
The Fusion of CLEC12A and MIR223HG Arises from a trans-Splicing Event in Normal and Transformed Human Cells

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. CLEC12A-MIR223HG Is Expressed in CML Patients and Healthy Controls
2.2. CLEC12A-MIR223HG Is Expressed in Normal Human Tissues and Leukemic Cell Lines
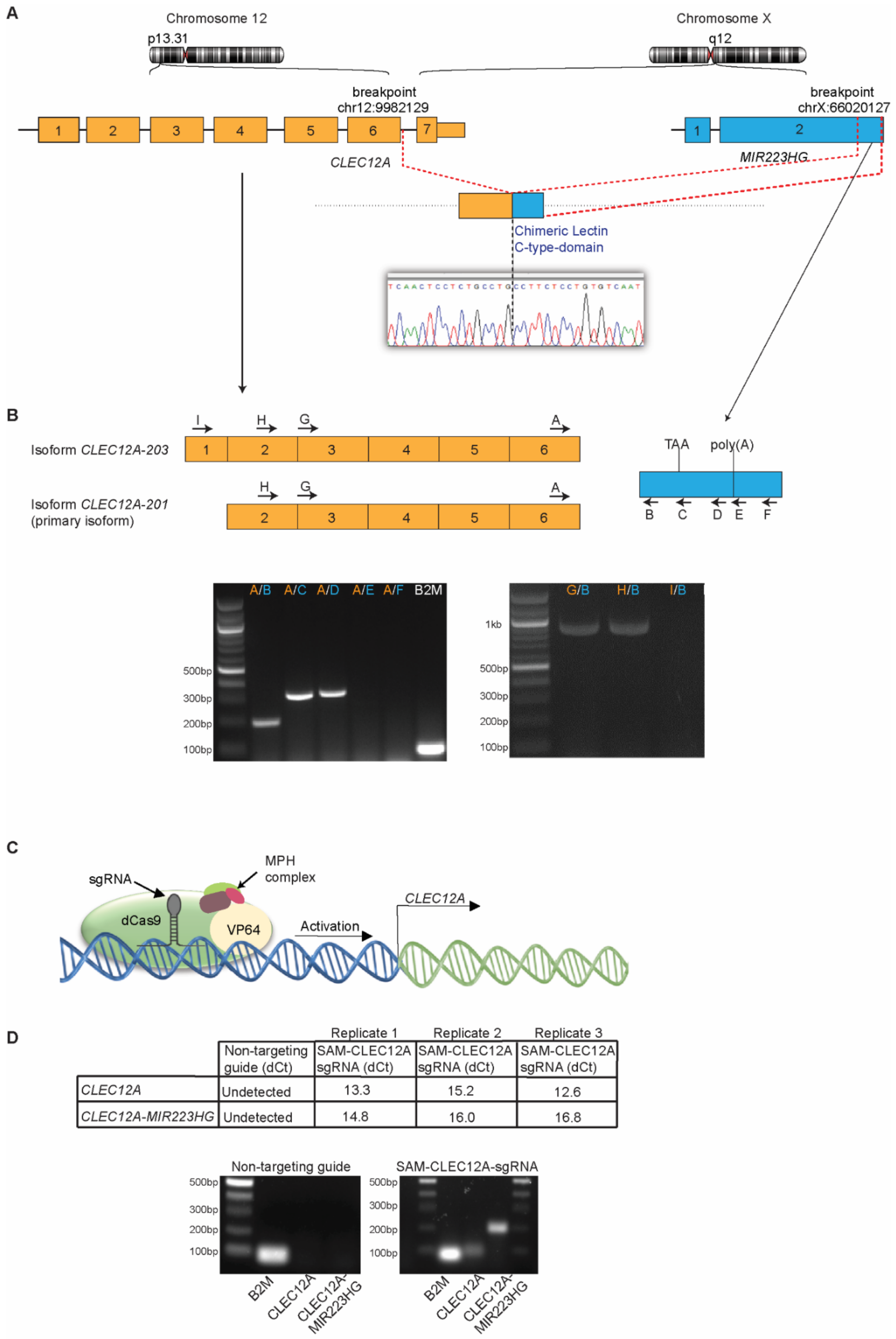
2.3. CLEC12A-MIR223HG Exhibits a Typical Fusion Transcript Architecture
2.4. CRISPR-Mediated Transcriptional Activation of CLEC12A Increases CLEC12A-MIR223HG Expression
2.5. CLEC12A-MIR223HG Encodes a Chimeric Protein That Is Distinct from CLEC12A
2.6. Increased CLEC12A-MIR223HG Expression following Cell Differentiation or Chemotherapy
3. Discussion
4. Materials and Methods
4.1. Clinical Samples and Bioinformatics Analysis
4.2. Cell Lines and Culture
4.3. Expression Vectors, Lentivirus Production, and Transduction
4.4. RNA Isolation and RT-PCR
4.5. Western Blotting
4.6. Immunofluorescence
4.7. Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS) Analysis
4.8. CRISPR-SAM-Mediated Activation of CLEC12A and Analysis of Chromosomal Translocations in HEK293 Cells
4.9. U-937 Monocyte Differentiation Assay
4.10. Cell Apoptosis with Annexin V Staining
4.11. Cell Viability Assay
4.12. Colony Formation-Methocult Assay
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jia, Y.; Xie, Z.; Li, H. Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer 2016, 2, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nambiar, M.; Kari, V.; Raghavan, S.C. Chromosomal translocations in cancer. Biochim. Biophys. Acta 2008, 1786, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apperley, J.F. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007, 8, 1018–1029. [Google Scholar] [CrossRef]
- Shaw, A.T.; Yeap, B.Y.; Solomon, B.J.; Riely, G.J.; Gainor, J.; Engelman, J.A.; Shapiro, G.I.; Costa, D.B.; Ou, S.H.; Butaney, M.; et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: A retrospective analysis. Lancet Oncol. 2011, 12, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Babiceanu, M.; Qin, F.; Xie, Z.; Jia, Y.; Lopez, K.; Janus, N.; Facemire, L.; Kumar, S.; Pang, Y.; Qi, Y.; et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016, 44, 2859–2872. [Google Scholar] [CrossRef] [Green Version]
- Velusamy, T.; Palanisamy, N.; Kalyana-Sundaram, S.; Sahasrabuddhe, A.A.; Maher, C.A.; Robinson, D.R.; Bahler, D.W.; Cornell, T.T.; Wilson, T.E.; Lim, M.S.; et al. Recurrent reciprocal RNA chimera involving YPEL5 and PPP1CB in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 3035–3040. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, J.; Ma, X.; Sklar, J. Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle 2009, 8, 218–222. [Google Scholar] [CrossRef]
- Lei, Q.; Li, C.; Zuo, Z.; Huang, C.; Cheng, H.; Zhou, R. Evolutionary Insights into RNA trans-Splicing in Vertebrates. Genome Biol. Evol. 2016, 8, 562–577. [Google Scholar] [CrossRef] [Green Version]
- Chwalenia, K.; Facemire, L.; Li, H. Chimeric RNAs in cancer and normal physiology. Wiley Interdiscip. Rev. RNA 2017, 8, e1427. [Google Scholar] [CrossRef]
