Design, Synthesis and Evaluation of Novel Derivatives of Curcuminoids with Cytotoxicity

 , ,
, ,  and
and
Abstract
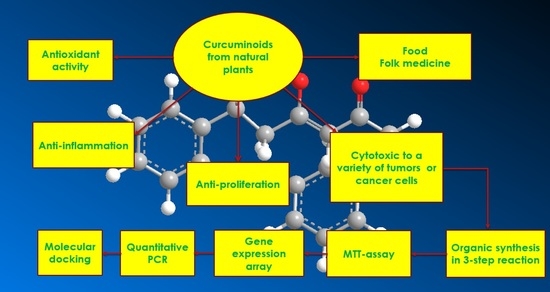
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Data
2.2.1. In Vitro Anti-Proliferative Activities
2.2.2. Cell Cycle and Apoptosis Analysis by Flow Cytometry
2.2.3. Cell Morphology
2.2.4. Gene Expression
Histogram Plot
Volcano Plot
Number of Differentially Expressed Genes
Principal Component Analysis (PCA)
Clustering Analysis
2.2.5. The p53 Signaling Pathway and Quantitative PCR (qPCR) Analysis for HepG2 and MCF-7 Cells Treated with Compounds 3 and MD12a
2.2.6. Docking Interaction of Compound MD12a with GADD45B
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of Starting Material Ring A Compounds: A1–15
3.1.2. General Procedure for the Synthesis of Starting Material Ring B Compounds: B1–4
3.1.3. General Procedure for the Synthesis of β′-Hydroxy-α,β-Unsaturated Ketones (1–8, 10–17 and MD1–12) and α,β-Unsaturated Ketones (1a, 2a, 3a, 4a, 5a, 6a, 7a, 12a, 14a and MD1a–12a)
3.1.4. General Procedure for the Synthesis of α,β-Unsaturated β-Diketones 19–24 and MD13
3.2. Biology
3.2.1. Cell Cultures
3.2.2. Anti-Proliferative Assay
3.2.3. Cell Cycle Analysis
3.2.4. Cell Apoptosis Analysis
3.2.5. Quantitative PCR Analysis
3.2.6. P53 Signaling Pathway Graph
3.3. Molecular Modeling
3.3.1. Ligand Preparation
3.3.2. Docking Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Preparation of the Starting Materials of A1–15

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Aldehyde | Abbreviation | Ring A Compound |
|---|---|---|---|
| 1 |  | Ph |  (A1) |
| 2 |  | 3,4-OMe-Ph |  (E)-4- (3,4-dimethoxyphenyl) but-3-en-2-one (A2) |
| 3 |  | 3,4,5-OMe-Ph |  (E)-4- (3,4,5-trimethoxyphenyl) but-3-en-2-one (A3) |
| 4 |  | 4-OH-Ph |  (E)-4- (4-hydroxyphenyl) but-3-en-2-one (A4) |
| 5 |  | 4-OMe-Ph |  (E)-4- (4-methoxyphenyl) but-3-en-2-one (A5) |
| 6 |  | 4-F-Ph |  (E)-4- (4-fluorophenyl) but-3en-2-one (A6) |
| 7 |  | 4-Cl-Ph |  (E)-4- (4-chlorophenyl) but-3-en-2-one (A7) |
| 8 |  | 2-pyr |  (E)-4- (pyridin-2-yl) but-3-en-2-one (A8) |
| 9 |  | 3-pyr |  (E)-4- (pyridin-3-yl) but-3-en-2-one (A9) |
| 10 |  | 4-pyr |  (E)-4- (pyridin-4-yl) but-3-en-2-one (A10) |
| 11 |  | 2-furan |  (E)-4- (furan-2-yl) but-3-en-2-one (A11) |
| 12 |  | 3-furan |  (E)-4- (furan-3-yl) but-3-en-2-one (A12) |
| 13 |  | 2-thiophene |  (E)-4- (thiophen-2-yl) but-3-en-2-one (A13) |
| 14 |  | 2-methyl-2-thiophene |  (E)-4- (5-methylthiophen-2-yl) but-3-en-2-one (A14) |
| 15 |  | 2-pyrrole |  (E)-4- (1H-pyrrol-2-yl) but-3-en-2-one (A15) |
