
MMP9 Differentially Regulates Proteins Involved in Actin Polymerization and Cell Migration during TGF-β-Induced EMT in the Lens


Abstract
:1. Introduction
2. Results
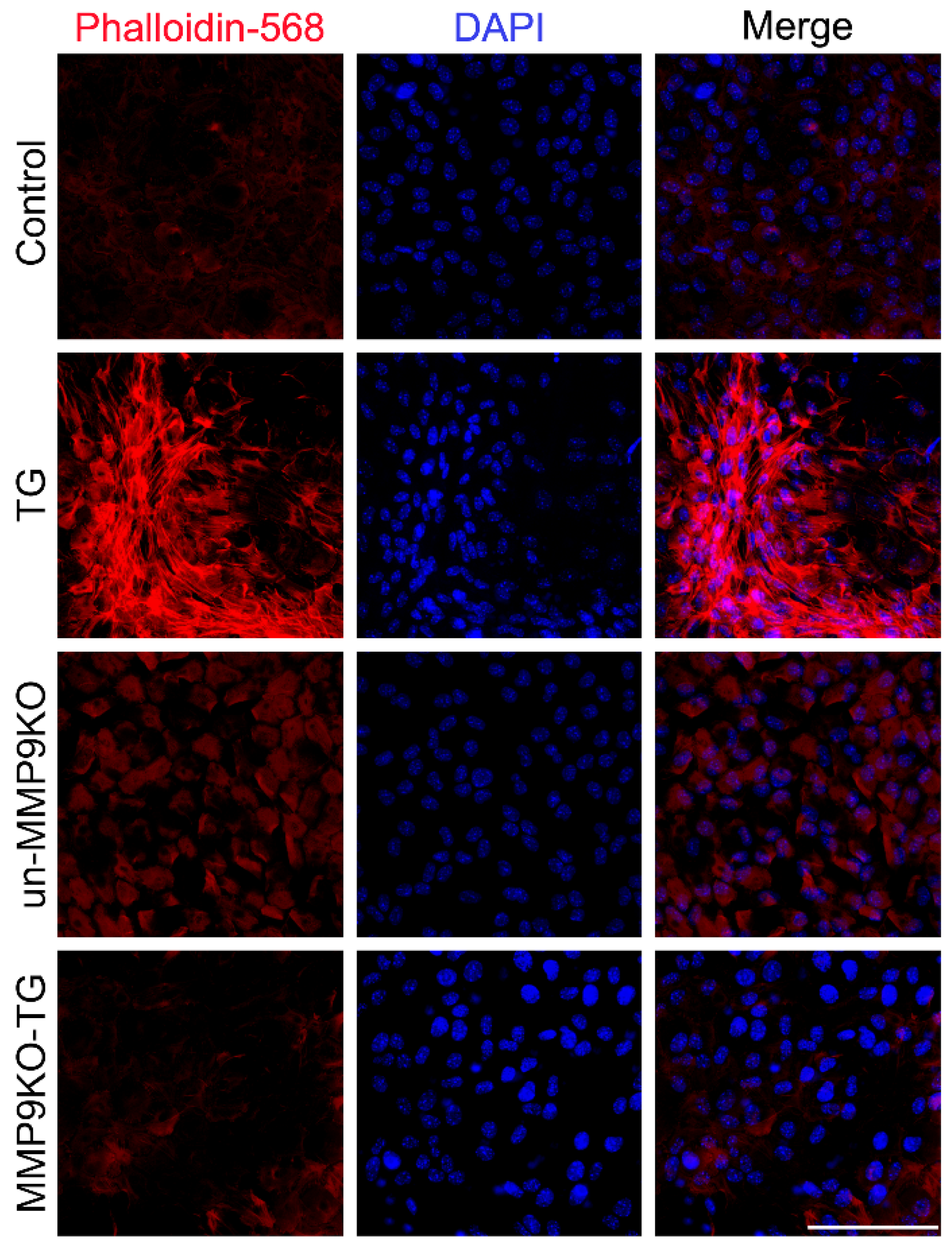
