howarth, K.D.; Blood, K.A.; Ng, B.L.; Beavis, J.C.; Chua, Y.; Cooke, S.L.; Raby, S.; Ichimura, K.; Collins, V.P.; Carter, N.P.; et al. Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes. Oncogene 2008, 27, 3345–3359. [Google Scholar] [CrossRef] [Green Version]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Baris, A.; Aladjem, M.I. Replication timing and nuclear structure. Curr. Opin. Cell Biol. 2018, 52, 43–50. [Google Scholar] [CrossRef]
- Marchal, C.; Sima, J.; Gilbert, D.M. Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.P.; Mahbubani, H.M.; Khoo, C.Y.; Blow, J.J. Purification of an MCM-containing complex as a component of the DNA replication licensing system. Nature 1995, 375, 418–421. [Google Scholar] [CrossRef]
- Boos, D.; Ferreira, P. Origin firing regulations to control genome replication timing. Genes 2019, 10, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacNeill, S.A. Structure and function of the GINS complex, a key component of the eukaryotic replisome. Biochem. J. 2010, 425, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [Green Version]
- Friedman, K.L.; Diller, J.D.; Ferguson, B.M.; Nyland, S.V.; Brewer, B.J.; Fangman, W.L. Multiple determinants controlling activation of yeast replication origins late in S phase. Genes Dev. 1996, 10, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Koren, A. Positive and negative regulation of DNA replication initiation. Trends Genet. 2020, 36, 868–879. [Google Scholar] [CrossRef]
- Shoaib, M.; Walter, D.; Gillespie, P.J.; Izard, F.; Fahrenkrog, B.; Lleres, D.; Lerdrup, M.; Johansen, J.V.; Hansen, K.; Julien, E.; et al. Histone H4K20 methylation mediated chromatin compaction threshold ensures genome integrity by limiting DNA replication licensing. Nat. Commun. 2018, 9, 3704. [Google Scholar] [CrossRef] [Green Version]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, D.M. Cell fate transitions and the replication timing decision point. J. Cell Biol. 2010, 191, 899–903. [Google Scholar] [CrossRef] [Green Version]
- Cvetic, C.; Walter, J.C. Eukaryotic origins of DNA replication: Could you please be more specific? Semin. Cell Dev. Biol. 2005, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, M.K.; Winzeler, E.A.; Collingwood, D.; Hunt, S.; Wodicka, L.; Conway, A.; Lockhart, D.J.; Davis, R.W.; Brewer, B.J.; Fangman, W.L. Replication dynamics of the yeast genome. Science 2001, 294, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Farago, M.; Rosenbluh, C.; Tevlin, M.; Fraenkel, S.; Schlesinger, S.; Masika, H.; Gouzman, M.; Teng, G.; Schatz, D.; Rais, Y.; et al. Clonal allelic predetermination of immunoglobulin-κ rearrangement. Nature 2012, 490, 561–565. [Google Scholar] [CrossRef]
- Mostoslavsky, R.; Singh, N.; Tenzen, T.; Goldmit, M.; Gabay, C.; Elizur, S.; Qi, P.; Reubinoff, B.E.; Chess, A.; Cedar, H.; et al. Asynchronous replication and allelic exclusion in the immune system. Nature 2001, 414, 221–225. [Google Scholar] [CrossRef]
- Chess, A. Random and non-random monoallelic expression. Neuropsychopharmacology 2013, 38, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chess, A.; Simon, I.; Cedar, H.; Axel, R. Allelic inactivation regulates olfactory receptor gene expression. Cell 1994, 78, 823–834. [Google Scholar] [CrossRef]
- Blumenfeld, B.; Masika, H.; Farago, M.; Yehuda, Y.; Halaseh, L.; Vardi, O.; Rapoport, R.; Levin-Klein, R.; Cedar, H.; Bergman, Y.; et al. Chromosomal coordination and differential structure of asynchronous replicating regions. Nat. Commun. 2021, 12, 1035. [Google Scholar] [CrossRef]