| 16 |  1H-indole-3-carbaldehyde | 3-indole |  (E)-4- (1H-indol-3-yl) but-3-en-2-one (A16) |
Appendix B. Preparation for Ring B
| Item | Ring B Compound |
|---|---|
| 1 |  (B1) |
| 2 |  (E)-4- (4-methoxyphenyl) but-3-en-2-one (B2) |
| 3 |  3- (3,4-dimethoxyphenyl) propanal (B3) |
| 4 |  3- (3,4,5-trimethoxyphenyl) propanal (B4) |
References
- Vogel, H.; Pelletier, J. Curcumin––Biological and medicinal properties. J. Pharm. 1815, 1, 289. [Google Scholar]
- Al-Abbasi, F.A.; Alghamdi, E.A.; Baghdadi, M.A.; Alamoudi, A.J.; El-Halawany, A.M.; El-Bassossy, H.M.; Aseeri, A.H.; Al-Abd, A.M. Gingerol synergizes the cytotoxic effects of doxorubicin against liver cancer cells and protects from its vascular toxicity. Molecules 2016, 21, 886. [Google Scholar] [CrossRef]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Ahn, K.S.; Murakami, A.; Sethi, G.; Limtrakul, P.; Badmaev, B.; Aggarwal, B.B. Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory and anti-proliferative responses through a ROS-independent mechanism. Carcinogenesis 2007, 28, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, T.; Farooqui, A.A. Curcumin: Historical Background, Chemistry, Pharmacological Action, and Potential Therapeutic Value. In Curcumin for Neurological and Psychiatric Disorders, 1st ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 23–44. [Google Scholar]
- Lee, D.Y.; Chun, Y.S.; Kim, J.K.; Lee, J.O.; Lee, Y.J.; Ku, S.K.; Shim, S.M. Curcumin Ameliorated Oxidative stress and inflammation-related muscle disorders in C2C12 myoblast cells. Antioxidants 2021, 10, 476. [Google Scholar] [CrossRef]
- Chen, D.; Nie, M.; Fan, M.-W.; Bian, Z. Anti-inflammatory activity of curcumin in macrophages stimulated by lipopolysaccharides from Porphyromonas gingivalis. Pharmacology 2008, 82, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Lee, M.J.; Kim, H.; Choi, S.K.; Kim, J.E.; Moon, H.I.; Park, W.H. Curcumin inhibits TNFalpha-induced lectin-like oxidised LDL receptor-1 (LOX-1) expression and suppresses the inflammatory response in human umbilical vein endothelial cells (HUVECs) by an antioxidant mechanism. J. Enzym. Inhib. Med. Chem. 2010, 25, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Willenbacher, E.; Khan, S.Z.; Mujica, S.C.A.; Trapani, D.; Hussain, S.; Wolf, D.; Willenbacher, W.; Spizzo, G.; Seeber, A. Curcumin: New insights into an ancient ingredient against cancer. Int. J. Mol. Sci. 2019, 20, 1808. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.L.; Huang, X.F.; Zhu, H.L. Curcumin and its formulations: Potential anti-cancer agents. Anticancer Agents Med. Chem. 2012, 12, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Padro’n, J.M.; Miranda, P.O.; Padro’n, J.I.; Martı’n, V.S. β′-Hydroxy-α,β-unsaturated ketones: A new pharmacophore for the design of anticancer drugs. Bioorg. Med. Chem. Lett. 2006, 16, 2266–2269. [Google Scholar] [CrossRef] [PubMed]
