
Early-Life Adversity Leaves Its Imprint on the Oral Microbiome for More Than 20 Years and Is Associated with Long-Term Immune Changes

 ,
,
Abstract
:1. Introduction
2. Results
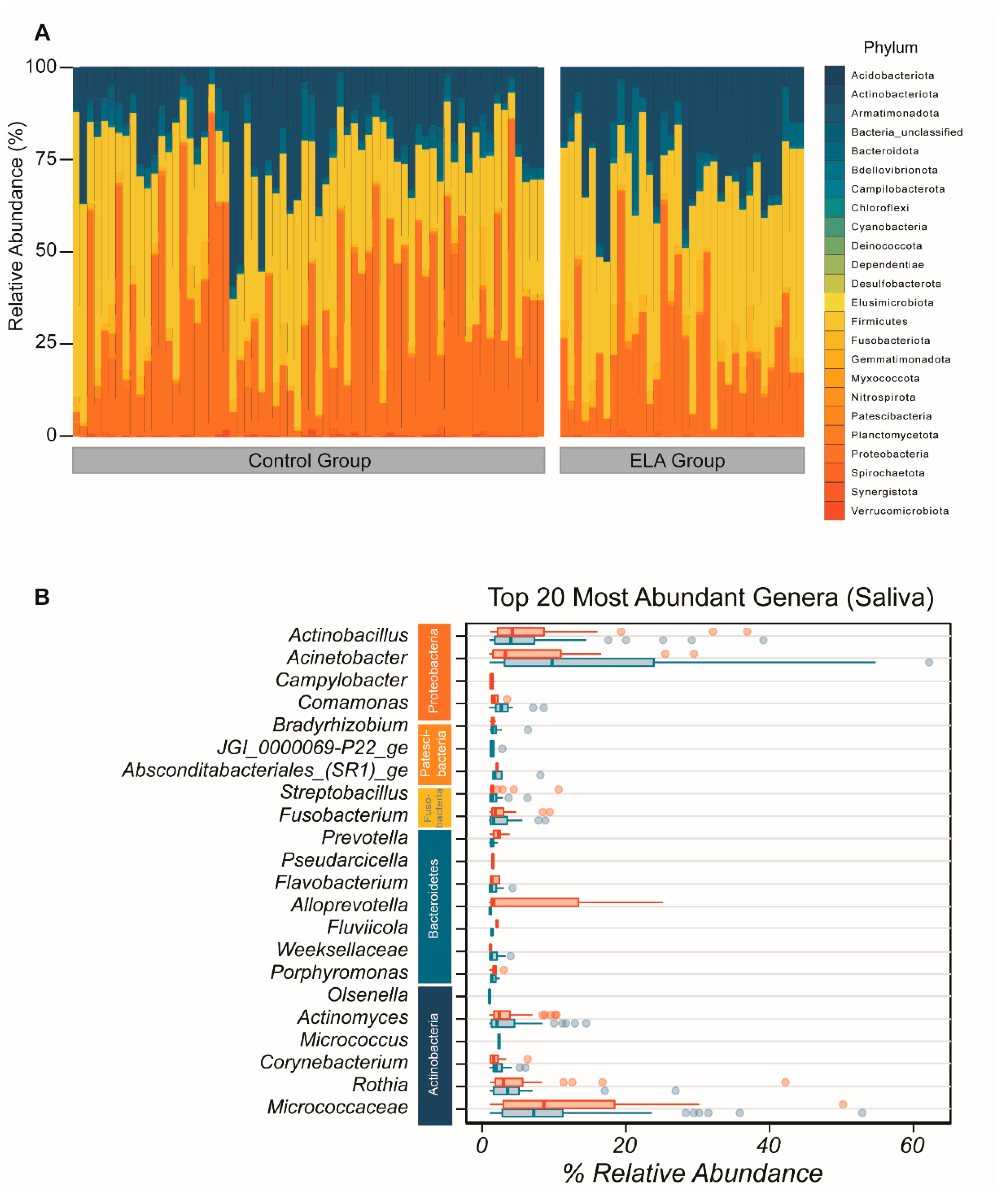
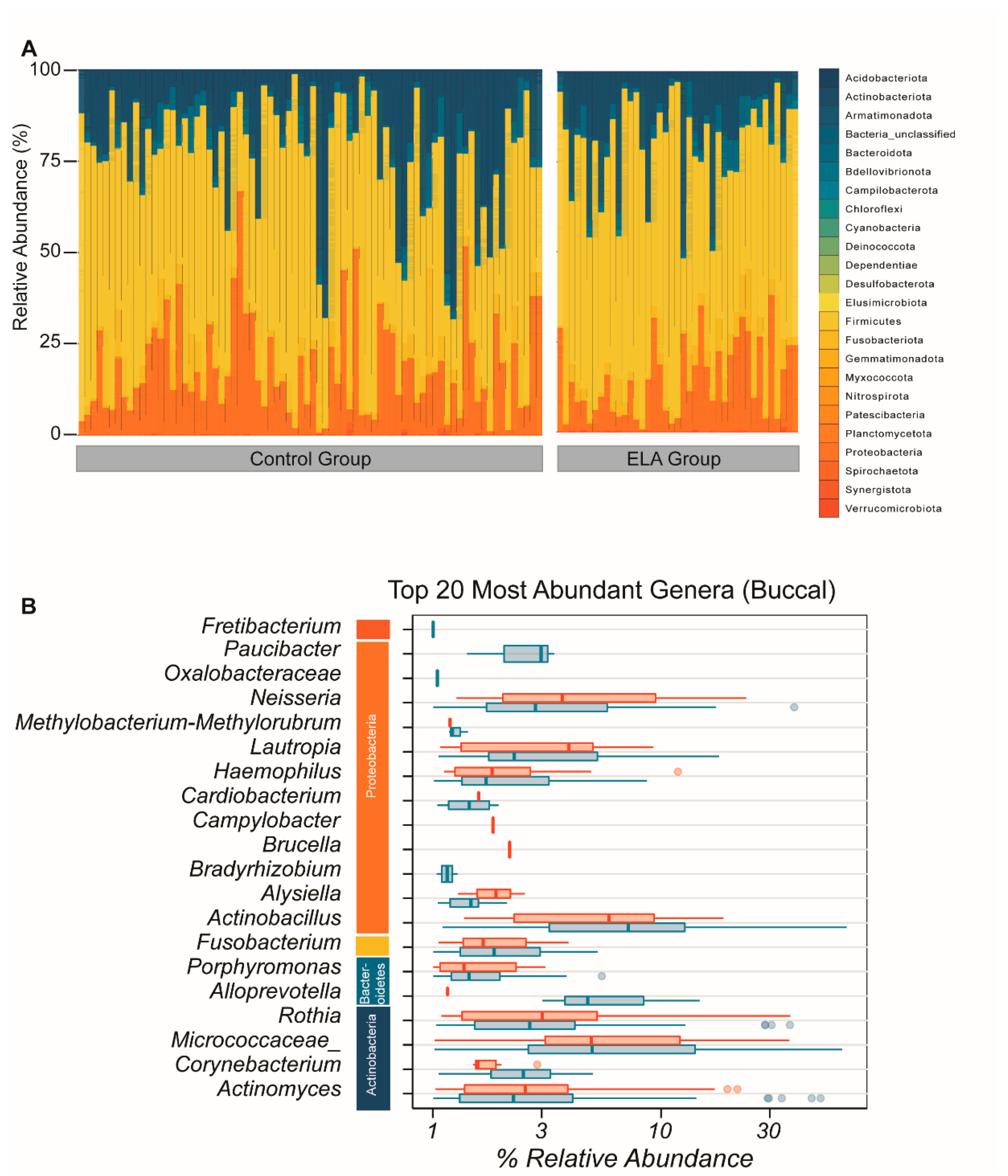
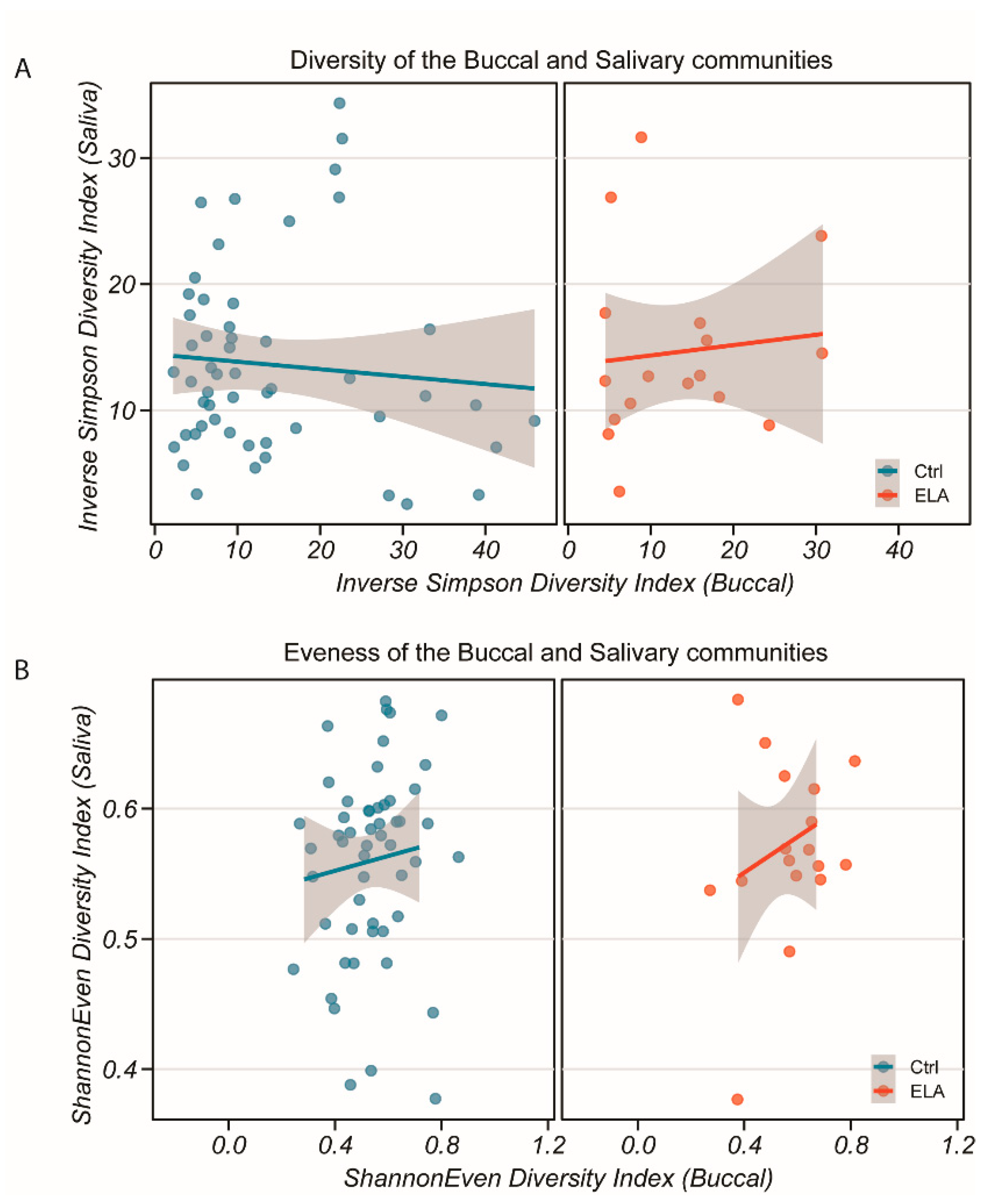
2.1. Microbial Diversity and Overall Microbial Composition
2.1.1. Salivary Microbiome (α- and β-Diversity)
2.1.2. Buccal Microbiome (α- and β-Diversity)
2.1.3. Salivary and Buccal Microbiomes Are Two Separate Entities
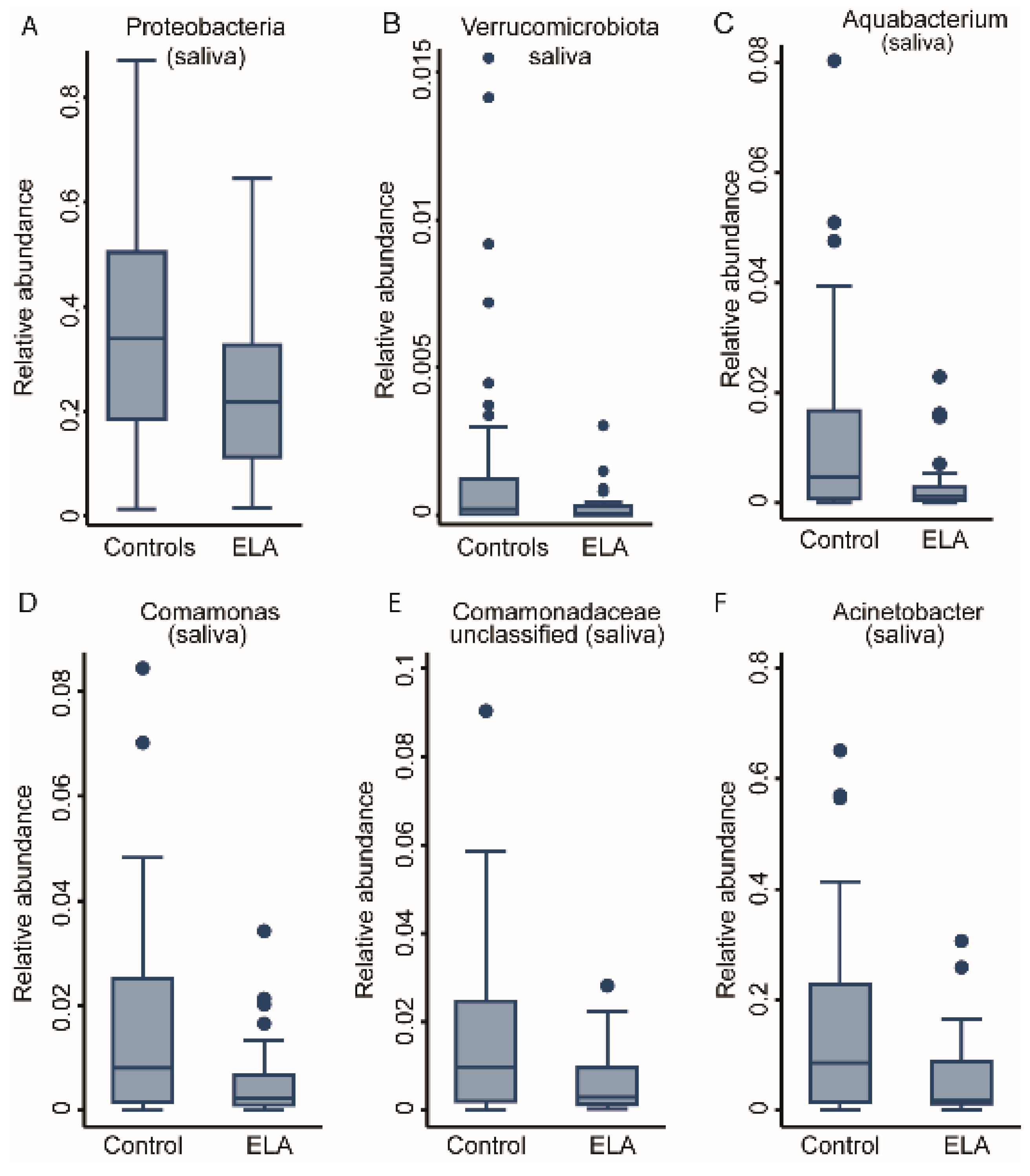
2.2. ELA Induces Differences in Specific Taxa in Both Salivary and Buccal Communities
2.3. Environmental Covariates
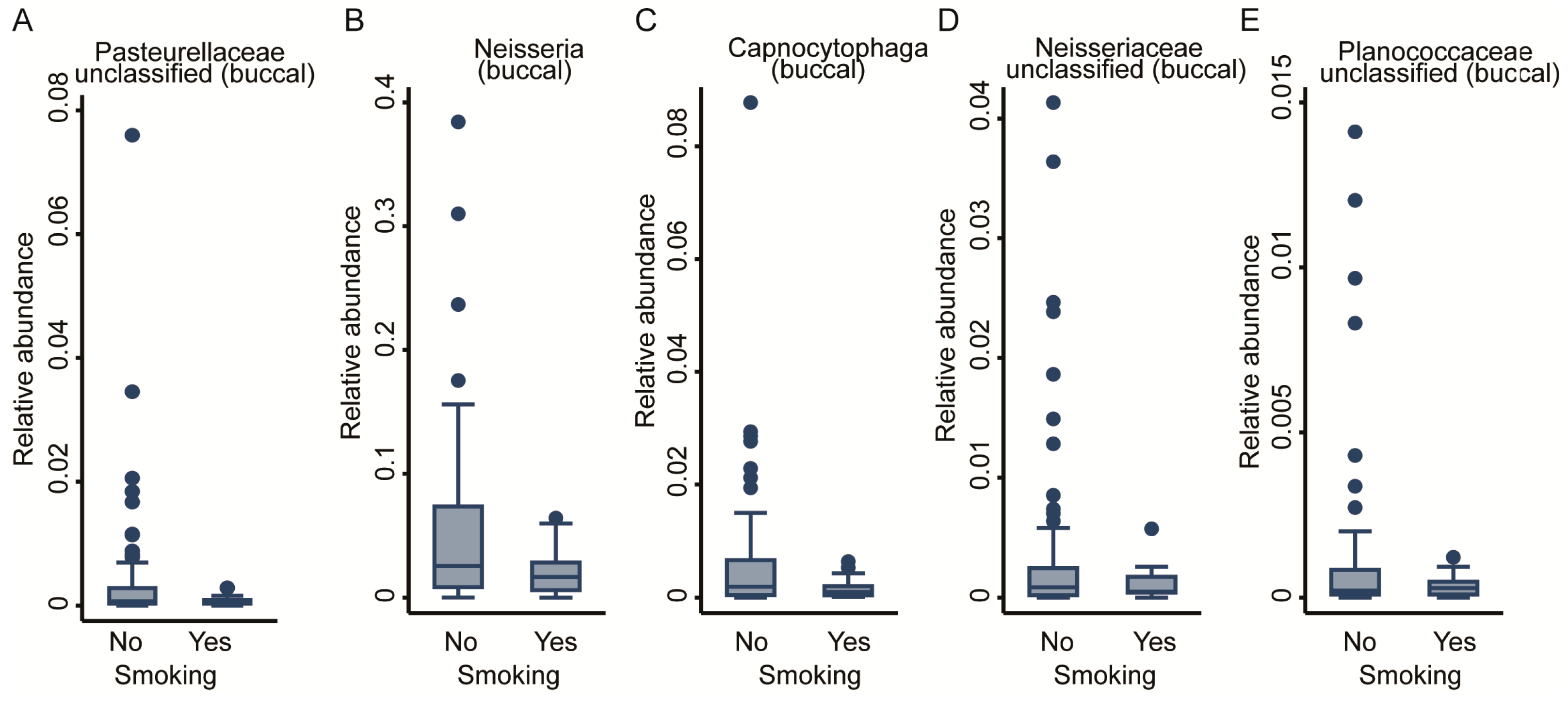
2.3.1. Smoking
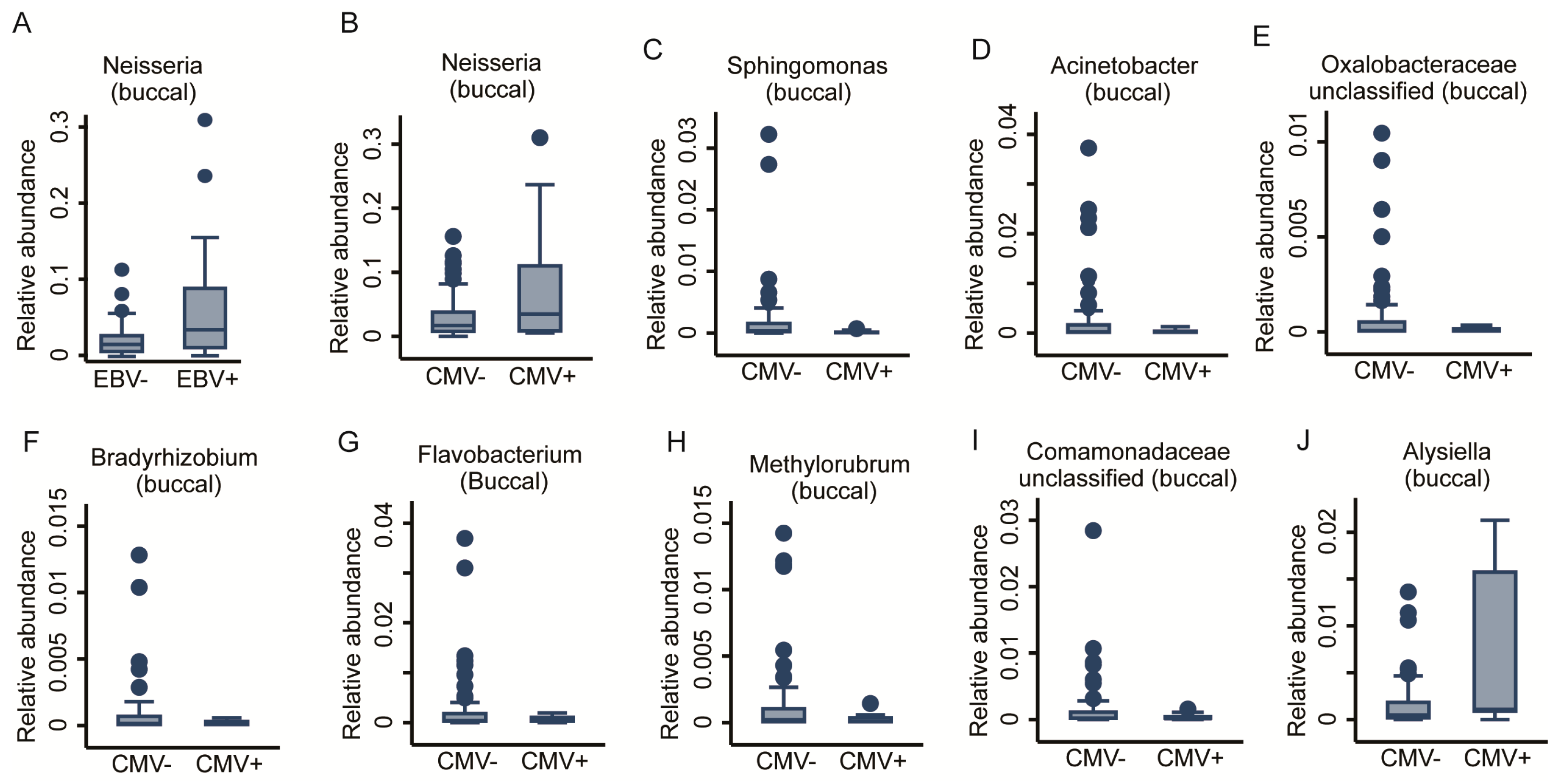
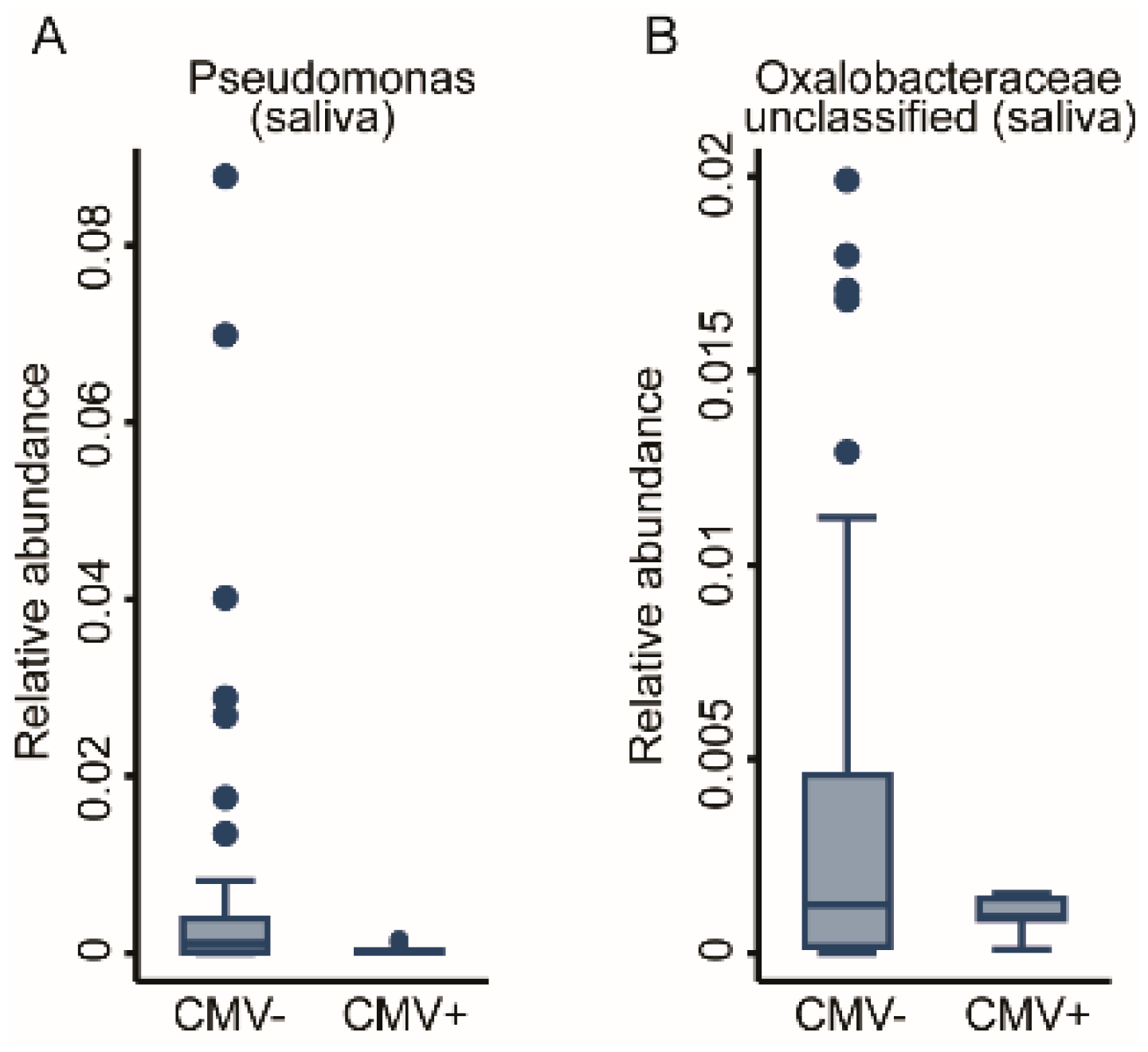
2.3.2. Prior Viral Infections
2.4. Fractional Regression Models of the Immune–Microbiome Interactions
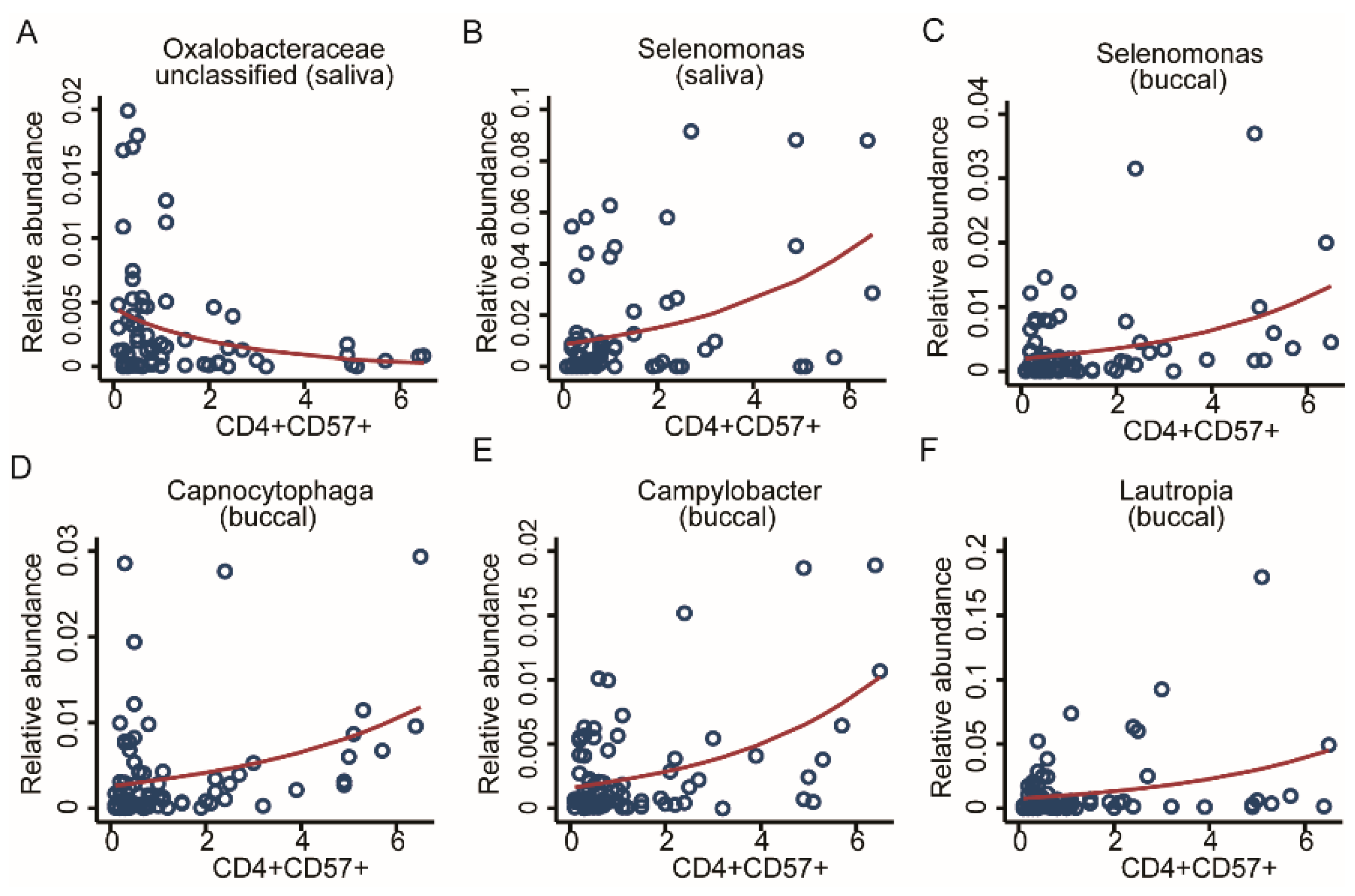
2.4.1. Association with CD4 T-Cell Immunosenescence
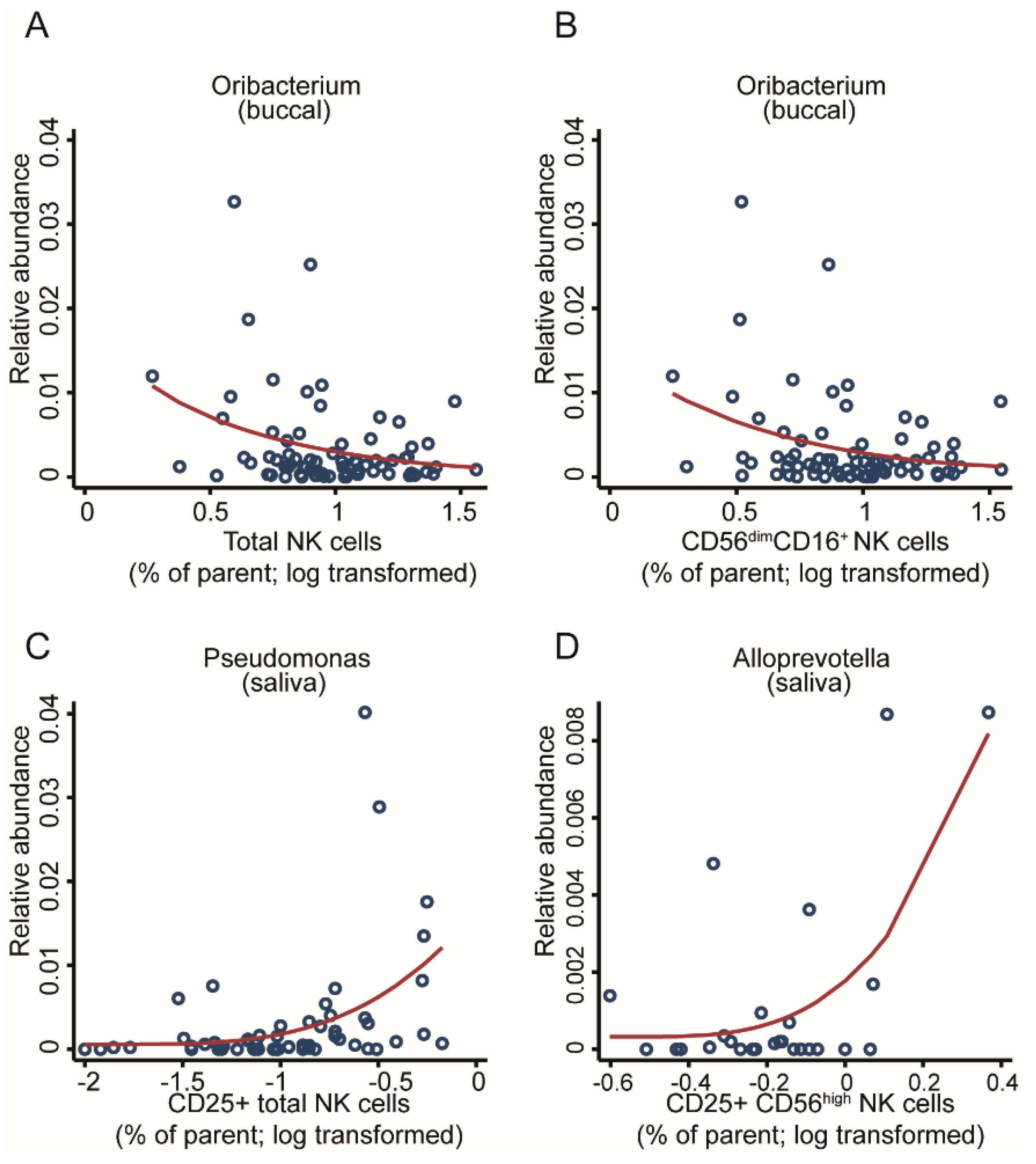
2.4.2. Association with NK Cell Activity
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grova, N.; Schroeder, H.; Olivier, J.L.; Turner, J.D. Epigenetic and Neurological Impairments Associated with Early Life Exposure to Persistent Organic Pollutants. Int. J. Genom. 2019, 2019, 2085496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.D. Holistic, personalized, immunology? The effects of socioeconomic status on the transcriptional milieu of immune cells. Pediatr. Pulmonol. 2018, 53, 696–697. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986, 1, 1077–1081. [Google Scholar] [CrossRef]
- Dube, S.R.; Fairweather, D.; Pearson, W.S.; Felitti, V.J.; Anda, R.F.; Croft, J.B. Cumulative childhood stress and autoimmune diseases in adults. Psychosom. Med. 2009, 71, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Wampach, L.; Heintz-Buschart, A.; Fritz, J.V.; Ramiro-Garcia, J.; Habier, J.; Herold, M.; Narayanasamy, S.; Kaysen, A.; Hogan, A.H.; Bindl, L.; et al. Birth mode is associated with earliest strain-conferred gut microbiome functions and immunostimulatory potential. Nat. Commun. 2018, 9, 5091. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Forster, S.C.; Tsaliki, E.; Vervier, K.; Strang, A.; Simpson, N.; Kumar, N.; Stares, M.D.; Rodger, A.; Brocklehurst, P.; et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 2019, 574, 117–121. [Google Scholar] [CrossRef]
- Reyman, M.; van Houten, M.A.; van Baarle, D.; Bosch, A.; Man, W.H.; Chu, M.; Arp, K.; Watson, R.L.; Sanders, E.A.M.; Fuentes, S.; et al. Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat. Commun. 2019, 10, 4997. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, A.; Yoo, J.Y.; Valeria Ozorio Dutra, S.; Morgan, K.H.; Groer, M. The Association between Early-Life Gut Microbiota and Long-Term Health and Diseases. J. Clin. Med. 2021, 10, 459. [Google Scholar] [CrossRef]
