
Phosphocatalytic Kinome Activity Profiling of Apoptotic and Ferroptotic Agents in Multiple Myeloma Cells

 , , and
, , and
Abstract
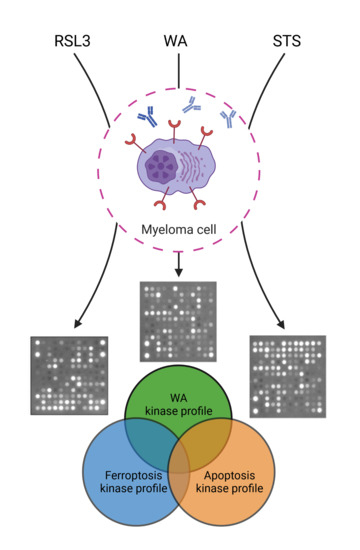
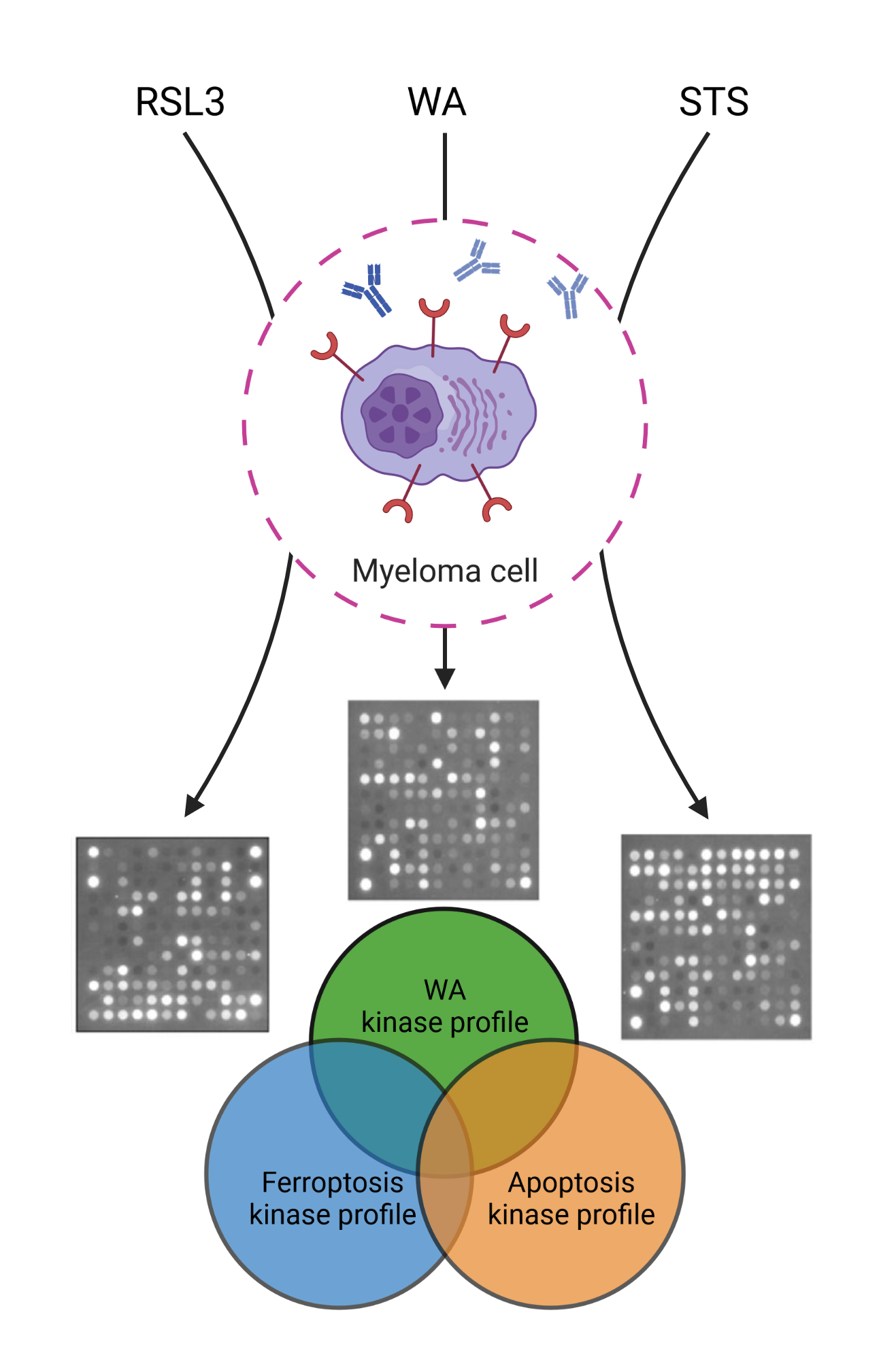
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
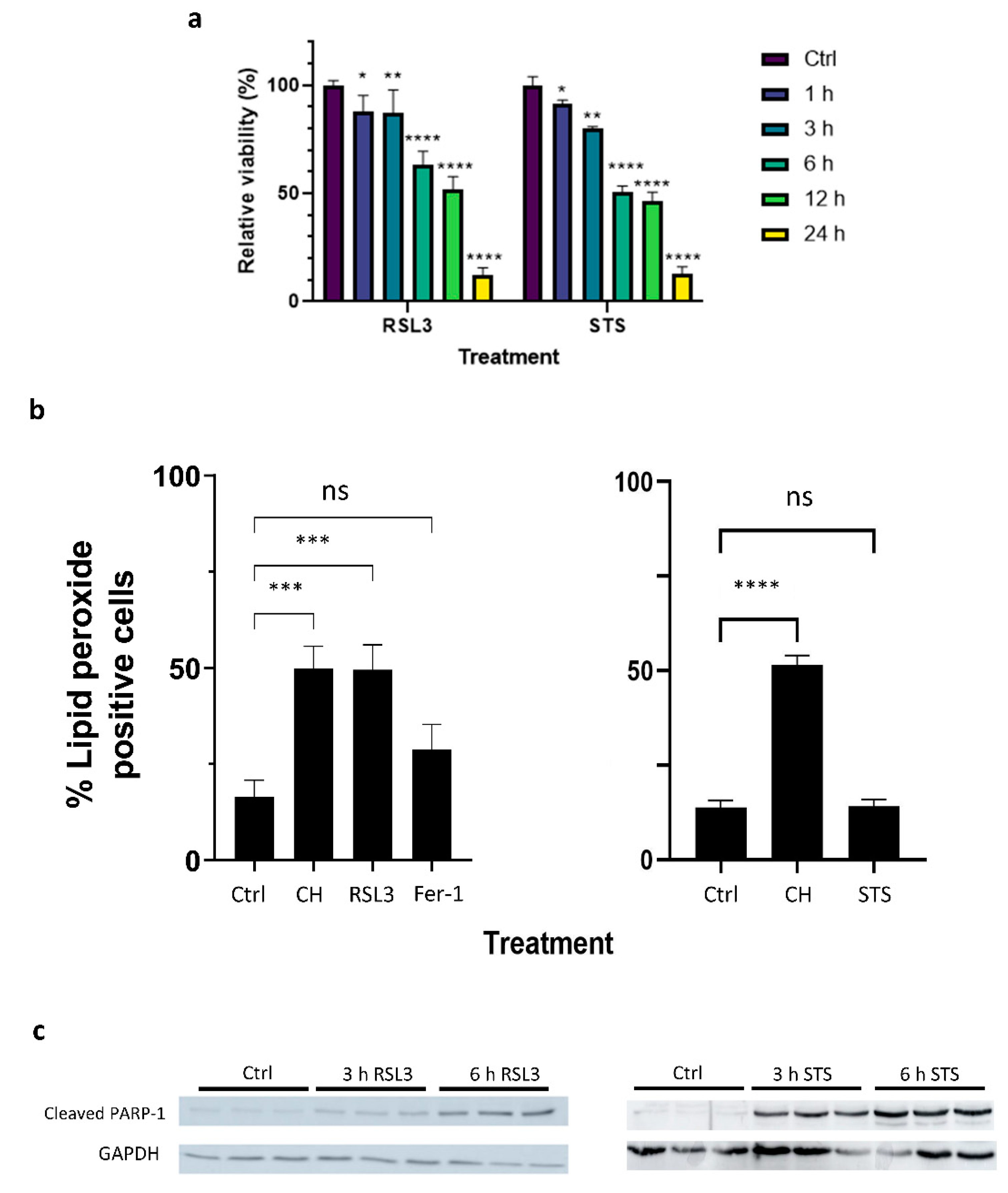
2.1. Both Apoptotic and Ferroptotic Cell Death Are Associated with a Progressive Inhibition of Protein Kinase Activity
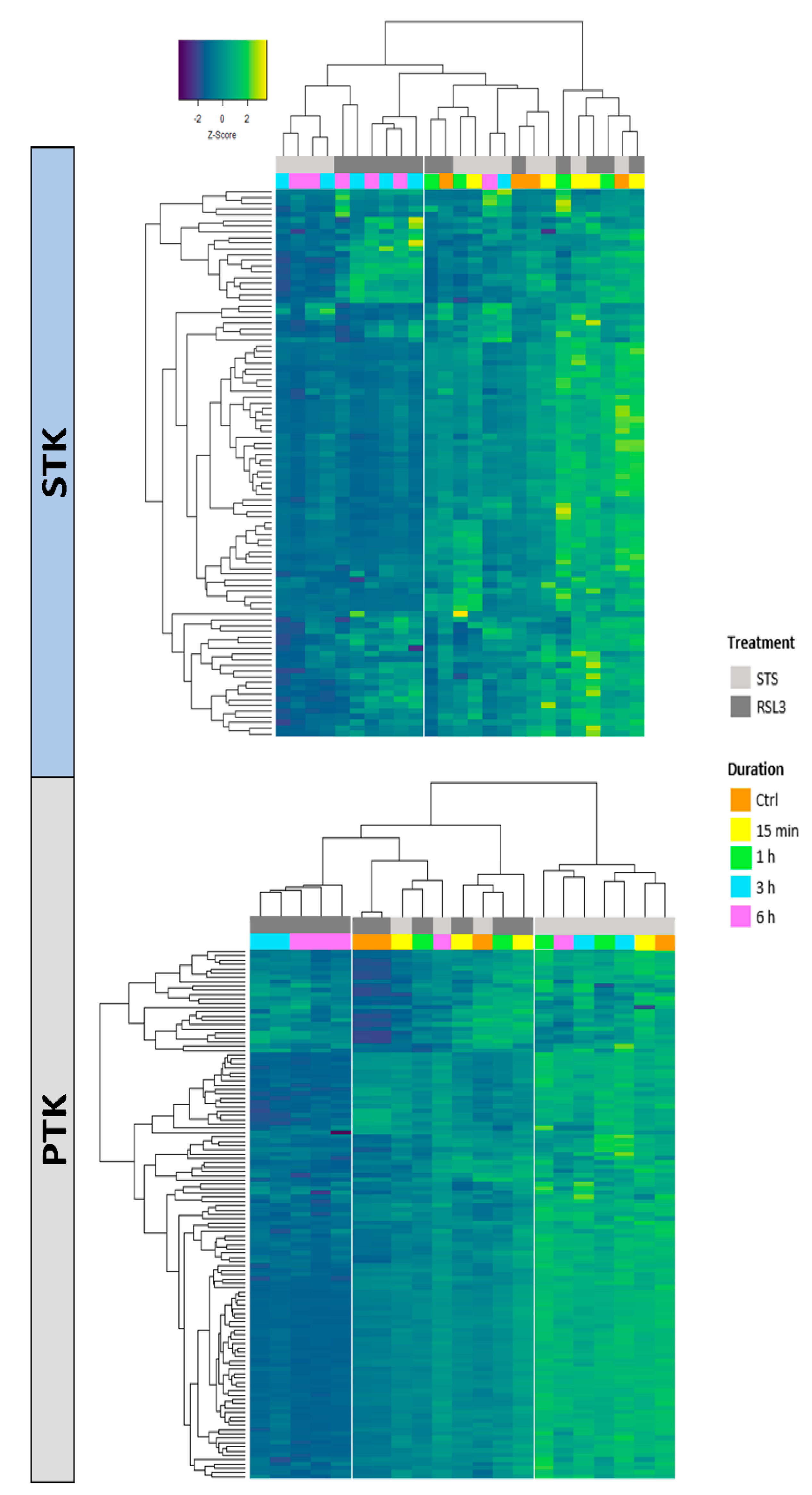
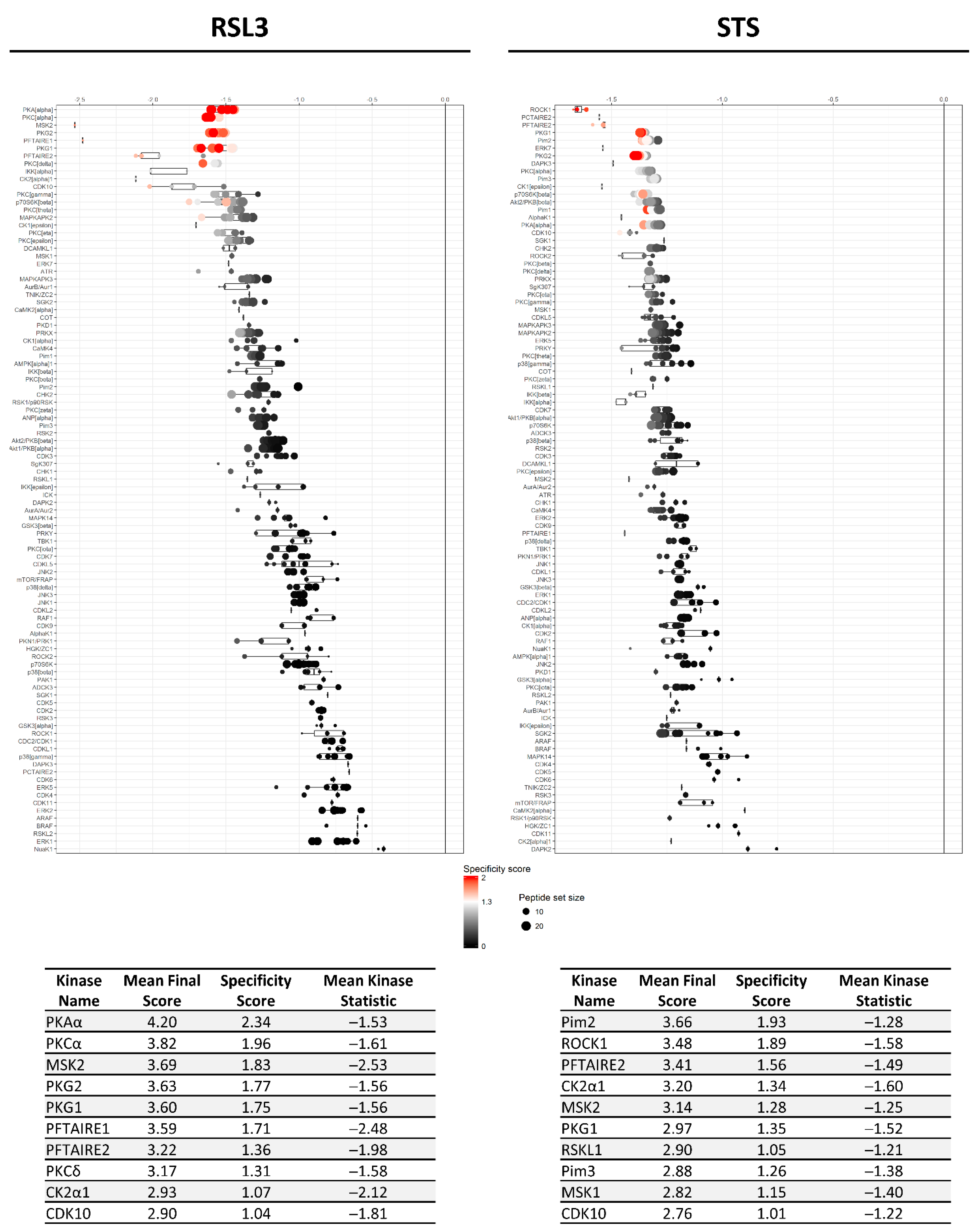
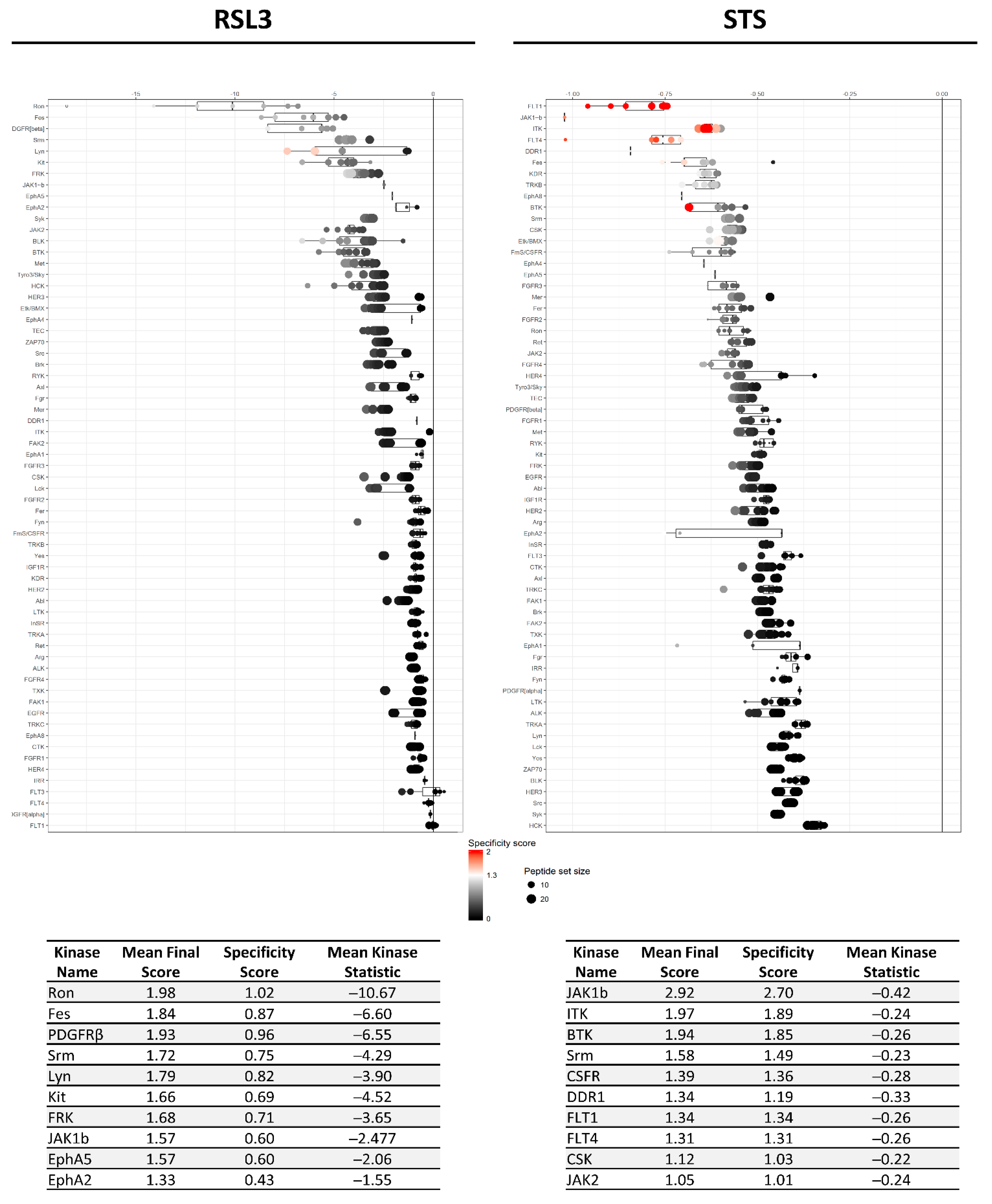
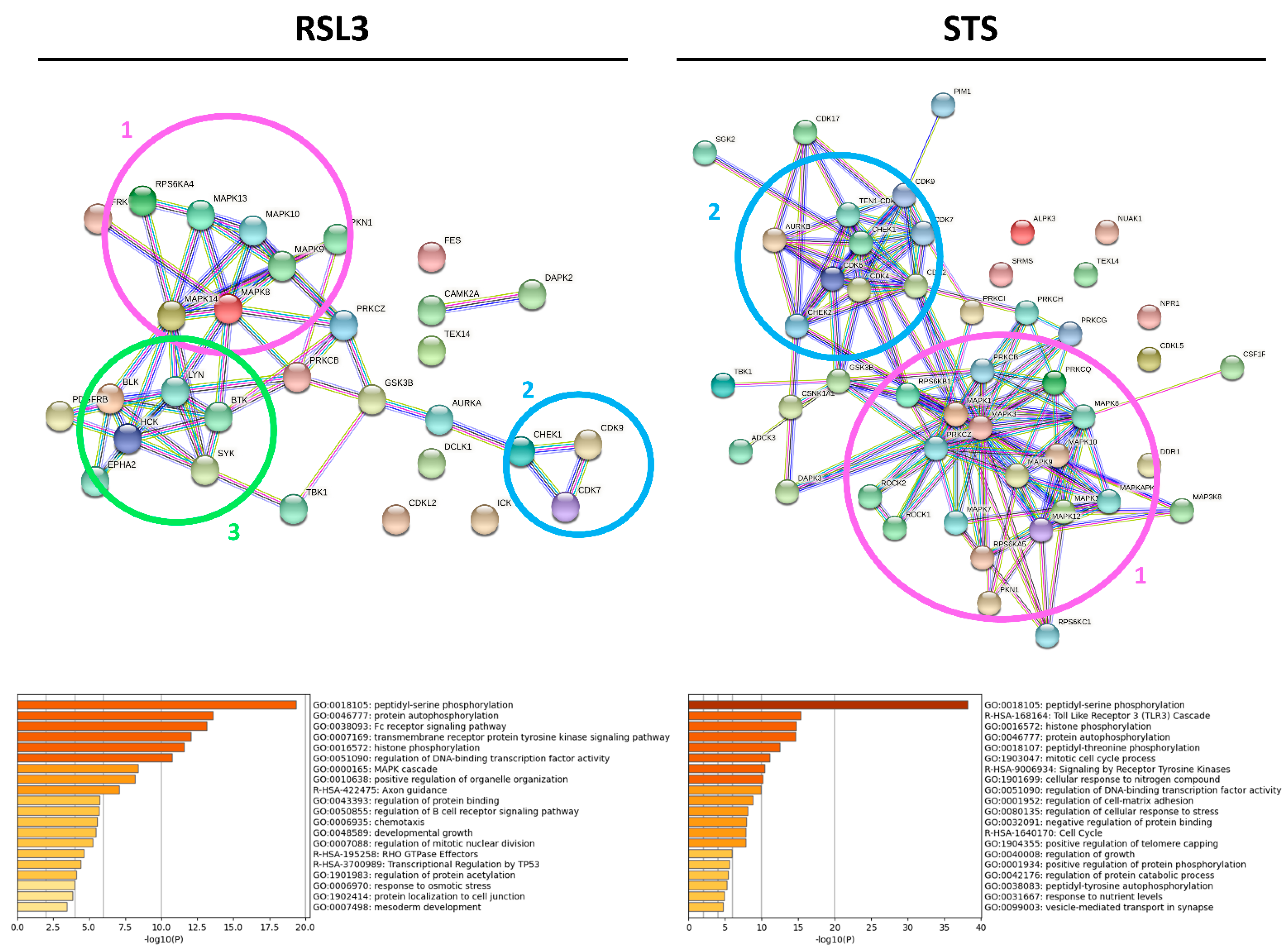
2.2. RSL3 and STS Treatment of MM1R Cells Mainly Affects CMGC and AGC Protein Kinases
2.3. Predicting the Cell Death Modality of Withaferin a Based on Kinome Profiling
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Viability Assays
4.2. Antibodies and Reagents
4.3. Lipid Peroxidation Assay
4.4. Protein Extraction and Western Blot Analysis
4.5. Kinase Activity Profiling Using PamChip® Peptide Microarrays
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef]