- Hadjadj, D.; Denecker, T.; Maric, C.; Fauchereau, F.; Baldacci, G.; Cadoret, J.C. Characterization of the replication timing program of 6 human model cell lines. Genom. Data 2016, 9, 113–117. [Google Scholar] [CrossRef]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.W.; Lyou, Y.; Townes, T.M.; Schübeler, D.; Gilbert, D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008, 6, e245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donley, N.; Thayer, M.J. DNA replication timing, genome stability and cancer: Late and/or delayed DNA replication timing is associated with increased genomic instability. Semin. Cancer Biol. 2013, 23, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Petrie, M.V.; He, Y.; Peace, J.M.; Chiolo, I.E.; Aparicio, O.M. Dynamic relocalization of replication origins by Fkh1 requires execution of DDK function and Cdc45 loading at origins. eLife 2019, 8, e45512. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nakato, R.; Katou, Y.; Shirahige, K.; Araki, H. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr. Biol. 2011, 21, 2055–2063. [Google Scholar] [CrossRef] [Green Version]
- Briu, L.M.; Maric, C.; Cadoret, J.C. Replication stress, genomic instability, and replication timing: A complex relationship. Int. J. Mol. Sci. 2021, 22, 4764. [Google Scholar] [CrossRef] [PubMed]
- Alver, R.C.; Chadha, G.S.; Gillespie, P.J.; Blow, J.J. Reversal of DDK-mediated MCM phosphorylation by Rif1-PP1 regulates replication initiation and replisome stability independently of ATR/Chk1. Cell Rep. 2017, 18, 2508–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pope, B.D.; Gilbert, D.M. The replication domain model: Regulating replicon firing in the context of large-scale chromosome architecture. J. Mol. Biol. 2013, 425, 4690–4695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, K.N.; Zhao, P.A.; Lyu, X.; Sasaki, T.; Bartlett, D.A.; Singh, A.M.; Tasan, I.; Zhang, M.; Watts, L.P.; Hiraga, S.I.; et al. Replication timing maintains the global epigenetic state in human cells. Science 2021, 372, 371–378. [Google Scholar] [CrossRef]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 2017, 169, 930–944.e922. [Google Scholar] [CrossRef] [Green Version]
- Oldach, P.; Nieduszynski, C.A. Cohesin-mediated genome architecture does not define DNA replication timing domains. Genes 2019, 10, 196. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Miura, H.; Takahashi, S.; Shibata, T.; Nagao, K.; Obuse, C.; Okumura, K.; Ogata, M.; Hiratani, I.; Takebayashi, S.I. Mapping replication timing domains genome wide in single mammalian cells with single-cell DNA replication sequencing. Nat. Protoc. 2020, 15, 4058–4100. [Google Scholar] [CrossRef]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Hayano, M.; Masai, H. Replication timing regulation of eukaryotic replicons: Rif1 as a global regulator of replication timing. Trends Genet. 2013, 29, 449–460. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Li, F.; Zhang, R.; Li, S.; Liu, H.; Qin, Z.S.; Sun, X. Integrative characterization of G-Quadruplexes in the three-dimensional chromatin structure. Epigenetics 2019, 14, 894–911. [Google Scholar] [CrossRef]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic instability in cancer: Teetering on the limit of tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef] [Green Version]
- Keszthelyi, A.; Minchell, N.E.; Baxter, J. The causes and consequences of topological Stress during DNA replication. Genes 2016, 7, 134. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Fujita, M. DNA damage responses that enhance resilience to replication stress. Cell. Mol. Life Sci. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ui, A.; Chiba, N.; Yasui, A. Relationship among DNA double-strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer Sci. 2020, 111, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Huang, J. DNA double-strand break repair pathway choice: The fork in the road. Genome Instab. Dis. 2020, 1, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J. Single-strand annealing in cancer. Int. J. Mol. Sci. 2021, 22, 2167. [Google Scholar] [CrossRef]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. 2017, 803–805, 51–55. [Google Scholar] [CrossRef]