- Nacu, S.; Yuan, W.; Kan, Z.; Bhatt, D.; Rivers, C.S.; Stinson, J.; Peters, B.A.; Modrusan, Z.; Jung, K.; Seshagiri, S.; et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genom. 2011, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Wang, L.; Wang, J.; Ittmann, M.M.; Li, W.; Yen, L. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9172–9177. [Google Scholar] [CrossRef] [Green Version]
- Annala, M.J.; Parker, B.C.; Zhang, W.; Nykter, M. Fusion genes and their discovery using high throughput sequencing. Cancer Lett. 2013, 340, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Qin, F.; Li, H. Chimeric RNAs and their implications in cancer. Curr. Opin. Genet. Dev. 2018, 48, 36–43. [Google Scholar] [CrossRef]
- Elfman, J.; Li, H. Chimeric RNA in Cancer and Stem Cell Differentiation. Stem Cells Int. 2018, 2018, 3178789. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Qin, F.; Kumar, S.; Elfman, J.; Lin, E.; Pham, L.P.; Yang, A.; Li, H. The landscape of chimeric RNAs in non-diseased tissues and cells. Nucleic Acids Res. 2020, 48, 1764–1778. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Singh, S.; Shi, X.; Xie, Z.; Lin, E.; Li, X.; Li, H. Functional heritage: The evolution of chimeric RNA into a gene. RNA Biol. 2020, 17, 125–134. [Google Scholar] [CrossRef]
- Qin, F.; Song, Z.; Babiceanu, M.; Song, Y.; Facemire, L.; Singh, R.; Adli, M.; Li, H. Discovery of CTCF-sensitive Cis-spliced fusion RNAs between adjacent genes in human prostate cells. PLoS Genet. 2015, 11, e1005001. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 2008, 321, 1357–1361. [Google Scholar] [CrossRef]
- Ma, X.; Wang, J.; Ma, C.X.; Gao, X.; Patriub, V.; Sklar, J.L. The JAZF1-SUZ12 fusion protein disrupts PRC2 complexes and impairs chromatin repression during human endometrial stromal tumorogenesis. Oncotarget 2017, 8, 4062–4078. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013, 3, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Schmitt-Ney, M.; Camussi, G. The PAX3-FOXO1 fusion protein present in rhabdomyosarcoma interferes with normal FOXO activity and the TGF-β pathway. PLoS ONE 2015, 10, e0121474. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Martín, B.; Palumbo, E.; Marco-Sola, S.; Griebel, T.; Ribeca, P.; Alonso, G.; Rastrojo, A.; Aguado, B.; Guigó, R.; Djebali, S. ChimPipe: Accurate detection of fusion genes and transcription-induced chimeras from RNA-seq data. BMC Genom. 2017, 18, 7. [Google Scholar] [CrossRef] [Green Version]
- Benelli, M.; Pescucci, C.; Marseglia, G.; Severgnini, M.; Torricelli, F.; Magi, A. Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 2012, 28, 3232–3239. [Google Scholar] [CrossRef] [Green Version]
- Davidson, N.M.; Majewski, I.J.; Oshlack, A. JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Li, H. Prediction, Characterization, and In Silico Validation of Chimeric RNAs. Methods Mol. Biol. 2020, 2079, 3–12. [Google Scholar] [CrossRef]