- Yang, I.; Corwin, E.J.; Brennan, P.A.; Jordan, S.; Murphy, J.R.; Dunlop, A. The Infant Microbiome: Implications for Infant Health and Neurocognitive Development. Nurs. Res. 2016, 65, 76–88. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.E.; Townsend, S.D. Temporal development of the infant gut microbiome. Open Biol. 2019, 9, 190128. [Google Scholar] [CrossRef] [Green Version]
- Backhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, M.; Raikkonen, K.; Eriksson, J.G. Early life stress and later health outcomes—Findings from the Helsinki Birth Cohort Study. Am. J. Hum. Biol. 2014, 26, 111–116. [Google Scholar] [CrossRef]
- Spitzer, C.; Wegert, S.; Wollenhaupt, J.; Wingenfeld, K.; Barnow, S.; Grabe, H.J. Gender-specific association between childhood trauma and rheumatoid arthritis: A case-control study. J. Psychosom. Res. 2013, 74, 296–300. [Google Scholar] [CrossRef]
- Tomasdottir, M.O.; Sigurdsson, J.A.; Petursson, H.; Kirkengen, A.L.; Krokstad, S.; McEwen, B.; Hetlevik, I.; Getz, L. Self Reported Childhood Difficulties, Adult Multimorbidity and Allostatic Load. A Cross-Sectional Analysis of the Norwegian HUNT Study. PLoS ONE 2015, 10, e0130591. [Google Scholar] [CrossRef] [Green Version]
- Gern, J.E.; Visness, C.M.; Gergen, P.J.; Wood, R.A.; Bloomberg, G.R.; O’Connor, G.T.; Kattan, M.; Sampson, H.A.; Witter, F.R.; Sandel, M.T.; et al. The Urban Environment and Childhood Asthma (URECA) birth cohort study: Design, methods, and study population. BMC Pulm. Med. 2009, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Herzog, J.I.; Schmahl, C. Adverse Childhood Experiences and the Consequences on Neurobiological, Psychosocial, and Somatic Conditions across the Lifespan. Front. Psychiatry 2018, 9, 420. [Google Scholar] [CrossRef]
- Mansuri, F.; Nash, M.C.; Bakour, C.; Kip, K. Adverse Childhood Experiences (ACEs) and Headaches among Children: A Cross-Sectional Analysis. Headache 2020, 60, 735–744. [Google Scholar] [CrossRef]
- Elwenspoek, M.M.C.; Kuehn, A.; Muller, C.P.; Turner, J.D. The effects of early life adversity on the immune system. Psychoneuroendocrinology 2017, 82, 140–154. [Google Scholar] [CrossRef]
- Elwenspoek, M.M.C.; Hengesch, X.; Leenen, F.A.D.; Sias, K.; Fernandes, S.B.; Schaan, V.K.; Meriaux, S.B.; Schmitz, S.; Bonnemberger, F.; Schachinger, H.; et al. Glucocorticoid receptor signaling in leukocytes after early life adversity. Dev. Psychopathol. 2020, 32, 853–863. [Google Scholar] [CrossRef]
- Holland, J.F.; Khandaker, G.M.; Dauvermann, M.R.; Morris, D.; Zammit, S.; Donohoe, G. Effects of early life adversity on immune function and cognitive performance: Results from the ALSPAC cohort. Soc. Psychiatry Psychiatr. Epidemiol. 2020, 55, 723–733. [Google Scholar] [CrossRef]
- Reid, B.M.; Coe, C.L.; Doyle, C.M.; Sheerar, D.; Slukvina, A.; Donzella, B.; Gunnar, M.R. Persistent skewing of the T-cell profile in adolescents adopted internationally from institutional care. Brain Behav. Immun. 2019, 77, 168–177. [Google Scholar] [CrossRef]