2.1. Absence of MMP9 Leads to Defects in TGF-β-Induced Actin Polymerization and Differential Expression of αSMA
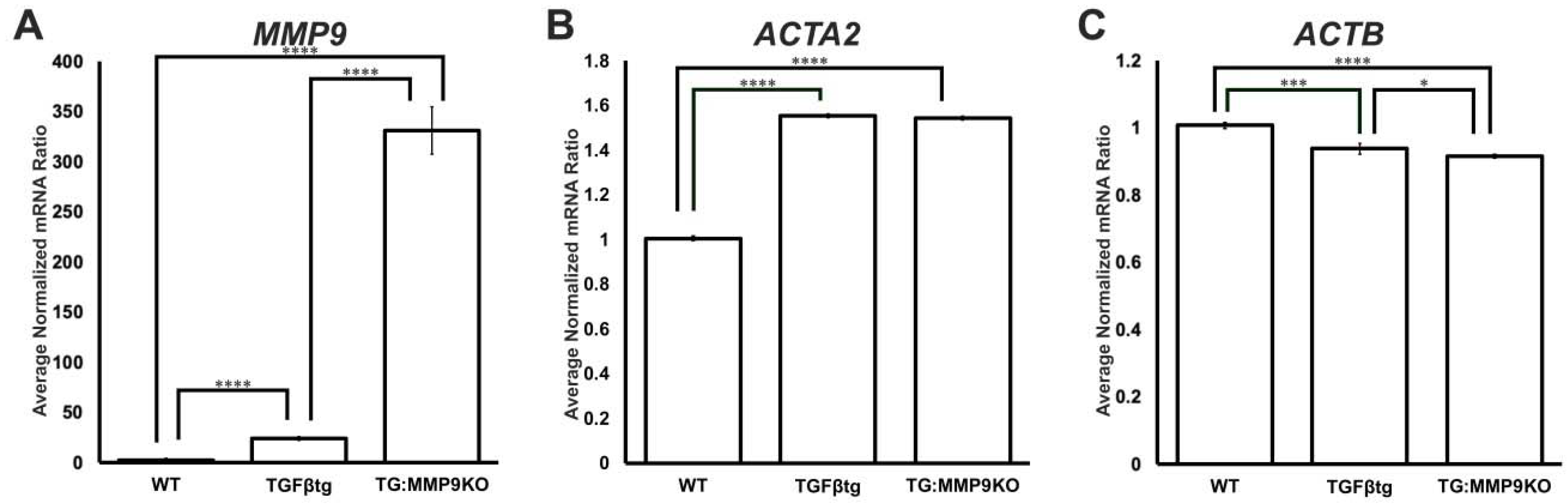
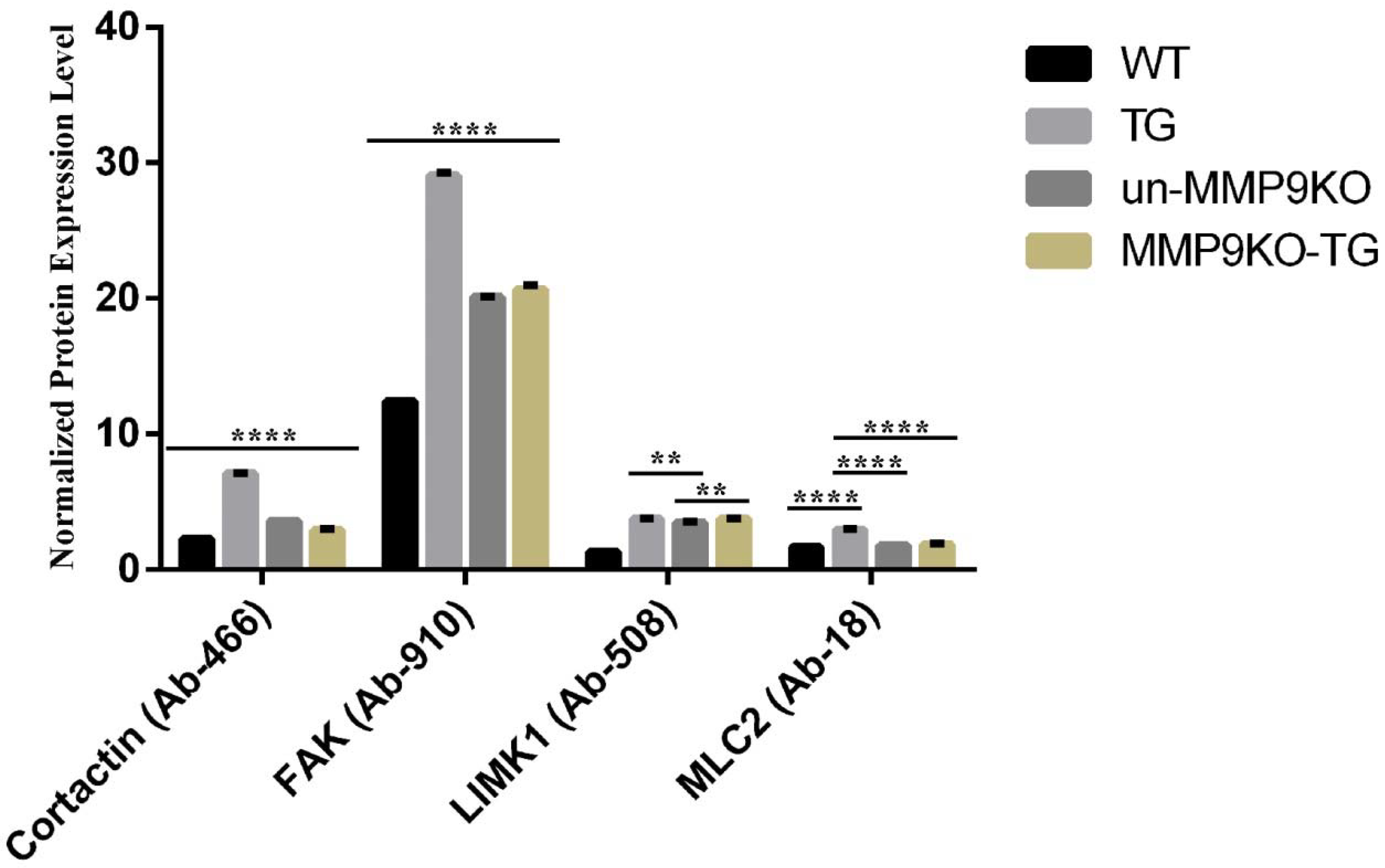