- Bernal, A.; Tusell, L. Telomeres: Implications for cancer development. Int. J. Mol. Sci. 2018, 19, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smogorzewska, A.; Karlseder, J.; Holtgreve-Grez, H.; Jauch, A.; de Lange, T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr. Biol. 2002, 12, 1635–1644. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.D.; Niedernhofer, L.; Kuster, B.; Mann, M.; Hoeijmakers, J.H.; de Lange, T. ERCC1/XPF removes the 3′ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol. Cell 2003, 12, 1489–1498. [Google Scholar] [CrossRef]
- Shubin, C.B.; Greider, C.W. The role of Rif1 in telomere length regulation is separable from its role in origin firing. eLife 2020, 9, e58066. [Google Scholar] [CrossRef]
- Shubin, C.B.; Mayangsari, R.; Swett, A.D.; Greider, C.W. Rif1 regulates telomere length through conserved HEAT repeats. Nucleic Acids Res. 2021, 49, 3967–3980. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, G.A.; Reinert, J.K.; Thomä, N.H.; Rass, U. Shepherding DNA ends: Rif1 protects telomeres and chromosome breaks. Microb. Cell 2018, 5, 327–343. [Google Scholar] [CrossRef]
- Adams, I.R.; McLaren, A. Identification and characterisation of mRif1: A mouse telomere-associated protein highly expressed in germ cells and embryo-derived pluripotent stem cells. Dev. Dyn. 2004, 229, 733–744. [Google Scholar] [CrossRef]
- Li, X.; Wang, M.; Zheng, W.; Huang, W.; Wang, Z.; Jin, K.; Liu, L.; Yu, Z. Dynamics of TRF1 organizing a single human telomere. Nucleic Acids Res. 2021, 49, 760–775. [Google Scholar] [CrossRef] [PubMed]
- McKerlie, M.; Zhu, X.D. Cyclin B-dependent kinase 1 regulates human TRF1 to modulate the resolution of sister telomeres. Nat. Commun. 2011, 2, 371. [Google Scholar] [CrossRef]
- McKerlie, M.; Walker, J.R.; Mitchell, T.R.; Wilson, F.R.; Zhu, X.D. Phosphorylated (pT371)TRF1 is recruited to sites of DNA damage to facilitate homologous recombination and checkpoint activation. Nucleic Acids Res. 2013, 41, 10268–10282. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Drané, P.; Brault, M.E.; Cui, G.; Meghani, K.; Chaubey, S.; Detappe, A.; Parnandi, N.; He, Y.; Zheng, X.F.; Botuyan, M.V.; et al. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 2017, 543, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef]
- Adkins, N.L.; Niu, H.; Sung, P.; Peterson, C.L. Nucleosome dynamics regulates DNA processing. Nat. Struct. Mol. Biol. 2013, 20, 836–842. [Google Scholar] [CrossRef] [Green Version]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Díaz, C.; Orthwein, A.; Leung, C.C.; Huang, H.; Landry, M.C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Bai, Y.; Zhao, M.; Zhou, M.; Shen, Q.; Yun, C.H.; Zhang, H.; Zhu, W.G.; Wang, J. Acetylation of 53BP1 dictates the DNA double strand break repair pathway. Nucleic Acids Res. 2018, 46, 689–703. [Google Scholar] [CrossRef] [Green Version]