- Elwenspoek, M.M.C.; Sias, K.; Hengesch, X.; Schaan, V.K.; Leenen, F.A.D.; Adams, P.; Meriaux, S.B.; Schmitz, S.; Bonnemberger, F.; Ewen, A.; et al. T Cell Immunosenescence after Early Life Adversity: Association with Cytomegalovirus Infection. Front. Immunol. 2017, 8, 1263. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, C.M.; White, M.J.; Goodier, M.R.; Riley, E.M. Functional Significance of CD57 Expression on Human NK Cells and Relevance to Disease. Front. Immunol. 2013, 4, 422. [Google Scholar] [CrossRef] [Green Version]
- Judge, S.J.; Murphy, W.J.; Canter, R.J. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front. Cell Infect. Microbiol. 2020, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Della Chiesa, M.; Falco, M.; Podesta, M.; Locatelli, F.; Moretta, L.; Frassoni, F.; Moretta, A. Phenotypic and functional heterogeneity of human NK cells developing after umbilical cord blood transplantation: A role for human cytomegalovirus? Blood 2012, 119, 399–410. [Google Scholar] [CrossRef]
- Lopez-Verges, S.; Milush, J.M.; Schwartz, B.S.; Pando, M.J.; Jarjoura, J.; York, V.A.; Houchins, J.P.; Miller, S.; Kang, S.M.; Norris, P.J.; et al. Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 14725–14732. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Xie, L.; Li, Y.; Wei, C. More than 9,000,000 unique genes in human gut bacterial community: Estimating gene numbers inside a human body. PLoS ONE 2009, 4, e6074. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Alammar, N.; Singh, R.; Nanavati, J.; Song, Y.; Chaudhary, R.; Mullin, G.E. Gut Microbial Dysbiosis in the Irritable Bowel Syndrome: A Systematic Review and Meta-Analysis of Case-Control Studies. J. Acad. Nutr. Diet. 2020, 120, 565–586. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.B.; Keating, D.J.; Young, R.L.; Wong, M.L.; Licinio, J.; Wesselingh, S. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.E.; Chen, E.; Shalowitz, M.U.; Story, R.E.; Leigh, A.K.K.; Ham, P.; Arevalo, J.M.G.; Cole, S.W. Divergent transcriptional profiles in pediatric asthma patients of low and high socioeconomic status. Pediatr. Pulmonol. 2018, 53, 710–719. [Google Scholar] [CrossRef]
- Zijlmans, M.A.; Korpela, K.; Riksen-Walraven, J.M.; de Vos, W.M.; de Weerth, C. Maternal prenatal stress is associated with the infant intestinal microbiota. Psychoneuroendocrinology 2015, 53, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Alcon-Giner, C.; Dalby, M.J.; Caim, S.; Ketskemety, J.; Shaw, A.; Sim, K.; Lawson, M.A.E.; Kiu, R.; Leclaire, C.; Chalklen, L.; et al. Microbiota Supplementation with Bifidobacterium and Lactobacillus Modifies the Preterm Infant Gut Microbiota and Metabolome: An Observational Study. Cell Rep. Med. 2020, 1, 100077. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, C.; Krolick, K.N.; Shi, H.; Zhu, J. Difference in post-stress recovery of the gut microbiome and its altered metabolism after chronic adolescent stress in rats. Sci. Rep. 2020, 10, 3950. [Google Scholar] [CrossRef] [Green Version]
- Toussaint, L.; Shields, G.S.; Dorn, G.; Slavich, G.M. Effects of lifetime stress exposure on mental and physical health in young adulthood: How stress degrades and forgiveness protects health. J. Health Psychol. 2016, 21, 1004–1014. [Google Scholar] [CrossRef] [Green Version]