- Sati, S.C.; Sati, N. Bioactive constituents and medicinal importance of genus. Alnus. Pharmacogn. Rev. 2011, 5, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maehara, S.; Ikeda, M.; Haraguchi, H.; Kitamura, C.; Nagoe., T.; Ohashi, K.; Shibuya, H. Microbial conversion of curcumin into colorless hydroderivatives by the endophytic fungus Diaporthe sp. associated with Curcuma longa. Chem. Pharm. Bull. 2011, 59, 1042–1044. [Google Scholar] [CrossRef]
- Hsieh, M.-T.; Chang, L.-C.; Hung, H.-Y.; Lin, H.-Y.; Shih, M.-H.; Tsai, C.-H.; Kuo, S.-C. New bis(hydroxymethyl) alkanoate curcuminoid derivatives exhibit activity against triple-negative breast cancer in-vitro and in-vivo. Eur. J. Med. Chem. 2017, 131, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.W.; Chia, S.L. Asymmetrical meta-methoxylated diarylpentanoids: Rational design, synthesis and anti-cancer evaluation in-vitro. Eur. J. Med. Chem. 2018, 157, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; He, T.; Chang, Y.; Chang, Y.; Zhao, Y.; Chen, X.; Bai, S.; Wang, L.; Shen, M.; She, G. The genus Alnus, a comprehensive outline of its chemical constituents and biological activities. Molecules 2017, 22, 1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badoni, R.; Semwal, D.K.; Kothiyal, S.K.; Rawat, U.J. Chemical Constituents and Biological Applications of Genus Symplocos. Asian Nat. Prod. Res. 2010, 12, 1069–1080. [Google Scholar] [CrossRef]
- León, L.G.; Carballo, R.M.; Vega-Hernández, M.C.; Miranda, P.O.; Martín, V.S.; Padrón, J.I.; Padrón, J.M. β′-Hydroxy-α,β-unsaturated ketones: A new pharmacophore for the design of anticancer drugs. Part 2. ChemMedChem 2008, 3, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.N.; Wiseman, L.J., Jr.; Slater, C.D. Replacement substituent constants for simple heterocycles. Tetrahedron 1989, 45, 4103–4112. [Google Scholar] [CrossRef]
- Benington, F.; Morin, R.D.; Khaled, M.A. An efficient procedure for the synthesis of trans-2-, -3-, and -4-pyridalacetones. Synthesis 1984, 7, 619–620. [Google Scholar] [CrossRef]
- Stork, G.; Rosen, P.; Goldman, N.L. The α-alkylation of enoleates from the lithium-ammonia reduction of α,β-unsaturated ketones. J. Am. Chem. Soc. 1961, 83, 2965–2966. [Google Scholar] [CrossRef]