- Grabarz, A.; Guirouilh-Barbat, J.; Barascu, A.; Pennarun, G.; Genet, D.; Rass, E.; Germann, S.M.; Bertrand, P.; Hickson, I.D.; Lopez, B.S. A role for BLM in double-strand break repair pathway choice: Prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 2013, 5, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakr, A.; Köcher, S.; Volquardsen, J.; Petersen, C.; Borgmann, K.; Dikomey, E.; Rothkamm, K.; Mansour, W.Y. Impaired 53BP1/RIF1 DSB mediated end-protection stimulates CtIP-dependent end resection and switches the repair to PARP1-dependent end joining in G1. Oncotarget 2016, 7, 57679–57693. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.M.; Chan, D.L.H.; Tio, M.; Patil, S.M.; Traina, T.A.; Robson, M.E.; Khasraw, M. PARP (Poly ADP-Ribose Polymerase) inhibitors for locally advanced or metastatic breast cancer. Cochrane Database Syst. Rev. 2021, 4, CD011395. [Google Scholar] [CrossRef]
- Setiaputra, D.; Escribano-Diaz, C.; Reinert, J.K.; Sadana, P.; Zong, D.; Callen, E.; Seebacher, J.; Nussenzweig, A.; Thomä, N.H.; Durocher, D. RIF1 acts in DNA repair through phosphopeptide recognition of 53BP1. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Aylon, Y.; Liefshitz, B.; Kupiec, M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004, 23, 4868–4875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiano, J.; Zolnerowich, N.; Wu, W.; Pavani, R.; Wang, C.; Li, H.; Zheng, L.; Shen, B.; Sleckman, B.P.; Chen, B.R.; et al. Role of 53BP1 in end protection and DNA synthesis at DNA breaks. Genes Dev. 2021, 35, 1356–1367. [Google Scholar] [CrossRef]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.Y.; Obuse, C.; Nishi, R.; et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.R.; Barral, P.; Vannier, J.B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 2021, 81, 2868. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wan, L.; Jiang, G.; Cao, L.; Xia, F.; Tian, T.; Zhu, X.; Wu, M.; Huen, M.S.Y.; Wang, Y.; et al. ATM controls the extent of DNA end resection by eliciting sequential posttranslational modifications of CtIP. Proc. Natl. Acad. Sci. USA 2021, 118, e2022600118. [Google Scholar] [CrossRef]
- Li, L.; Poon, H.Y.; Hildebrandt, M.R.; Monckton, E.A.; Germain, D.R.; Fahlman, R.P.; Godbout, R. Role for RIF1-interacting partner DDX1 in BLM recruitment to DNA double-strand breaks. DNA Repair 2017, 55, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callen, E.; Di Virgilio, M.; Kruhlak, M.J.; Nieto-Soler, M.; Wong, N.; Chen, H.T.; Faryabi, R.B.; Polato, F.; Santos, M.; Starnes, L.M.; et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 2013, 153, 1266–1280. [Google Scholar] [CrossRef] [Green Version]
- Escribano-Diaz, C.; Durocher, D. DNA repair pathway choice—A PTIP of the hat to 53BP1. EMBO Rep. 2013, 14, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Alternative non-homologous end-joining: Error-prone DNA repair as cancer’s Achilles’ heel. Cancers 2021, 13, 1392. [Google Scholar] [CrossRef]
- Han, J.; Ruan, C.; Huen, M.S.Y.; Wang, J.; Xie, A.; Fu, C.; Liu, T.; Huang, J. BRCA2 antagonizes classical and alternative nonhomologous end-joining to prevent gross genomic instability. Nat. Commun. 2017, 8, 1470. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Aroumougame, A.; Lobrich, M.; Li, Y.; Chen, D.; Chen, J.; Gong, Z. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014, 28, 2693–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isobe, S.Y.; Nagao, K.; Nozaki, N.; Kimura, H.; Obuse, C. Inhibition of RIF1 by SCAI allows BRCA1-mediated repair. Cell Rep. 2017, 20, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Feng, S.; Ning, S.; Liu, J.; Zhao, H.; Xu, Y.; Shang, J.; Li, K.; Li, Q.; Guo, R.; et al. An OB-fold complex controls the repair pathways for DNA double-strand breaks. Nat. Commun. 2018, 9, 3925. [Google Scholar] [CrossRef]
- Findlay, S.; Heath, J.; Luo, V.M.; Malina, A.; Morin, T.; Coulombe, Y.; Djerir, B.; Li, Z.; Samiei, A.; Simo-Cheyou, E.; et al. SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 2018, 37, e100158. [Google Scholar] [CrossRef]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J.L. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 2015, 521, 537–540. [Google Scholar] [CrossRef] [Green Version]