- Duran-Pinedo, A.E.; Solbiati, J.; Frias-Lopez, J. The effect of the stress hormone cortisol on the metatranscriptome of the oral microbiome. NPJ Biofilms Microbiomes 2018, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Boustedt, K.; Roswall, J.; Dahlen, G.; Dahlgren, J.; Twetman, S. Salivary microflora and mode of delivery: A prospective case control study. BMC Oral Health 2015, 15, 155. [Google Scholar] [CrossRef] [Green Version]
- Chu, D.M.; Ma, J.; Prince, A.L.; Antony, K.M.; Seferovic, M.D.; Aagaard, K.M. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 2017, 23, 314–326. [Google Scholar] [CrossRef] [Green Version]
- Mason, M.R.; Chambers, S.; Dabdoub, S.M.; Thikkurissy, S.; Kumar, P.S. Characterizing oral microbial communities across dentition states and colonization niches. Microbiome 2018, 6, 67. [Google Scholar] [CrossRef]
- Mark Welch, J.L.; Dewhirst, F.E.; Borisy, G.G. Biogeography of the Oral Microbiome: The Site-Specialist Hypothesis. Annu. Rev. Microbiol. 2019, 73, 335–358. [Google Scholar] [CrossRef]
- Carpenter, G.H. Salivary Factors that Maintain the Normal Oral Commensal Microflora. J. Dent. Res. 2020, 99, 644–649. [Google Scholar] [CrossRef]
- David, L.A.; Materna, A.C.; Friedman, J.; Campos-Baptista, M.I.; Blackburn, M.C.; Perrotta, A.; Erdman, S.E.; Alm, E.J. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 2014, 15, R89. [Google Scholar] [CrossRef] [Green Version]
- Shaw, L.; Ribeiro, A.L.R.; Levine, A.P.; Pontikos, N.; Balloux, F.; Segal, A.W.; Roberts, A.P.; Smith, A.M. The Human Salivary Microbiome Is Shaped by Shared Environment Rather than Genetics: Evidence from a Large Family of Closely Related Individuals. mBio 2017, 8, e01237-17. [Google Scholar] [CrossRef] [Green Version]
- Marsh, P.D. Dental plaque as a biofilm and a microbial community—Implications for health and disease. BMC Oral Health 2006, 6 (Suppl. S1), S14. [Google Scholar] [CrossRef] [Green Version]
- Zaura, E.; Brandt, B.W.; Teixeira de Mattos, M.J.; Buijs, M.J.; Caspers, M.P.; Rashid, M.U.; Weintraub, A.; Nord, C.E.; Savell, A.; Hu, Y.; et al. Same Exposure but Two Radically Different Responses to Antibiotics: Resilience of the Salivary Microbiome versus Long-Term Microbial Shifts in Feces. mBio 2015, 6, e01693-15. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.; Peura, S.; Hammar, U.; Vicenzi, S.; Hedman, A.; Almqvist, C.; Andolf, E.; Pershagen, G.; Dicksved, J.; Bertilsson, S.; et al. Oral Microbiota Development in Early Childhood. Sci. Rep. 2019, 9, 19025. [Google Scholar] [CrossRef]
- Abeles, S.R.; Jones, M.B.; Santiago-Rodriguez, T.M.; Ly, M.; Klitgord, N.; Yooseph, S.; Nelson, K.E.; Pride, D.T. Microbial diversity in individuals and their household contacts following typical antibiotic courses. Microbiome 2016, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Almeida-Santos, A.; Martins-Mendes, D.; Gaya-Vidal, M.; Perez-Pardal, L.; Beja-Pereira, A. Characterization of the Oral Microbiome of Medicated Type-2 Diabetes Patients. Front. Microbiol. 2021, 12, 610370. [Google Scholar] [CrossRef]