- Shangpliang, O.R.; Wanniang, K.; Kshiar, B.; Marpna, I.D.; Lipon, T.M.; Mizar, P.; Myrboh, B. PTSA-catalyzed reaction of alkyl/aryl methyl ketones with aliphatic alcohols in the presence of selenium dioxide: A protocol for the generation of an α-ketoacetals library. ACS Omega 2019, 4, 6035–6043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, T.J.; Basutto, J.A.; Bower, J.F.; Rathi, A. Heteroaromatic synthesis via olefin cross-metathesis: Entry to polysubstituted pyridines. Org. Lett. 2011, 13, 1036–1039. [Google Scholar] [CrossRef]
- Trost, B.M.; Jonasson, C.; Wucher, M. Atom economy. Aldol-Type products by Vanadium-catalyzed additions of allenic alcohols and aldehydes. J. Am. Chem. Soc. 2001, 123, 12736–12737. [Google Scholar] [CrossRef] [PubMed]
- Schrag, J.D.; Jiralerspong, S.; Banville, M.; Jaramillo, M.L.; O’Connor-McCourt, M.D. The crystal structure and dimerization interface of GADD45gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 6566–6571. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.B.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]


















 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | Ring A | R1 | R2 | Compound | Ring A | R1 | R2 |
| 1 [23] | Ph | H | H | 22 | Ph | 3,4,5-OMe | H |
| 1a | Ph | H | H | 23 | Ph | 4-F | H |
| 2 | Ph | 3,4-OMe | H | 24 | Ph | 4-Cl | H |
| 2a | Ph | 3,4-OMe | H | MD1 | Ph | 4-OH | 4-OMe |
| 3 | Ph | 3,4,5-OMe | H | MD1a | Ph | 4-OH | 4-OMe |
| 3a | Ph | 3,4,5-OMe | H | MD2 | Ph | 4-OMe | 4-OMe |
| 4 | Ph | 4-OMe | H | MD2a | Ph | 4-OMe | 4-OMe |
| 4a | Ph | 4-OMe | H | MD3 | Ph | 3,4-OMe | 4-OMe |
| 5 | Ph | 4-OH | H | MD3a | Ph | 3,4-OMe | 4-OMe |
| 5a | Ph | 4-OH | H | MD4 | Ph | 3,4,5-OMe | 4-OMe |
| 6 | Ph | 4-F | H | MD4a | Ph | 3,4,5-OMe | 4-OMe |
| 6a | Ph | 4-F | H | MD5 | Ph | 4-OH | 3,4-OMe |
| 7 | Ph | 4-Cl | H | MD5a | Ph | 4-OH | 3,4-OMe |
| 7a | Ph | 4-C1 | H | MD6 | Ph | 4-OMe | 3,4-OMe |
| 8 | 2-pyridine | H | MD6a | Ph | 4-OMe | 3,4-OMe | |
| 10 | 3-pyridine | H | MD7 [12] | Ph | 3,4-OMe | 3,4-OMe | |
| 11 | 4-pyridine | H | MD7a | Ph | 3,4-OMe | 3,4-OMe | |
| 12 | 2-furan | H | MD8 | Ph | 3,4,5-OMe | 3,4-OMe | |
| 12a | 2-furan | H | MD8a | Ph | 3,4,5-OMe | 3,4-OMe | |
| 13 | 3-furan | H | MD9 | Ph | 4-OH | 3,4,5-OMe | |
| 14 | 2-thiophene | H | MD9a | Ph | 4-OH | 3,4,5-OMe | |
| 14a | 2-thiophene | H | MD10 | Ph | 4-OMe | 3,4,5-OMe | |
| 15 | 2-thiophene | 2-methyl | H | MD10a | Ph | 4-OMe | 3,4,5-OMe |
| 16 | 2-pyrrole | H | MD11 | Ph | 3,4-OMe | 3,4,5-OMe | |
| 17 | 3-indole | H | MD11a | Ph | 3,4-OMe | 3,4,5-OMe | |
| 19 [1] | Ph | H | MD12 | Ph | 3,4,5-OMe | 3,4,5-OMe | |
| 20 | Ph | 4-OMe | H | MD12a | Ph | 3,4,5-OMe | 3,4,5-OMe |
| 21 | Ph | 3.4-OMe | H | MD13 | Ph | 3,4,5-OMe | 4-OMe |