2.2. MMP9 Differentially Regulates Cytoskeletal Components Involved in Actin Polymerization
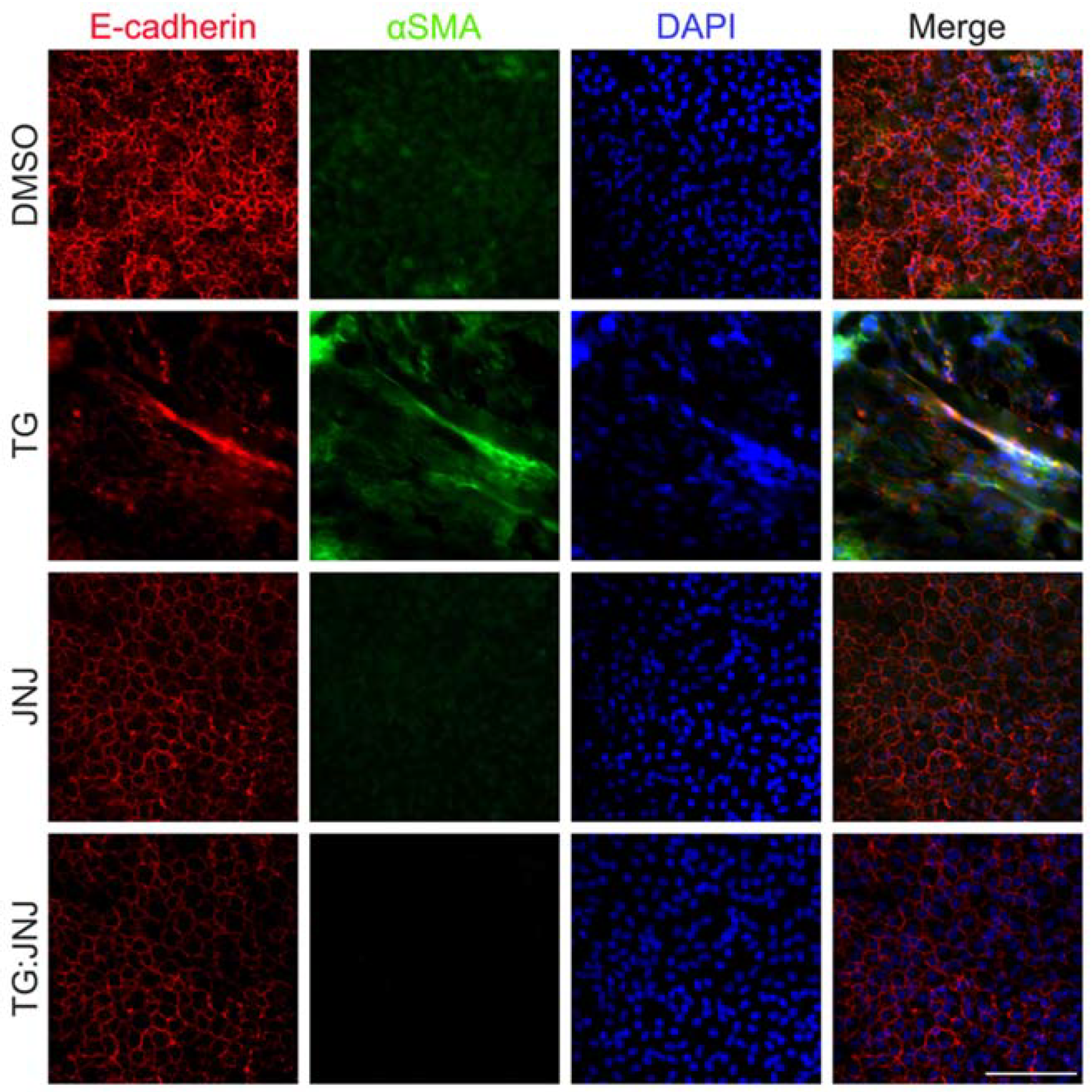
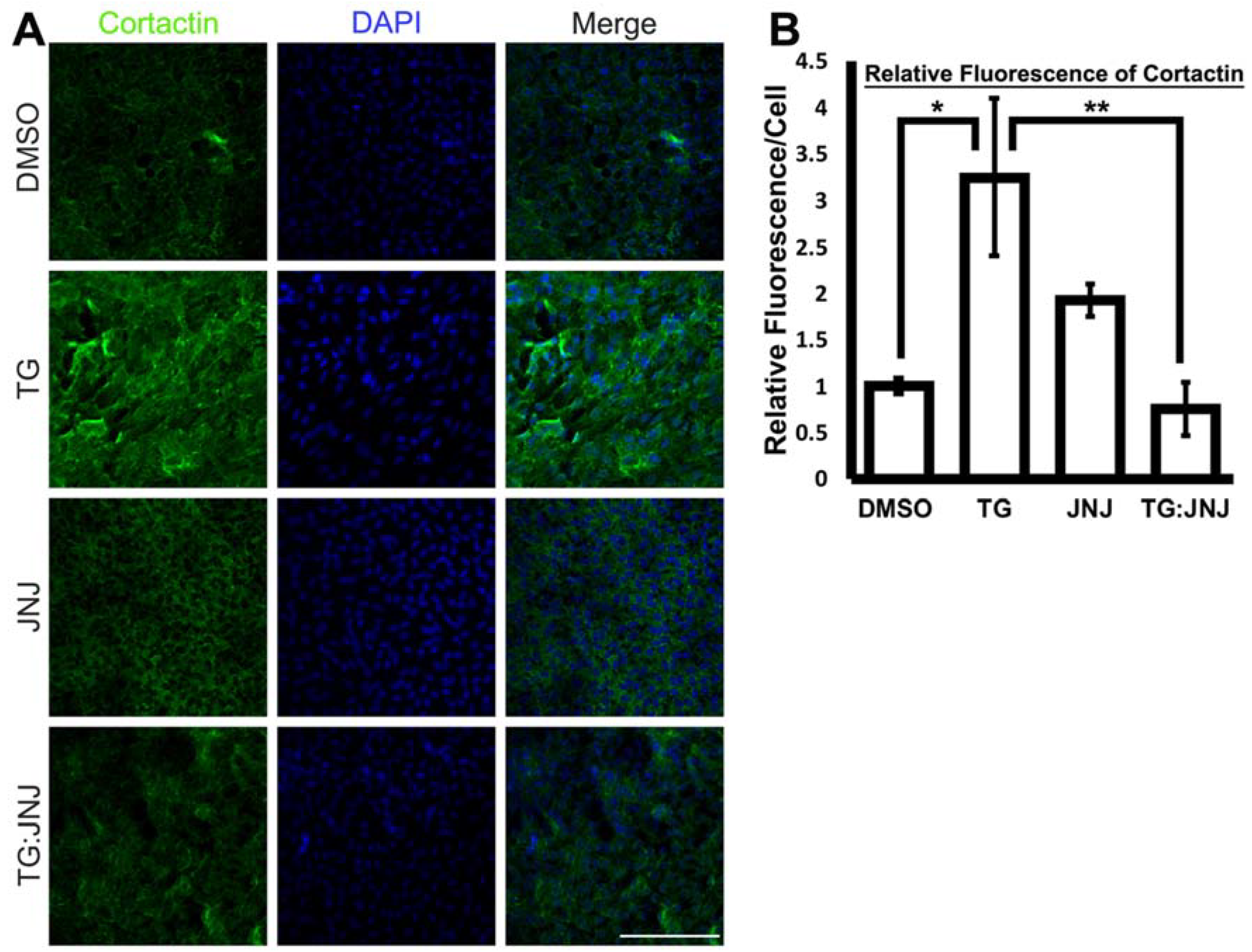
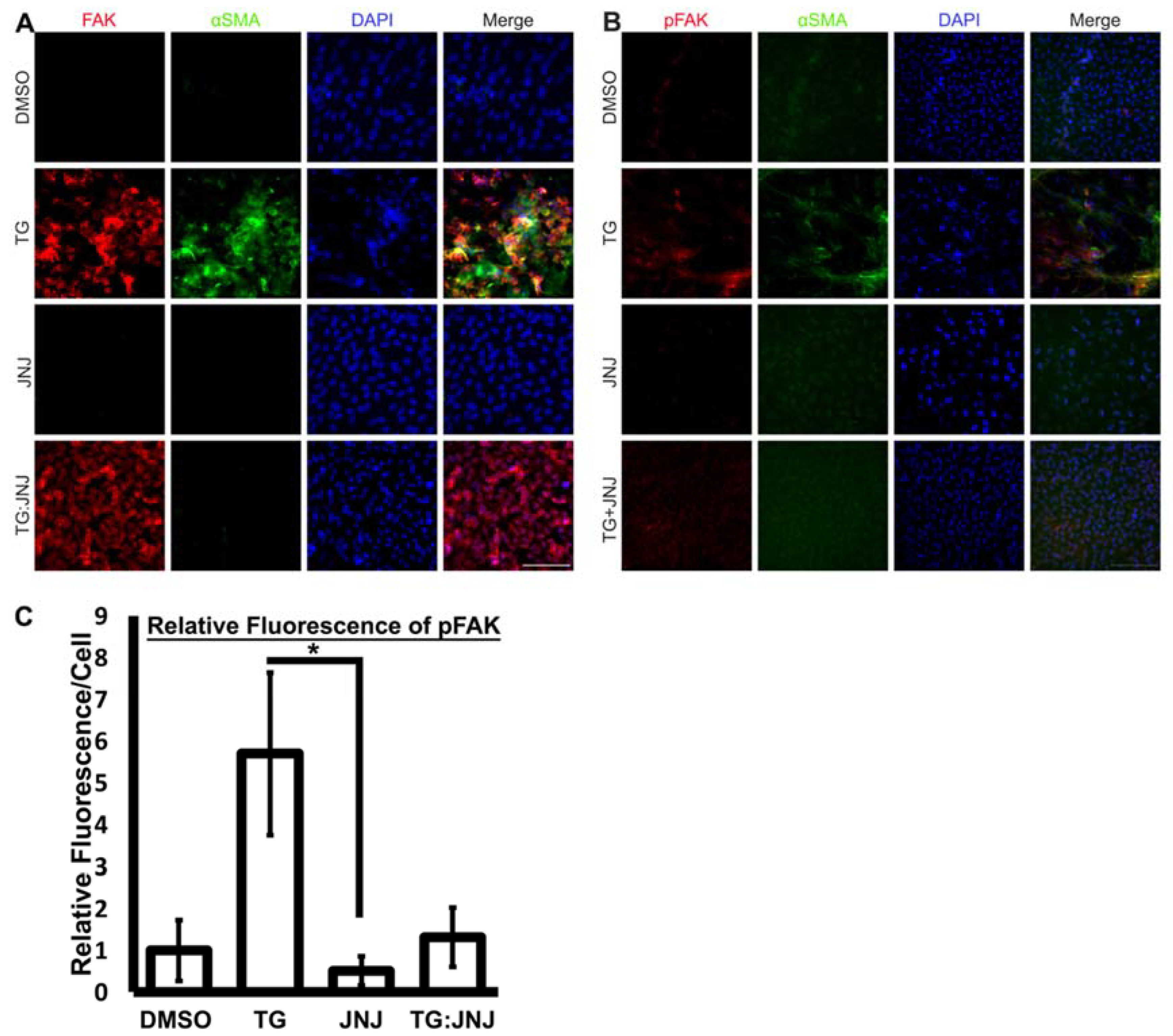
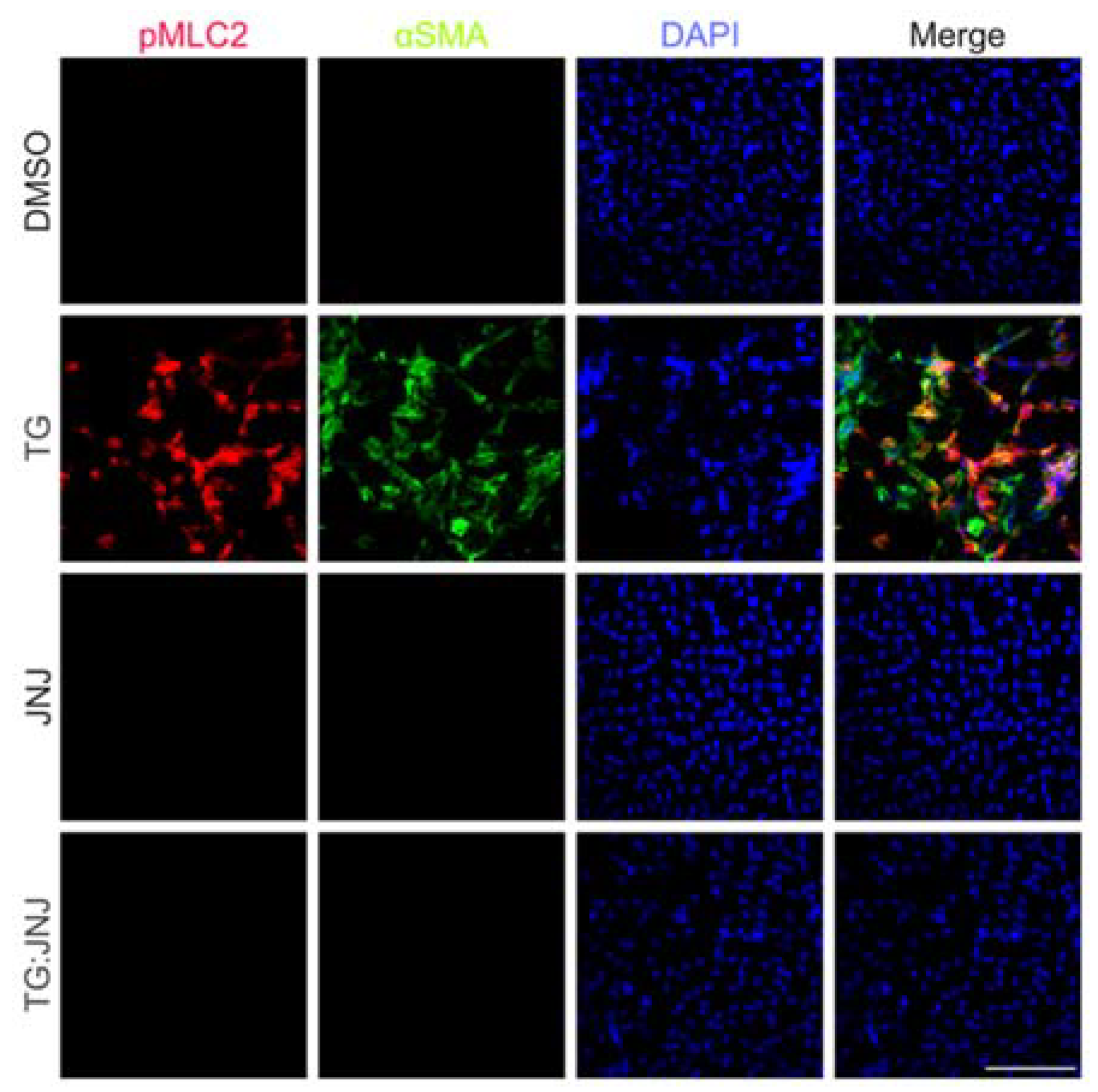