- Beharry, Z.; Mahajan, S.; Zemskova, M.; Lin, Y.-W.; Tholanikunnel, B.G.; Xia, Z.; Smith, C.D.; Kraft, A.S. The Pim protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 528–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, A.R.; Black, J.D. Protein kinase C signaling and cell cycle regulation. Front. Immunol. 2013, 27, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-X.; Zhang, Y.; Park, H.W.; Jewell, J.L.; Chen, Q.; Deng, Y.; Pan, D.; Taylor, S.S.; Lai, Z.-C.; Guan, K.-L. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013, 27, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinehara, M.; Kawamura, S.; Tateyama, D.; Suga, M.; Matsumura, H.; Mimura, S.; Hirayama, N.; Hirata, M.; Uchio-Yamada, K.; Kohara, A.; et al. Protein Kinase C Regulates Human Pluripotent Stem Cell Self-Renewal. PLoS ONE 2013, 8, e54122. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Declercq, W.; Berghe, T.V.; Vandenabeele, P. RIP Kinases at the Crossroads of Cell Death and Survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Ravanan, P.; Talwar, P. Death Associated Protein Kinase 1 (DAPK1): A Regulator of Apoptosis and Autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Logie, E.; Chirumamilla, C.; Perez-Novo, C.; Shaw, P.; Declerck, K.; Palagani, A.; Rangarajan, S.; Cuypers, B.; De Neuter, N.; Turabe, F.M.H.U.; et al. Covalent Cysteine Targeting of Bruton’s Tyrosine Kinase (BTK) Family by Withaferin-A Reduces Survival of Glucocorticoid-Resistant Multiple Myeloma MM1 Cells. Cancers 2021, 13, 1618. [Google Scholar] [CrossRef]
- Wang, L.; Lin, N.; Li, Y. The PI3K/AKT signaling pathway regulates ABCG2 expression and confers resistance to chemotherapy in human multiple myeloma. Oncol. Rep. 2019, 41, 1678–1690. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the mul-tidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Tao, J. The B-cell receptor orchestrates environment-mediated lymphoma survival and drug resistance in B-cell malignancies. Oncogene 2014, 33, 