| Cell Line | IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|
| Compound | Compound Abbreviation | HeLaS3 | Kbvin | NCI-H460 | NCI-H460/MX20 | MCF7 | HepG2 |
| 1 | Ph-b’-OH | >40 | >40 | >40 | >40 | >40 | >40 |
| 1a | Ph-a’ = b’ | 5.72 ± 0.05 | 13.69 ± 0.02 | 26.48 ± 0.29 | 35.15 ± 0.27 | 14.25 ± 1.31 | 10.19 ± 0.70 |
| 2 | 3,4-OMe-Ph-b’-OH | 26.42 ± 0.23 | 38.36 ± 0.34 | >40 | >40 | 33.15 ± 0.42 | 37.08 ± 0.075 |
| 2a | 3,4-OMe-Ph-a’ = b’ | 5.52 ± 0.39 | 8.09 ± 0.44 | 13.78 ± 0.23 | 26.11 ± 1.22 | 9.87 ± 0.08 | 8.85 ± 0.61 |
| 3 | 3,4,5-OMe-Ph-b’-OH | 5.88 ± 0.05 | 5.84 ± 0.31 | 7.25 ± 0.47 | 8.25 ± 0.01 | 7.59 ± 0.13 | 7.46 ± 0.68 |
| 3a | 3,4,5-OMe-Ph-a’ = b’ | 5.51 ± 0.03 | 5.96 ± 0.31 | 13.24 ± 0.58 | 31.46 ± 0.58 | 8.09 ± 0.33 | 7.33 ± 0.52 |
| 4 | 4-OMe-Ph-b’-OH | 29.67 ± 0.007 | 30.2 ± 1.33 | >40 | >40 | 29.84 ± 2.16 | 31.11 ± 0.45 |
| 4a | 4-OMe-Ph-a’ = b’ | 11.07 ± 1.71 | 31.76 ± 1.86 | 32.28 ± 0.36 | >40 | 19.82 ± 1.80 | 31.86 ± 2.23 |
| 5 | 4-OH-Ph-b’-OH | 31.98 ± 0.32 | >40 | >40 | >40 | >40 | >40 |
| 5a | 4-OH-Ph-a’ = b’ | 6.25 ± 0.40 | 10.38 ± 1.85 | 16.88 ± 0.24 | 35.44 ± 2.08 | 8.52 ± 0.16 | 17.55 ± 0.33 |
| 6 | 4-F-Ph-b’-OH | >40 | >40 | >40 | >40 | >40 | >40 |
| 6a | 4-F-Ph-a’ = b’ | 5.90 ± 0.009 | 23.98 ± 0.13 | 31.25 ± 0.29 | 34.38 ± 0.002 | 15.48 ± 0.03 | 14.26 ± 0.64 |
| 7 | 4-Cl-Ph-b’-OH | 31.73 ± 0.66 | >40 | >40 | >40 | 30.43 ± 0.31 | >40 |
| 7a | 4-Cl-Ph-a’ = b’ | 5.29 ± 0.51 | 18.90 ± 1.32 | 30.31 ± 0.49 | 33.37 ± 0.81 | 15.14 ± 0.15 | 12.84 ± 1.21 |
| 8 | 2-pyr-b’-OH | 22.60 ± 0.85 | >40 | >40 | >40 | 37.51 ± 1.33 | >40 |
| 9 | 3-pyr-b’-di-OH | >40 | >40 | >40 | >40 | 38.18 ± 0.65 | >40 |
| 10 | 3-pyr-b’-OH | 23.29 ± 0.28 | 34.54 ± 0.13 | >40 | >40 | 33.41 ± 1.55 | 33.71 ± 0.81 |
| 11 | 4-pyr-b’-OH | 9.67 ± 0.38 | 11.53 ± 0.004 | 15.83 ± 0.15 | 29.95 ± 0.48 | 12.45 ± 0.45 | 8.26 ± 1.03 |
| 12 | 2-f-b’-OH | >40 | >40 | >40 | >40 | >40 | >40 |
| 12a | 2-f-a’ = b’ | 16.10 ± 0.83 | 27.97 ± 0.59 | >40 | >40 | 31.69 ± 0.15 | 34.41 ± 1.55 |
| 13 | 3-f-b’-OH | 27.34 ± 5.87 | 26.16 ± 4.56 | 34.27 ± 0.01 | >40 | 15.65 ± 0.27 | 29.62 ± 0.30 |
| 14 | 2-t-b’-OH | 15.41 ± 0.19 | 20.39 ± 1.05 | 28.63 ± 0.36 | 34.04 ± 1.32 | 18.93 ± 0.89 | 19.73 ± 0.13 |
| 14a | 2-t-a’ = b’ | >40 | >40 | >40 | >40 | >40 | >40 |
| 15 | 2-methyl-t-b’-OH | 29.99 ± 0.58 | 29.48 ± 0.22 | >40 | >40 | 19.24 ± 1.54 | 31.27 ± 2.69 |
| 16 | 2-pyrrole-b’-OH | >40 | >40 | >40 | >40 | >40 | >40 |
| 17 | 3-indole-b’-OH | 32.73 ± 0.64 | 28.13 ± 0.94 | 33.87 ± 1.62 | 39.58 ± 0.18 | 33.77 ± 1.53 | 31.99 ± 1.79 |
| 18 | 4-F-Ph-b’-Cl | 7.05 ± 0.26 | 17.34 ± 0.29 | 30.01 ± 0.54 | >40 | 24.53 ± 1.32 | 21.27 ± 3.15 |