- Balamurali, D.; Gorohovski, A.; Detroja, R.; Palande, V.; Raviv-Shay, D.; Frenkel-Morgenstern, M. ChiTaRS 5.0: The comprehensive database of chimeric transcripts matched with druggable fusions and 3D chromatin maps. Nucleic Acids Res. 2020, 48, D825–D834. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, U.; Shah, J.S.; Dhungel, B.P.; Monteuuis, G.; Luu, P.L.; Petrova, V.; Metierre, C.; Nair, S.S.; Bailey, C.G.; Saunders, V.A.; et al. Widespread Aberrant Alternative Splicing despite Molecular Remission in Chronic Myeloid Leukaemia Patients. Cancers 2020, 12, 3738. [Google Scholar] [CrossRef]
- Bill, M.; B van Kooten Niekerk, P.; S Woll, P.; Laine Herborg, L.; Stidsholt Roug, A.; Hokland, P.; Nederby, L. Mapping the CLEC12A expression on myeloid progenitors in normal bone marrow; implications for understanding CLEC12A-related cancer stem cell biology. J. Cell. Mol. Med. 2018, 22, 2311–2318. [Google Scholar] [CrossRef] [Green Version]
- Toft-Petersen, M.; Nederby, L.; Kjeldsen, E.; Kerndrup, G.B.; Brown, G.D.; Hokland, P.; Stidsholt Roug, A. Unravelling the relevance of CLEC12A as a cancer stem cell marker in myelodysplastic syndrome. Br. J. Haematol. 2016, 175, 393–401. [Google Scholar] [CrossRef]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for acute myeloid leukemia therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.S.; Willment, J.A.; Pyz, E.; Dennehy, K.M.; Reid, D.M.; Dri, P.; Gordon, S.; Wong, S.Y.; Brown, G.D. Human MICL (CLEC12A) is differentially glycosylated and is down-regulated following cellular activation. Eur. J. Immunol. 2006, 36, 2159–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, K.; Castiñeiras-Vilariño, M.; Höckendorf, U.; Hannesschläger, N.; Lemeer, S.; Kupka, D.; Meyermann, S.; Lech, M.; Anders, H.J.; Kuster, B.; et al. Clec12a is an inhibitory receptor for uric acid crystals that regulates inflammation in response to cell death. Immunity 2014, 40, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukao, T.; Fukuda, Y.; Kiga, K.; Sharif, J.; Hino, K.; Enomoto, Y.; Kawamura, A.; Nakamura, K.; Takeuchi, T.; Tanabe, M. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell 2007, 129, 617–631. [Google Scholar] [CrossRef] [Green Version]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; De Marchis, M.L.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar] [CrossRef] [Green Version]
- Fatica, A.; Rosa, A.; Fazi, F.; Ballarino, M.; Morlando, M.; De Angelis, F.G.; Caffarelli, E.; Nervi, C.; Bozzoni, I. MicroRNAs and hematopoietic differentiation. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Song, Y.; Zhang, Y.; Facemire, L.; Frierson, H.; Li, H. Role of CTCF in Regulating SLC45A3-ELK4 Chimeric RNA. PLoS ONE 2016, 11, e0150382. [Google Scholar] [CrossRef]
- Fang, W.; Wei, Y.; Kang, Y.; Landweber, L.F. Detection of a common chimeric transcript between human chromosomes 7 and 16. Biol. Direct 2012, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Rickman, D.S.; Pflueger, D.; Moss, B.; VanDoren, V.E.; Chen, C.X.; de la Taille, A.; Kuefer, R.; Tewari, A.K.; Setlur, S.R.; Demichelis, F.; et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009, 69, 2734–2738. [Google Scholar] [CrossRef] [Green Version]