- Mukherjee, C.; Moyer, C.O.; Steinkamp, H.M.; Hashmi, S.B.; Beall, C.J.; Guo, X.; Ni, A.; Leys, E.J.; Griffen, A.L. Acquisition of oral microbiota is driven by environment, not host genetics. Microbiome 2021, 9, 54. [Google Scholar] [CrossRef]
- Jakubovics, N.S. Saliva as the Sole Nutritional Source in the Development of Multispecies Communities in Dental Plaque. Microbiol. Spectr. 2015, 3, 263–277. [Google Scholar] [CrossRef] [Green Version]
- Elwenspoek, M.M.C.; Hengesch, X.; Leenen, F.A.D.; Schritz, A.; Sias, K.; Schaan, V.K.; Meriaux, S.B.; Schmitz, S.; Bonnemberger, F.; Schachinger, H.; et al. Proinflammatory T Cell Status Associated with Early Life Adversity. J. Immunol. 2017, 199, 4046–4055. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, S.B.; Patil, N.D.; Meriaux, S.B.; Theresine, M.; Leenen, F.A.D.; Elwenspoek, M.M.C.; Zimmer, J.; Turner, J.D. Unbiased Screening Identifies Functional Differences in NK Cells after Early Life Psycho-Social Stress. Front. Immunol. 2021, 12, 674532. [Google Scholar] [CrossRef]
- Reid, B.M.; Horne, R.; Donzella, B.; Szamosi, J.C.; Coe, C.L.; Foster, J.A.; Gunnar, M.R. Microbiota-immune alterations in adolescents following early life adversity: A proof of concept study. Dev. Psychobiol. 2021, 63, 851–863. [Google Scholar] [CrossRef]
- Prodan, A.; Levin, E.; Nieuwdorp, M. Does disease start in the mouth, the gut or both? eLife 2019, 8, e45931. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.S.; Hayward, M.R.; Coelho, L.P.; Li, S.S.; Costea, P.I.; Voigt, A.Y.; Wirbel, J.; Maistrenko, O.M.; Alves, R.J.; Bergsten, E.; et al. Extensive transmission of microbes along the gastrointestinal tract. Elife 2019, 8, e42693. [Google Scholar] [CrossRef]
- Utter, D.R.; Mark Welch, J.L.; Borisy, G.G. Individuality, Stability, and Variability of the Plaque Microbiome. Front. Microbiol. 2016, 7, 564. [Google Scholar] [CrossRef]
- Belstrom, D.; Holmstrup, P.; Bardow, A.; Kokaras, A.; Fiehn, N.E.; Paster, B.J. Temporal Stability of the Salivary Microbiota in Oral Health. PLoS ONE 2016, 11, e0147472. [Google Scholar] [CrossRef]
- Stahringer, S.S.; Clemente, J.C.; Corley, R.P.; Hewitt, J.; Knights, D.; Walters, W.A.; Knight, R.; Krauter, K.S. Nurture trumps nature in a longitudinal survey of salivary bacterial communities in twins from early adolescence to early adulthood. Genome Res. 2012, 22, 2146–2152. [Google Scholar] [CrossRef] [Green Version]
- Gomez, A.; Espinoza, J.L.; Harkins, D.M.; Leong, P.; Saffery, R.; Bockmann, M.; Torralba, M.; Kuelbs, C.; Kodukula, R.; Inman, J.; et al. Host Genetic Control of the Oral Microbiome in Health and Disease. Cell Host Microbe 2017, 22, 269–278.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Premaraj, T.S.; Vella, R.; Chung, J.; Lin, Q.; Panier, H.; Underwood, K.; Premaraj, S.; Zhou, Y. Ethnic variation of oral microbiota in children. Sci. Rep. 2020, 10, 14788. [Google Scholar] [CrossRef] [PubMed]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, K.M.; Gulati, A.S. The “Gum-Gut” Axis in Inflammatory Bowel Diseases: A Hypothesis-Driven Review of Associations and Advances. Front. Immunol. 2021, 12, 620124. [Google Scholar] [CrossRef]