| 19 | Ph-a’ = b’-OH | >40 | >40 | >40 | >40 | >40 | >40 |
| 20 | 4-OMe-Ph-a’ = b’-OH | >40 | >40 | >40 | >40 | >40 | >40 |
| 21 | 3,4-OMe-Ph-a’ = b’-OH | 20.37 ± 1.34 | 22.82 ± 3.28 | >40 | >40 | 17.85 ± 0.40 | 38.85 ± 3.09 |
| 22 | 3,4,5-OMe-Ph-a’ = b’-OH | 20.89 ± 3.60 | 21.65 ± 3.99 | >40 | >40 | 19.52 ± 0.93 | >40 |
| 23 | 4-F-Ph-a’ = b’-OH | >40 | >40 | >40 | >40 | >40 | >40 |
| 24 | 4-Cl-Ph-a’ = b’-OH | >40 | >40 | >40 | >40 | >40 | >40 |
| MD1 | 4-OH-4-OMe-b’-OH | 23.8 ± 3.14 | 27.6 ± 1.44 | 31 ± 1.32 | >40 | 22.01 ± 0.36 | 25.88 ± 4.04 |
| MD1a | 4-OH-4-OMe-a’ = b’ | 24.4 ± 0.77 | 29 ± 2.94 | >40 | >40 | 25.28 ± 0.96 | 27.62 ± 0.37 |
| MD2 | 4-OMe-4-OMe-b’-OH | 7 ± 0.05 | 32.1 ± 0.58 | 23.9 ± 0.03 | >40 | 17.91 ± 0.29 | 26.15 ± 0.49 |
| MD2a | 4-OMe-4-OMe-a’ = b’ | 6.1 ± 0.04 | 7.7 ± 0.37 | 19.2 ± 0.14 | 35.7 ± 1.45 | 7.41 ± 0.23 | 7.33 ± 0.15 |
| MD3 | 3,4-OMe-4-OMe-b’-OH | 29 ± 1.14 | 34 ± 0.71 | >40 | >40 | 33.59 ± 0.14 | 31.28 ± 1.5 |
| MD3a | 3,4-OMe-4-OMe-a’ = b’ | 7 ± 0.37 | 9.3 ± 0.01 | 20.9 ± 1.4 | >40 | 8.54 ± 0.75 | 19.2 ± 7.35 |
| MD4 | 3,4,5-OMe-4-OMe-b’-OH | 25.9 ± 3.47 | 21.9 ± 0.47 | >40 | >40 | 29.71 ± 0.99 | >40 |
| MD4a | 3,4,5-OMe-4-OMe-a’ = b’ | 6 ± 0.51 | 4.6 ± 0.03 | 18.9 ± 0.62 | 17.9 ± 1.63 | 5.35 ± 0.41 | 5.49 ± 0.1 |
| MD5 | 4-OH-3,4-OMe-b’-OH | 9.2 ± 0.48 | 14.4 ± 2.19 | 38.7 ± 26.05 | >40 | 21.06 ± 3.92 | 23.13 ± 1.92 |
| MD5a | 4-OH-3,4-OMe-a’ = b’ | 6.6 ± 0.87 | 5 ± 0.08 | 16.4 ± 0.81 | 21.3 ± 0.32 | 6.36 ± 0.41 | 5.29 ± 0.17 |
| MD6 | 4-OMe-3,4-OMe-b’-OH | 5.6 ± 0.53 | 9.4 ± 0.26 | 25.6 ± 2.45 | 25.6 ± 1.95 | 8.87 ± 0.47 | 18.22 ± 0.61 |
| MD6a | 4-OMe-3,4-OMe-a’ = b’ | 5.7 ± 0.64 | 5.3 ± 0.06 | 20.6 ± 2.29 | 22.6 ± 0.78 | 6.24 ± 0.13 | 8.46 ± 0.36 |
| MD7 | 3,4-OMe-3,4-OMe-b’-OH | 4.68 ± 0.47 | 8.54 ± 0.33 | 18.8 ± 1.21 | 2.5 ± 1.05 | 4.64 ± 0.72 | 4.3 ± 0.35 |
| MD7a | 3,4-OMe-3,4-OMe-a’ = b’ | 5.13 ± 0.17 | 4.91 ± 0.09 | 4.3 ± 0.4 | 1.2 ± 0.65 | 5.35 ± 0.43 | 3.8 ± 0.51 |
| MD8 | 3,4,5-OMe-3,4-OMe-b’-OH | 19.34 ± 0.13 | 15.32 ± 2.6 | 20.7 ± 3.23 | 9.7 ± 1.07 | 6.29 ± 0.7 | 8.51 ± 1.52 |
| MD8a | 3,4,5-OMe-3,4-OMe-a’ = b’ | 5.43 ± 0.06 | 5.54 ± 0.06 | 5.2 ± 0.92 | 2.7 ± 1.12 | 4.93 ± 0.55 | 4.91 ± 0.43 |
| MD9 | 4-OH-3,4,5-OMe-b’-OH | 34.63 ± 2.26 | 35.51 ± 1.39 | >40 | 34.3 ± 4.83 | 21.41 ± 2.39 | 28.57 ± 2.34 |
| MD9a | 4-OH-3,4,5-OMe-a’ = b’ | 7.07 ± 0.13 | 16.44 ± 0.29 | 17.2 ± 1.39 | 5.3 ± 0.75 | 8.55 ± 0.77 | 8.03 ± 2.77 |
| MD10 | 4-OMe-3,4,5-OMe-b’-OH | 24.23 ± 0.14 | 27.76 ± 2.74 | >40 | 32.7 ± 9.25 | 24.55 ± 2.51 | 22.4 ± 0.76 |