2.3. A MMP9-Specific Inhibitor of Activation Prevented EMT in Rat LECs by Differentially Regulating Cytoskeletal Components Involved in Actin Polymerization
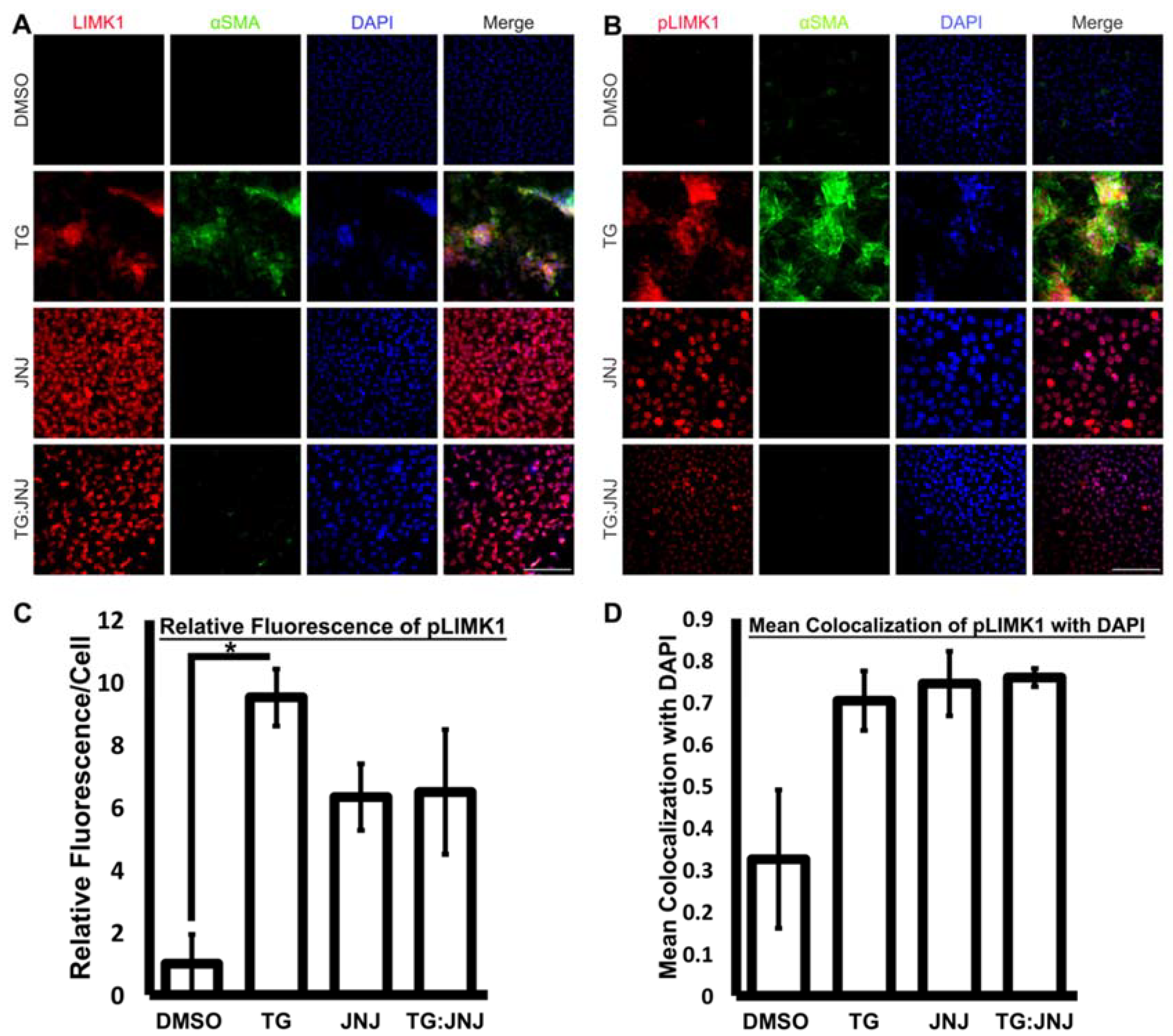
2.4. MMP9 Deficiency Notably Reduced MRTF-A Translocation to the Nucleus
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Obtaining and Culturing LEC Explants
4.3. Treatments of LEC Explants with TGF-β and JNJ0966
4.4. Nanostring
4.5. Cytoskeletal Protein Array Using Mouse LEC Explants
4.6. Immunofluorescence Staining
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/blindness-and-visual-impairment (accessed on 10 June 2021).