- Isobe, S.Y.; Hiraga, S.I.; Nagao, K.; Sasanuma, H.; Donaldson, A.D.; Obuse, C. Protein phosphatase 1 acts as a RIF1 effector to suppress DSB resection prior to Shieldin action. Cell Rep. 2021, 36, 109383. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Kim, W.; Gao, H.; Liu, C.; Zhang, Y.; Chen, Y.; Deng, M.; Zhou, Q.; Huang, J.; Hu, Q.; et al. ASTE1 promotes shieldin-complex-mediated DNA repair by attenuating end resection. Nat. Cell Biol. 2021, 23, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef]
- Luijsterburg, M.S.; Typas, D.; Caron, M.C.; Wiegant, W.W.; van den Heuvel, D.; Boonen, R.A.; Couturier, A.M.; Mullenders, L.H.; Masson, J.Y.; van Attikum, H. A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. eLife 2017, 6, e20922. [Google Scholar] [CrossRef] [Green Version]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of homologous recombination and the checkpoint by ATR. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef]
- Panier, S.; Durocher, D. Push back to respond better: Regulatory inhibition of the DNA double-strand break response. Nat. Rev. Mol. Cell Biol. 2013, 14, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, M.; de Krijger, I.; Serrat, J.; Moatti, N.; Fortunato, D.; Hoekman, L.; Bleijerveld, O.B.; Altelaar, A.F.M.; Jacobs, J.J.L. H4K20me2 distinguishes pre-replicative from post-replicative chromatin to appropriately direct DNA repair pathway choice by 53BP1-RIF1-MAD2L2. Cell Cycle 2018, 17, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Cheok, C.F. Dynamics of RIF1 SUMOylation is regulated by PIAS4 in the maintenance of Genomic Stability. Sci. Rep. 2017, 7, 17367. [Google Scholar] [CrossRef] [Green Version]
- Batenburg, N.L.; Walker, J.R.; Noordermeer, S.M.; Moatti, N.; Durocher, D.; Zhu, X.D. ATM and CDK2 control chromatin remodeler CSB to inhibit RIF1 in DSB repair pathway choice. Nat. Commun. 2017, 8, 1921. [Google Scholar] [CrossRef] [PubMed]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.B.; Lukas, J.; Lukas, C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Spies, J.; Lukas, C.; Somyajit, K.; Rask, M.B.; Lukas, J.; Neelsen, K.J. 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 2019, 21, 487–497. [Google Scholar] [CrossRef]
- Kraus, F.; Miron, E.; Demmerle, J.; Chitiashvili, T.; Budco, A.; Alle, Q.; Matsuda, A.; Leonhardt, H.; Schermelleh, L.; Markaki, Y. Quantitative 3D structured illumination microscopy of nuclear structures. Nat. Protoc. 2017, 12, 1011–1028. [Google Scholar] [CrossRef]
- Wegel, E.; Göhler, A.; Lagerholm, B.C.; Wainman, A.; Uphoff, S.; Kaufmann, R.; Dobbie, I.M. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Sci. Rep. 2016, 6, 27290. [Google Scholar] [CrossRef] [Green Version]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef] [PubMed]
- Ochs, F.; Karemore, G.; Miron, E.; Brown, J.; Sedlackova, H.; Rask, M.B.; Lampe, M.; Buckle, V.; Schermelleh, L.; Lukas, J.; et al. Stabilization of chromatin topology safeguards genome integrity. Nature 2019, 574, 571–574. [Google Scholar] [CrossRef]
- Kondratick, C.M.; Washington, M.T.; Spies, M. Making choices: DNA replication fork recovery mechanisms. Semin. Cell Dev. Biol. 2021, 113, 27–37. [Google Scholar] [CrossRef]