- Gomez, A.; Nelson, K.E. The Oral Microbiome of Children: Development, Disease, and Implications beyond Oral Health. Microb. Ecol. 2017, 73, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Kolltveit, K.M.; Tronstad, L.; Olsen, I. Systemic diseases caused by oral infection. Clin. Microbiol. Rev. 2000, 13, 547–558. [Google Scholar] [CrossRef]
- Li, S.S.; Zhu, A.; Benes, V.; Costea, P.I.; Hercog, R.; Hildebrand, F.; Huerta-Cepas, J.; Nieuwdorp, M.; Salojarvi, J.; Voigt, A.Y.; et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 2016, 352, 586–589. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Bouskra, D.; Brezillon, C.; Berard, M.; Werts, C.; Varona, R.; Boneca, I.G.; Eberl, G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008, 456, 507–510. [Google Scholar] [CrossRef]
- Bry, L.; Falk, P.G.; Midtvedt, T.; Gordon, J.I. A model of host-microbial interactions in an open mammalian ecosystem. Science 1996, 273, 1380–1383. [Google Scholar] [CrossRef]
- Henriksen, R.E.; Thuen, F. Marital Quality and Stress in Pregnancy Predict the Risk of Infectious Disease in the Offspring: The Norwegian Mother and Child Cohort Study. PLoS ONE 2015, 10, e0137304. [Google Scholar] [CrossRef] [Green Version]
- Fragkou, P.C.; Karaviti, D.; Zemlin, M.; Skevaki, C. Impact of Early Life Nutrition on Children’s Immune System and Noncommunicable Diseases Through Its Effects on the Bacterial Microbiome, Virome and Mycobiome. Front. Immunol. 2021, 12, 644269. [Google Scholar] [CrossRef]
- Ong, S.; Rose, N.R.; Cihakova, D. Natural killer cells in inflammatory heart disease. Clin. Immunol. 2017, 175, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Day, J.; Souza-Fonseca Guimaraes, F.; Wicks, I.P.; Louis, C. Natural killer cells in inflammatory autoimmune diseases. Clin. Transl. Immunol. 2021, 10, e1250. [Google Scholar] [CrossRef]
- Poli, A.; Michel, T.; Patil, N.; Zimmer, J. Revisiting the Functional Impact of NK Cells. Trends Immunol. 2018, 39, 460–472. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dahling, S.; Kastenmuller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8(+) T Cells. Immunity 2019, 51, 285–297. [Google Scholar] [CrossRef]
- Souza-Fonseca-Guimaraes, F.; Adib-Conquy, M.; Cavaillon, J.M. Natural killer (NK) cells in antibacterial innate immunity: Angels or devils? Mol. Med. 2012, 18, 270–285. [Google Scholar] [CrossRef]
- Theresine, M.; Patil, N.D.; Zimmer, J. Airway Natural Killer Cells and Bacteria in Health and Disease. Front. Immunol. 2020, 11, 585048. [Google Scholar] [CrossRef]
- Przemska-Kosicka, A.; Childs, C.E.; Maidens, C.; Dong, H.; Todd, S.; Gosney, M.A.; Tuohy, K.M.; Yaqoob, P. Age-Related Changes in the Natural Killer Cell Response to Seasonal Influenza Vaccination Are Not Influenced by a Synbiotic: A Randomised Controlled Trial. Front. Immunol. 2018, 9, 591. [Google Scholar] [CrossRef]