4107–4113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boussac, H.; Bruyer, A.; Jourdan, M.; Maes, A.; Robert, N.; Gourzones, C.; Vincent, L.; Seckinger, A.; Cartron, G.; Hose, D.; et al. Kinome expression profiling to target new therapeutic avenues in multiple myeloma. Haematologica 2020, 105, 784–795. [Google Scholar] [CrossRef]
- Richardson, P.G.; Bensinger, W.I.; Huff, C.A.; Costello, C.L.; Lendvai, N.; Berdeja, J.G.; Anderson, L.D., Jr.; Siegel, D.S.; Lebovic, D.; Jagannath, S.; et al. Ibrutinib alone or with dexamethasone for relapsed or relapsed and refractory multiple myeloma: Phase 2 trial results. Br. J. Haematol. 2018, 180, 821–830. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer. Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Lind, J.; Czernilofsky, F.; Vallet, S.; Podar, K. Emerging protein kinase inhibitors for the treatment of multiple myeloma. Expert Opin. Emerg. Drugs 2019, 24, 133–152. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Bebber, C.M.; Müller, F.; Prieto Clemente, L.; Weber, J.; Von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [Green Version]
- Poltorack, C.D.; Dixon, S.J. Understanding the role of cysteine in ferroptosis: Progress & paradoxes. FEBS J. 2021. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and Cancer: 2020 Vision. Cancer Res. 2020, 80, 5435–5448. [Google Scholar] [CrossRef] [PubMed]
- Combs, J.A.; DeNicola, G.M. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers 2019, 11, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Seki, T.; Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009, 61, 290–302. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Berghe, T.V. Targeting Ferroptosis to Iron out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Chen, P.-H.; Wu, J.; Ding, C.-K.C.; Lin, C.-C.; Pan, S.; Bossa, N.; Xu, Y.; Yang, W.-H.; Mathey-Prevot, B.; Chi, J.-T. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020, 27, 1008–1022. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer. Res. 2014, 34, 6417–6422. [Google Scholar]