- Morsink, L.M.; Walter, R.B.; Ossenkoppele, G.J. Prognostic and therapeutic role of CLEC12A in acute myeloid leukemia. Blood Rev. 2019, 34, 26–33. [Google Scholar] [CrossRef]
- Li, K.; Neumann, K.; Duhan, V.; Namineni, S.; Hansen, A.L.; Wartewig, T.; Kurgyis, Z.; Holm, C.K.; Heikenwalder, M.; Lang, K.S.; et al. The uric acid crystal receptor Clec12A potentiates type I interferon responses. Proc. Natl. Acad. Sci. USA 2019, 116, 18544–18549. [Google Scholar] [CrossRef] [Green Version]
- Dintzis, S.M.; Velculescu, V.E.; Pfeffer, S.R. Receptor extracellular domains may contain trafficking information. Studies of the 300-kDa mannose 6-phosphate receptor. J. Biol. Chem. 1994, 269, 12159–12166. [Google Scholar] [CrossRef]
- Drake, M.T.; Shenoy, S.K.; Lefkowitz, R.J. Trafficking of G protein-coupled receptors. Circ. Res. 2006, 99, 570–582. [Google Scholar] [CrossRef]
- Luo, H.; Zhao, L.; Ji, X.; Zhang, X.; Jin, Y.; Liu, W. Glycosylation affects the stability and subcellular distribution of human PAT1 protein. FEBS Lett. 2017, 591, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Marshall, A.S.; Willment, J.A.; Lin, H.H.; Williams, D.L.; Gordon, S.; Brown, G.D. Identification and characterization of a novel human myeloid inhibitory C-type lectin-like receptor (MICL) that is predominantly expressed on granulocytes and monocytes. J. Biol. Chem. 2004, 279, 14792–14802. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, S.; Xiao, W.; Li, W.; Wang, L.; Yang, S.; Wang, W.; Xu, L.; Liao, S.; Liu, W.; et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef] [Green Version]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Fröhlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021, 31, 448–460. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.T.; Quinlan, A.R.; Hall, I.M. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat. Methods 2015, 12, 966–968. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhungel, B.P.; Monteuuis, G.; Giardina, C.; Tabar, M.S.; Feng, Y.; Metierre, C.; Ho, S.; Nagarajah, R.; Fontaine, A.R.M.; Shah, J.S.; et al. The Fusion of CLEC12A and MIR223HG Arises from a trans-Splicing Event in Normal and Transformed Human Cells. Int. J. Mol. Sci. 2021, 22, 12178. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212178
Dhungel BP, Monteuuis G, Giardina C, Tabar MS, Feng Y, Metierre C, Ho S, Nagarajah R, Fontaine ARM, Shah JS, et al. The Fusion of CLEC12A and MIR223HG Arises from a trans-Splicing Event in Normal and Transformed Human Cells. International Journal of Molecular Sciences. 2021; 22(22):12178. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212178
Chicago/Turabian StyleDhungel, Bijay P., Geoffray Monteuuis, Caroline Giardina, Mehdi S. Tabar, Yue Feng, Cynthia Metierre, Sarah Ho, Rajini Nagarajah, Angela R. M. Fontaine, Jaynish S. Shah, and et al. 2021. "The Fusion of CLEC12A and MIR223HG Arises from a trans-Splicing Event in Normal and Transformed Human Cells" International Journal of Molecular Sciences 22, no. 22: 12178. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212178