- Lee, J.Y.; Divaris, K. The ethical imperative of addressing oral health disparities: A unifying framework. J. Dent. Res. 2014, 93, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Coman, V.; Vodnar, D.C. Gut microbiota and old age: Modulating factors and interventions for healthy longevity. Exp. Gerontol. 2020, 141, 111095. [Google Scholar] [CrossRef]
- Verma, D.; Garg, P.K.; Dubey, A.K. Insights into the human oral microbiome. Arch. Microbiol. 2018, 200, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Koren, N.; Zubeidat, K.; Saba, Y.; Horev, Y.; Barel, O.; Wilharm, A.; Heyman, O.; Wald, S.; Eli-Berchoer, L.; Shapiro, H.; et al. Maturation of the neonatal oral mucosa involves unique epithelium-microbiota interactions. Cell Host Microbe 2021, 29, 197–209.e5. [Google Scholar] [CrossRef] [PubMed]
- Bellando-Randone, S.; Russo, E.; Venerito, V.; Matucci-Cerinic, M.; Iannone, F.; Tangaro, S.; Amedei, A. Exploring the Oral Microbiome in Rheumatic Diseases, State of Art and Future Prospective in Personalized Medicine with an AI Approach. J. Pers. Med. 2021, 11, 625. [Google Scholar] [CrossRef] [PubMed]
- Abusleme, L.; Moutsopoulos, N.M. IL-17: Overview and role in oral immunity and microbiome. Oral Dis. 2017, 23, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Moutsopoulos, N.M. Regulation of host-microbe interactions at oral mucosal barriers by type 17 immunity. Sci. Immunol. 2020, 5, eaau4594. [Google Scholar] [CrossRef]
- Konkel, J.E.; O’Boyle, C.; Krishnan, S. Distal Consequences of Oral Inflammation. Front. Immunol. 2019, 10, 1403. [Google Scholar] [CrossRef] [Green Version]
- De Aquino, S.G.; Talbot, J.; Sonego, F.; Turato, W.M.; Grespan, R.; Avila-Campos, M.J.; Cunha, F.Q.; Cirelli, J.A. The aggravation of arthritis by periodontitis is dependent of IL-17 receptor A activation. J. Clin. Periodontol. 2017, 44, 881–891. [Google Scholar] [CrossRef]
- Dutzan, N.; Kajikawa, T.; Abusleme, L.; Greenwell-Wild, T.; Zuazo, C.E.; Ikeuchi, T.; Brenchley, L.; Abe, T.; Hurabielle, C.; Martin, D.; et al. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci. Transl. Med. 2018, 10, eaat0797. [Google Scholar] [CrossRef] [Green Version]
- De Aquino, S.G.; Abdollahi-Roodsaz, S.; Koenders, M.I.; van de Loo, F.A.; Pruijn, G.J.; Marijnissen, R.J.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.A.; de Molon, R.S.; et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J. Immunol. 2014, 192, 4103–4111. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, M.T.; Mitchell, T.J.; Lord, J.M. Immune ageing and susceptibility to Streptococcus pneumoniae. Biogerontology 2016, 17, 449–465. [Google Scholar] [CrossRef]
- Dimitrov, D.V.; Hoeng, J. Systems approaches to computational modeling of the oral microbiome. Front. Physiol. 2013, 4, 172. [Google Scholar] [CrossRef] [Green Version]