- Eldred, J.A.; Dawes, L.J.; Wormstone, I.M. The lens as a model for fibrotic disease. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 1301–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, S.M.; Vasavada, A.R.; Johar, S.R.K.; Vasavada, V.A.; Vasavada, V.A. Post-Operative Capsular Opacification: A Review. Int. J. Mol. Sci. 2007, 3, 237–250. [Google Scholar]
- Wormstone, I.M. Posterior Capsule Opacification: A Cell Biological Perspective. Exp. Eye Res. 2002, 74, 337–347. [Google Scholar] [CrossRef]
- Šikić, H.; Shi, Y.; Lubura, S.; Bassnett, S. A full lifespan model of vertebrate lens growth. R. Soc. Open Sci. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- West-Mays, J.A.; Sheardown, H. Posterior Capsule Opacification. In Ocular Disease Mechanisms and Management, 1st ed.; Albert, D.M., Levin, L.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 238–242. [Google Scholar]
- Massague, J. TGF-β signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Miyazono, K. Positive and negative regulation of TGF-β signaling. J. Cell Sci. 2000, 113, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Stockis, J.; Colau, D.; Coulie, P.G.; Lucas, S. Membrane protein GARP is a receptor for latent TGF-β on the surface of activated human Treg. Eur. J. Immunol. 2009, 39, 3315–3322. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Cell Surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumour invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Korol, A.; Pino, G.; Dwivedi, D.; Robertson, J.V.; Deschamps, P.A.; West-Mays, J.A. Matrix Metalloproteinase-9–Null Mice Are Resistant to TGF-β–Induced Anterior Subcapsular Cataract Formation. Am. J. Pathol. 2014, 184, 2001–2012. [Google Scholar] [CrossRef] [Green Version]
- Taiyab, A.; Holms, J.; West-Mays, J.A. β-Catenin/Smad3 Interaction Regulates Transforming Growth Factor-β-Induced Epithelial to Mesenchymal Transition in the Lens. Int. J. Mol. Sci. 2019, 20, 2078. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.-H.D.; Chen, D.-Q.D.; Wang, Y.-N.D.; Feng, Y.-L.D.; Cao, G.D.; Vaziri, N.D.; Zhao, Y.-Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem.-Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarino, M.; Tosoni, A.; Nebuloni, M. Direct contribution of epithelium to organ fibrosis: Epithelial-mesenchymal transition. Hum. Pathol. 2009, 40, 1365–1376. [Google Scholar] [CrossRef]
- Lovicu, F.; Shin, E.; McAvoy, J. Fibrosis in the lens. Sprouty regulation of TGFβ signaling prevents lens EMT leading to cataract. Exp. Eye Res. 2016, 142, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korol, A.; Taiyab, A.; West-Mays, J.A. RhoA/ROCK Signaling Regulates TGFβ-Induced Epithelial-Mesenchymal Transition of Lens Epithelial Cells through MRTF-A. Mol. Med. 2016, 22, 713–723. [Google Scholar] [CrossRef]
- Saika, S.; Kono-Saika, S.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Flanders, K.C.; Yoo, J.; Anzano, M.; Liu, C.-Y.; et al. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am. J. Pathol. 2004, 164, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Stamenkovic, I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef]
- Dwivedi, D.J.; Pino, G.; Banh, A.; Nathu, Z.; Howchin, D.; Margetts, P.; Sivak, J.G.; West-Mays, J.A. Matrix Metalloproteinase Inhibitors Suppress Transforming Growth Factor-β-Induced Subcapsular Cataract Formation. Am. J. Pathol. 2006, 168, 69–79. [Google Scholar] [CrossRef]
- Hsu, J.Y.C.; Bourguignon, L.Y.W.; Adams, C.M.; Peyrollier, K.; Zhang, H.; Fandel, T.; Cun, C.L.; Werb, Z.; Noble-Haeusslein, L.J. Matrix Metalloproteinase-9 Facilitates Glial Scar Formation in the Injured Spinal Cord. J. Neurosci. 2008, 28, 13467–13477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathu, Z.; Dwivedi, D.J.; Reddan, J.R.; Sheardown, H.; Margetts, P.J.; West-Mays, J.A. Temporal changes in MMP mRNA expression in the lens epithelium during anterior subcapsular cataract formation. Exp. Eye Res. 2009, 88, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Bamburg, J.R.; Bernstein, B.W. Roles of ADF/cofilin in actin polymerization and beyond. F1000 Biol. Rep. 2010, 2, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.T. Focal adhesion Kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prunier, C.; Prudent, R.; Kapur, R.; Sadoul, K.; Lafanechère, L. LIM kinases: Cofilin and beyond. Oncotarget 2017, 8, 41749–41763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, F.; Lyon, R.C.; Chen, J. Functions of myosin light chain-2 (myl2) in cardiac muscle and disease. Gene 2015, 569, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.; Zhang, Y.-M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974. [Google Scholar] [CrossRef] [Green Version]
- Banh, A.; Deschamps, P.A.; Vijayan, M.M.; Sivak, J.G.; West-Mays, J.A. The role of Hsp70 and Hsp90 in TGF-β-induced epithelial-to-mesenchymal transition in rat lens epithelial explants. Mol. Vis. 2007, 13, 2248–2262. [Google Scholar] [PubMed]
- Grigera, P.R.; Jeffery, E.D.; Martin, K.H.; Shabanowitz, J.; Hunt, D.F.; Parsons, J.T. FAK phosphorylation sites mapped by mass spectrometry. J. Cell Sci. 2005, 118, 4931–4935. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, K.; Nagata, K.; Maekawa, M.; Ishizaki, T.; Narumiya, S.; Mizuno, K. Rho-associated Kinase ROCK Activates LIM-kinase 1 by Phosphorylation at Threonine 508 within the Activation Loop. J. Biol. Chem. 2000, 275, 3577–3582. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, C.; Treisman, R.; Bailly, M.; Khaw, P.T. The Role of the MRTF-A/SRF Pathway in Ocular Fibrosis. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4560–4567. [Google Scholar] [CrossRef] [Green Version]