- Scully, R.; Elango, R.; Panday, A.; Willis, N.A. Recombination and restart at blocked replication forks. Curr. Opin. Genet. Dev. 2021, 71, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Mayle, R.; Campbell, I.M.; Beck, C.R.; Yu, Y.; Wilson, M.; Shaw, C.A.; Bjergbaek, L.; Lupski, J.R.; Ira, G. DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 2015, 349, 742–747. [Google Scholar] [CrossRef] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Li, Q.; Dudás, K.; Tick, G.; Haracska, L. Coordinated cut and bypass: Replication of interstrand crosslink-containing DNA. Front. Cell Dev. Biol. 2021, 9, 699966. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.; Saadat, N.; Myung, B.; Shekhar, M.P. Crosstalk between translesion synthesis, Fanconi anemia network, and homologous recombination repair pathways in interstrand DNA crosslink repair and development of chemoresistance. Mutat. Res. Rev. Mutat. Res. 2015, 763, 258–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralf, C.; Hickson, I.D.; Wu, L. The Bloom’s syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 2006, 281, 22839–22846. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ning, S.; Wei, Z.; Xu, R.; Xu, X.; Xing, M.; Guo, R.; Xu, D. 53BP1 and BRCA1 control pathway choice for stalled replication restart. eLife 2017, 6, e30523. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Keeney, S. Regulating the formation of DNA double-strand breaks in meiosis. Genes Dev. 2008, 22, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Hu, Y.F.; Zhong, H.; Nye, A.C.; Belmont, A.S.; Li, R. BRCA1-induced large-scale chromatin unfolding and allele-specific effects of cancer-predisposing mutations. J. Cell Biol. 2001, 155, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Mattoo, A.R.; Pandita, R.K.; Chakraborty, S.; Charaka, V.; Mujoo, K.; Hunt, C.R.; Pandita, T.K. MCL-1 Depletion impairs DNA double-strand break repair and reinitiation of stalled DNA replication forks. Mol. Cell. Biol. 2017, 37, e00535-16. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Kobayashi, J.; Kanemaki, M.T.; Komatsu, K. RIF1 controls replication initiation and homologous recombination repair in a radiation dose-dependent manner. J. Cell Sci. 2020, 133, jcs240036. [Google Scholar] [CrossRef]
- Wallis, A.B.A.; Nieduszynski, C.A. Investigating the role of Rts1 in DNA replication initiation. Wellcome Open Res. 2018, 3, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Li, X.; Zhao, Y.; Yang, Q.; Kong, B. 53BP1 suppresses tumor growth and promotes susceptibility to apoptosis of ovarian cancer cells through modulation of the Akt pathway. Oncol. Rep. 2012, 27, 1251–1257. [Google Scholar] [CrossRef] [Green Version]
- Ward, I.M.; Difilippantonio, S.; Minn, K.; Mueller, M.D.; Molina, J.R.; Yu, X.; Frisk, C.S.; Ried, T.; Nussenzweig, A.; Chen, J. 53BP1 cooperates with p53 and functions as a haploinsufficient tumor suppressor in mice. Mol. Cell. Biol. 2005, 25, 10079–10086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, R.A. How TP53 (almost) became an oncogene. J. Mol. Cell Biol. 2019, 11, 531–533. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blasiak, J.; Szczepańska, J.; Sobczuk, A.; Fila, M.; Pawlowska, E. RIF1 Links Replication Timing with Fork Reactivation and DNA Double-Strand Break Repair. Int. J. Mol. Sci. 2021, 22, 11440. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111440
Blasiak J, Szczepańska J, Sobczuk A, Fila M, Pawlowska E. RIF1 Links Replication Timing with Fork Reactivation and DNA Double-Strand Break Repair. International Journal of Molecular Sciences. 2021; 22(21):11440. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111440
Chicago/Turabian StyleBlasiak, Janusz, Joanna Szczepańska, Anna Sobczuk, Michal Fila, and Elzbieta Pawlowska. 2021. "RIF1 Links Replication Timing with Fork Reactivation and DNA Double-Strand Break Repair" International Journal of Molecular Sciences 22, no. 21: 11440. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111440