- Hengesch, X.; Elwenspoek, M.M.C.; Schaan, V.K.; Larra, M.F.; Finke, J.B.; Zhang, X.; Bachmann, P.; Turner, J.D.; Vogele, C.; Muller, C.P.; et al. Blunted endocrine response to a combined physical-cognitive stressor in adults with early life adversity. Child. Abuse Negl. 2018, 85, 137–144. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microbial. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Papke, L.E.; Wooldridge, J.M. Econometric methods for fractional response variables with an application to 401 (k) plan participation rates. J. Appl. Econom. 1996, 11, 619–632. [Google Scholar] [CrossRef] [Green Version]
- Baldini, F.; Hertel, J.; Sandt, E.; Thinnes, C.C.; Neuberger-Castillo, L.; Pavelka, L.; Betsou, F.; Krüger, R.; Thiele, I. Parkinson’s disease-associated alterations of the gut microbiome predict disease-relevant changes in metabolic functions. BMC Biol. 2020, 18, 62. [Google Scholar] [CrossRef]
- Benjamini, Y. Discovering the false discovery rate. J. R. Stat. Soc. Ser. B 2010, 72, 405–416. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Saliva | Buccal | |||||
|---|---|---|---|---|---|---|
| Number of Associations (FDR < 0.05) | Positively Associated Taxa | Negatively Associated Taxa | Number of Associations (FDR < 0.05) | Positively Associated Taxa | Negatively Associated Taxa | |
| Smoking | 0 | - | - | 5 | - | Pasteurellaceae (unclassified), Neisseria, Capnocytophaga, Neisseriaceae (unc 1), Planococcaceae (unc) |
| ELA vs. control | 4 | - | Aquabacterium, Comamonas, Comamonadaceae (unc), Acinetobacter | 0 | - | - |
| Anti-CMV seropositive | 2 | - | Pseudomonas, Oxalobaceraceae (unc) | 9 | Alysiella, Neisseria | Sphingomonas, Acinetobacter, Oxalobacteraceae (unc), Bradyrhizobium, Flavobacterium, Methylorubrum, Comamonadaceae (unc) |
| Anti-EBV seropositive | 0 | - | - | 1 | Neisseria | - |
| HSV | 0 | - | - | 0 | - | - |
| CD4+ CD57+ | 2 | Selenomonas | Oxalobaceraceae (unc) | 4 | Selenomonas, Capnocytophaga, Campylobacter, Lautropia | - |
| CD8+ CD57+ | 0 | - | - | 0 | - | - |
| Total CTLs | 0 | - | - | 0 | - | - |
| Total Th cells | 0 | - | - | 0 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charalambous, E.G.; Mériaux, S.B.; Guebels, P.; Muller, C.P.; Leenen, F.A.D.; Elwenspoek, M.M.C.; Thiele, I.; Hertel, J.; Turner, J.D. Early-Life Adversity Leaves Its Imprint on the Oral Microbiome for More Than 20 Years and Is Associated with Long-Term Immune Changes. Int. J. Mol. Sci. 2021, 22, 12682. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312682
Charalambous EG, Mériaux SB, Guebels P, Muller CP, Leenen FAD, Elwenspoek MMC, Thiele I, Hertel J, Turner JD. Early-Life Adversity Leaves Its Imprint on the Oral Microbiome for More Than 20 Years and Is Associated with Long-Term Immune Changes. International Journal of Molecular Sciences. 2021; 22(23):12682. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312682
Chicago/Turabian StyleCharalambous, Eleftheria G., Sophie B. Mériaux, Pauline Guebels, Claude P. Muller, Fleur A. D. Leenen, Martha M. C. Elwenspoek, Ines Thiele, Johannes Hertel, and Jonathan D. Turner. 2021. "Early-Life Adversity Leaves Its Imprint on the Oral Microbiome for More Than 20 Years and Is Associated with Long-Term Immune Changes" International Journal of Molecular Sciences 22, no. 23: 12682. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312682