- Crider, B.J.; Risinger, G.M.; Haaksma, C.J.; Howard, E.W.; Tomasek, J.J. Myocardin-related transcription factors A and B are key regulators of TGF-β1-induced fibroblast to myofibroblast differentiation. J. Investig. Dermatol. 2011, 131, 2378–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming Growth Factor-β1 Mediates Epithelial to Mesenchymal Transdifferentiation through a RhoA-dependent Mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Daly, R. Cortactin signalling and dynamic actin networks. Biochem. J. 2004, 382, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Weed, S.A.; Karginov, A.V.; Schafer, D.A.; Weaver, A.M.; Kinley, A.W.; Cooper, J.A.; Parsons, J.T. Cortactin Localization to Sites of Actin Assembly in Lamellipodia Requires Interactions with F-Actin and the Arp2/3 Complex. J. Cell Biol. 2000, 151, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Mizumo, K. Nuclear export of LIM-kinase 1, mediated by two leucine-rich nuclear-export signals within the PDZ domain. Biochem. J. 1999, 338, 793–798. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.V.; Koto, K.; Gutierrez-Hartmann, A. Nuclear and cytoplasmic LIMK1 enhances human breast cancer progression. Mol. Cancer 2011, 10, 75. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Liu, C.; Zhi, Y.; Wu, L.; Jiang, M.; Zhang, Y.; Zhou, R.; Zhao, L. LIMK1 nuclear translocation promotes hepatocellular carcinoma progression by increasing p-ERK nuclear shuttling and by activating c-Myc signalling upon EGF stimulation. Oncogene 2021, 40, 2581–2595. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, Y.A.; Hu, W.; Cubero, F.J.; Haas, U.; Freimuth, J.; Tacke, F.; Trautwein, C.; Liedtke, C. Overexpression of c-myc in hepatocytes promotes activation of hepatic stellate cells and facilitates the onset of liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 1765–1775. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.T.S. Nuclear FAK: A new mode of gene regulation from cellular adhesions. Mol. Cells 2013, 36, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Nelson, C.M. New Insights into the Regulation of Epithelial–Mesenchymal Transition and Tissue Fibrosis. Int. Rev. Cell Mol. Biol. 2012, 294, 171–221. [Google Scholar] [CrossRef]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Thatcher, J.E.; Sutherland, L.B.; Kinoshita, H.; Gerard, R.D.; Richardson, J.A.; DiMaio, J.M.; Sadek, H.; Kuwahara, K.; Olson, E.N. Myocardin-Related Transcription Factor-A Controls Myofibroblast Activation and Fibrosis in Response to Myocardial Infarction. Circ. Res. 2010, 107, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.; Korol, A.; West-Mays, J.A. Nuclear translocation of myocardin-related transcription factor-A during transforming growth factor beta-induced epithelial to mesenchymal transition of lens epithelial cells. Mol. Vis. 2013, 19, 1017–1028. [Google Scholar] [PubMed]
- Shiwen, X.; Stratton, R.; Nikitorowicz-Buniak, J.; Ahmed-Abdi, B.; Ponticos, M.; Denton, C.; Abraham, D.; Takahashi, A.; Suki, B.; Layne, M.D.; et al. A Role of Myocardin Related Transcription Factor-A (MRTF-A) in Scleroderma Related Fibrosis. PLoS ONE 2015, 10, e0126015. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, C.; Owen, N.; Lees, J.; Tagalakis, A.D.; Hart, S.L.; Webster, A.R.; Orengo, C.A.; Khaw, P.T. Genome-wide RNA-Sequencing analysis identifies a distinct fibrosis gene signature in the conjunctiva after glaucoma surgery. Sci. Rep. 2017, 7, 5644. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody List | Fold Change Between Samples | |||
|---|---|---|---|---|
| TG/Control | MMP9KO-TG/un-MMP9KO | TG /un-MMP9KO | TG /MMP9KO-TG | |
| Cofilin (Ab-S3) | 2.27 | 0.93 | 1.58 | 1.69 |
| Cortactin (Ab-Y466) | 3.11 | 0.80 | 1.96 | 2.45 |
| FAK (Ab-Y910) | 2.34 | 1.03 | 1.45 | 1.41 |
| FAK (Ab-pY861) | 1.27 | 0.71 | 1.11 | 1.41 |
| Filamin A (Ab-S2152) | 1.59 | 1.16 | 1.52 | 1.31 |
| LIMK1 (Ab-T508) | 2.85 | 1.08 | 1.07 | 1.00 |
| LIMK1 (Ab-pT508) | 1.26 | 1.17 | 1.25 | 1.07 |
| MLC2 (Ab-S18) | 1.74 | 1.06 | 1.65 | 1.56 |
| MLC2 (Ab-pS18) | 1.57 | 0.72 | 0.69 | 0.95 |
| Rac1/CDC42 (Ab-S71) | 1.28 | 0.76 | 1.05 | 1.38 |
| Rho/Rac guanine nucleotide exchange factor (Ab-pS885) | 1.31 | 1.22 | 1.25 | 1.03 |
| VASP (Ab-157) | 2.08 | 1.11 | 1.35 | 1.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.Z.; Taiyab, A.; West-Mays, J.A. MMP9 Differentially Regulates Proteins Involved in Actin Polymerization and Cell Migration during TGF-β-Induced EMT in the Lens. Int. J. Mol. Sci. 2021, 22, 11988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111988
Liu ZZ, Taiyab A, West-Mays JA. MMP9 Differentially Regulates Proteins Involved in Actin Polymerization and Cell Migration during TGF-β-Induced EMT in the Lens. International Journal of Molecular Sciences. 2021; 22(21):11988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111988
Chicago/Turabian StyleLiu, Zi Zhen (Ginny), Aftab Taiyab, and Judith A. West-Mays. 2021. "MMP9 Differentially Regulates Proteins Involved in Actin Polymerization and Cell Migration during TGF-β-Induced EMT in the Lens" International Journal of Molecular Sciences 22, no. 21: 11988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111988