| MD10a | 4-OMe-3,4,5-OMe-a’ = b’ | 18.28 ± 1.14 | 20.57 ± 0.07 | 16.9 ± 4.7 | 17.4 ± 2.75 | 8.57 ± 0.38 | 21.65 ± 1.57 |
| MD11 | 3,4-OMe-3,4,5-OMe-b’-OH | 5.7 ± 0.28 | 14.7 ± 0.99 | 22.7 ± 0.35 | 20.44 ± 1.31 | 6.11 ± 0.38 | 5.32 ± 0.94 |
| MD11a | 3,4-OMe-3,4,5-OMe-a’ = b’ | 6.2 ± 0.03 | 8.8 ± 1.19 | 20.5 ± 1.42 | 21.47 ± 0.66 | 6.37 ± 0.02 | 5.99 ± 1.07 |
| MD12 | 3,4,5-OMe-3,4,5-OMe-b’-OH | 15.9 ± 3.77 | 22.6 ± 0.73 | >40 | >40 | 23.72 ± 0.44 | 19.38 ± 6.68 |
| MD12a | 3,4,5-OMe-3,4,5-OMe-a’ = b’ | 4.6 ± 0.8 | 4.2 ± 0.16 | 4.5 ± 0.12 | 5.63 ± 0.14 | 4.85 ± 0.3 | 4.49 ± 0.73 |
| MD13 | 3,4,5-OMe-4-OMe-a’ = b’-OH | 12.3 ± 0.92 | 18.7 ± 1.1 | 36.7 ± 1 | 33.63 ± 2.45 | 20.46 ± 1.8 | 9.09 ± 1.44 |
| Cells Treated with Different Concentrations of Compounds/Time Treatment | Cell Cycle Distribution (Mean ± SEMs) a | |||
|---|---|---|---|---|
| a | ||||
| MCF-7 (Compound 3)/24 h treatment | Sub-G1 (%) | G1 (%) | S (%) | G2/M (%) |
| control b | 8.67 ± 3.53 | 48.1 ± 3.53 | 17 ± 1.83 | 19.95 ± 6.85 |
| 5 μM | 6.335 ± 0.48 | 50.9 ± 0.98 | 12.55 ± 2.61 | 19.1 ± 4.10 |
| 10 μM | 7.955 ± 0.16 | 50.1 ± 3.39 | 12.9 ± 0.14 | 23.1 ± 10.88 |
| 20μM | 15.02 ± 10.57 | 50.8 ± 7.07 | 14.05 ± 5.02 | 14.125 ± 0.03 |
| MCF-7 (Compound 3)/48 h treatment | Sub-G1 (%) | G1 (%) | S (%) | G2/M (%) |
| control | 5.72 ± 5.08 | 63.73 ± 8.61 | 12.84 ± 2.7 | 15.56 ± 1.37 |
| 5 μM | 5.95 ± 4.91 | 63.43 ± 10.04 | 13.22 ± 5.04 | 14.6 ± 1.37 |
| 10 μM | 6.35 ± 1.97 | 60.13 ± 9.00 | 13.16 ± 0.77 | 17.66 ± 8.95 |
| 20 μM | 10.07 ± 5.69 | 60.26 ± 8.92 | 12.56 ± 2.66 | 14.96 ± 2.45 |
| b | ||||
| MCF-7 (MD12a)/24 h treatment | Sub-G1 (%) | G1 (%) | S (%) | G2/M (%) |
| Control | 8.67 ± 3.53 | 48.1 ± 3.53 | 17 ± 1.83 | 19.95 ± 6.85 |
| 5 μM | 14.35 ± 9.40 | 53.2 ± 17.18 | 8.8 ± 1.97 | 17 ± 3.81 |
| 10 μM | 50.3 ± 25.88 | 36.4 ± 17.88 | 14.25 ± 3.18 | 4.55 ± 4.16 |
| 20 μM | 85.4 ± 4.38 | 20.9 ± 4.17 | 0.25 ± 0.016 | 0.30 ± 0.29 |
| MCF-7 (MD12a)/48 h treatment | Sub-G1 (%) | G1 (%) | S (%) | G2/M (%) |
| Control | 5.72 ± 5.08 | 63.73 ± 8.61 | 12.84 ± 2.7 | 15.56 ± 1.37 |
| 5 μM | 28.06 ± 5.25 | 44.93 ± 6.50 | 18.03 ± 4.80 | 9.77 ± 6.05 |
| 10 μM | 65 ± 8.20 | 22.4 ± 9.00 | 11.44 ± 2.75 | 6.45 ± 9.58 |
| 20 μM | 98 ± 1.83 | 1.75 ± 1.78 | 0.21 ± 0.36 | 1.30 ± 2.16 |
| c | ||||
| HepG2 (Compound 3)/24 h treatment | Sub-G1 (%) | G1 (%) | S (%) | G2/M (%) |
| control | 1.39 ± 0.67 | 56.5 ± 6.1 | 13.73 ± 3.82 | 23.03 ± 4.66 |
| 5 μM | 2.47 ± 1.78 | 57.4 ± 3.95 | 12.83 ± 3.78 | 23.1 ± 5.04 |
| 10 μM | 0.90 ± 0.55 | 57.3 ± 4.04 | 14.54 ± 8.19 | 22.53 ± 6.01 |
| 20 μM | 0.96 ± 0.76 | 56.4 ± 3.74 | 13.01 ± 5.52 | 23.93 ± 5.16 |
| HepG2 (Compound 3)/48 h treatment | Sub-G1 (%) | G1 (%) | S (%) | G2/M (%) |
| control | 6.00 ± 3.86 | 55.56 ± 5.05 | 11.73 ± 3.32 | 21.63 ± 2.91 |
| 5 μM | 6.97 ± 6.86 | 53.23 ± 8.28 | 15.45 ± 8.86 | 19.3 ± 4.17 |
| 10 μM | 9.76 ± 11.72 | 50.56 ± 11.02 | 16.19 ± 8.62 | 18.23 ± 5.25 |
| 20 μM | 8.08 ± 7.54 | 53.86 ± 8.87 | 16.5 ± 8.85 | 17.26 ± 5.26 |
| d | ||||
| HepG2 (MD12a)/24 h treatment | Sub-G1 (%) | G1 (%) | S (%) | G2/M (%) |
| control | 1.39 ± 0.67 | 56.5 ± 6.1 | 13.73 ± 3.82 | 23.03 ± 4.66 |
| 5 μM | 1.06 ± 0.56 | 57.53 ± 4.90 | 14.22 ± 3.80 | 21.13 ± 3.81 |
| 10 μM | 1.79 ± 1.41 | 56.3 ± 7.13 | 14.66 ± 5.15 | 21.33 ± 3.97 |
| 20 μM | 3.90 ± 2.57 | 53.2 ± 3.26 | 17.16 ± 2.54 | 20.2 ± 4.15 |
| HepG2 (MD12a)/48 h treatment | Sub-G1 (%) | G1 (%) | S (%) | G2/M (%) |
| control | 6.00 ± 3.86 | 55.56 ± 5.05 | 11.73 ± 3.32 | 21.63 ± 2.91 |
| 5 μM | 13.35 ± 14.34 | 48.45 ± 1.20 | 18.9 ± 3.81 | 15.70 ± 9.75 |
| 10 μM | 26.95 ± 7.84 | 47.45 ± 7.42 | 15.2 ± 5.37 | 9.00 ± 5.50 |
| 20 μM | 75.95 ± 20.01 | 16.12 ± 9.72 | 6.14 ± 8.14 | 1.25 ± 1.33 |
| Item | Comparison | Up-Regulated Gene | Down-Regulated Gene |
|---|---|---|---|
| 1 | HepG2 (Comp.3)/HepG2 Control | 1089 | 729 |
| 2 | HepG2 (MD12a)/HepG2 Control | 796 | 1094 |
| 3 | MCF-7 (Comp.3)/MCF-7 Control | 1166 | 1524 |
| 4 | MCF-7 (MD12a)/MCF-7 Control | 2620 | 2193 |
| Gene Name | Primer Name | Primer Sequence |
|---|---|---|
| GADD45B | H1-GADD45B (H)-F | ACTTGGTTGGTCCTTGTCTGC |
| H1-GADD45B (H)-R | ACTGGGAGTTCATGGGTACAGA | |
| SESN2 | H2-SESN2 (H)-F | GGCATGGGGATCTGGGTTCTA |
| H2-SESN2 (H)-R | ATATGGAACTGGTAGCAAAGCC | |
| BBC3 | H15-BBC3 (H)-F | TGTGAATCCTGTGCTCTGCCC |
| H15-BBC3 (H)-R | GCTTCAGCCAAAATCTCCCAC | |
| GAPDH | GAPDH-F | CATGGAAGTATGACAACAGCCT |
| GAPDH-R | AGTCCTTCCACGATACCAAAGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-Y.; Lien, J.-C.; Chen, C.-Y.; Hung, C.-C.; Lin, H.-C. Design, Synthesis and Evaluation of Novel Derivatives of Curcuminoids with Cytotoxicity. Int. J. Mol. Sci. 2021, 22, 12171. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212171
Chen C-Y, Lien J-C, Chen C-Y, Hung C-C, Lin H-C. Design, Synthesis and Evaluation of Novel Derivatives of Curcuminoids with Cytotoxicity. International Journal of Molecular Sciences. 2021; 22(22):12171. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212171
Chicago/Turabian StyleChen, Chen-Yin, Jin-Cherng Lien, Chien-Yu Chen, Chin-Chuan Hung, and Hui-Chang Lin. 2021. "Design, Synthesis and Evaluation of Novel Derivatives of Curcuminoids with Cytotoxicity" International Journal of Molecular Sciences 22, no. 22: 12171. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212171