- Bahlis, N.J.; Miao, Y.; Koc, O.N.; Lee, K.; Boise, L.H.; Gerson, S.L. N-benzoylstaurosporine (PKC412) inhibits Akt kinase inducing apoptosis in multiple myeloma cells. Leuk. Lymphoma. 2005, 46, 899–908. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Yuan, C.-X.; Zhou, Z.-W.; Yang, Y.-X.; He, Z.-X.; Zhang, X.; Wang, D.; Yang, T.; Wang, N.-J.; Zhao, R.J. Inhibition of mitotic Aurora kinase A by alisertib induces apoptosis and autophagy of human gastric cancer AGS and NCI-N78 cells. Drug Des. Dev. Ther. 2015, 9, 487–508. [Google Scholar] [CrossRef] [Green Version]
- Zekri, A.; Ghaffari, S.H.; Yaghmaie, M.; Estiar, M.A.; Alimoghaddam, K.; Modarressi, M.H.; Ghavamzadeh, A. Inhibitor of Aurora Kinase B Induces Differentially Cell Death and Polyploidy via DNA Damage Response Pathways in Neurological Malignancy: Shedding New Light on the Challenge of Resistance to AZD1152-HQPA. Mol. Neurobiol. 2016, 53, 1808–1823. [Google Scholar] [CrossRef]
- Geering, B. Death-associated protein kinase 2: Regulator of apoptosis, autophagy and inflammation. Int. J. Biochem. Cell Biol. 2015, 65, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Kake, S.; Ohama, T.; Usui, T.; Yamawaki, H.; Sato, K. Death-associated protein kinase 3 controls the tumor progression of A549 cells through ERK MAPK/c-Myc signaling. Oncol. Rep. 2017, 37, 1100–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.H.; Lee, D.H.; Lee, Y.S.; Jo, M.J.; Jeong, Y.A.; Kwon, W.T.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Molecular crosstalk between ferroptosis and apoptosis: Emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 2017, 8, 115164–115178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Eid, S.; Turk, S.; Volkamer, A.; Rippmann, F.; Fulle, S. KinMap: A web-based tool for interactive navigation through human kinome data. BMC Bioinform. 2017, 18, 16. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Hassannia, B.; Logie, E.; Vandenabeele, P.; Berghe, T.V.; Berghe, W.V. Withaferin A: From ayurvedic folk medicine to preclinical anti-cancer drug. Biochem. Pharmacol. 2020, 173, 113602. [Google Scholar] [CrossRef]
- Heyninck, K.; Lahtela-Kakkonen, M.; Van der Veken, P.; Haegeman, G.; Vanden Berghe, W. Withaferin A inhibits NF-kappaB activation by targeting cysteine 179 in IKKbeta. Biochem Pharmacol. 2014, 91, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Y.; Cheryan, V.T.; Wu, W.; Cui, C.Q.; Polin, L.A.; Pass, H.I.; Dou, Q.P.; Rishi, A.K.; Wali, A. Withaferin A inhibits the proteasome activity in mesothelioma in vitro and in vivo. PLoS ONE 2012, 7, e41214. [Google Scholar]
- Yu, Y.; Hamza, A.; Zhang, T.; Gu, M.; Zou, P.; Newman, B.; Li, Y.; Gunatilaka, A.L.; Zhan, C.-G.; Sun, D. Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem. Pharmacol. 2010, 79, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, H.N. Kinase inhibitors as potential agents in the treatment of multiple myeloma. Oncotarget 2016, 7, 81926–81968. [Google Scholar] [CrossRef] [Green Version]
- John, L.; Krauth, M.; Podar, K.; Raab, M.-S. Pathway-Directed Therapy in Multiple Myeloma. Cancers 2021, 13, 1668. [Google Scholar] [CrossRef]
- Chong, P.S.Y.; Chng, W.-J.; de Mel, S. STAT3: A Promising Therapeutic Target in Multiple Myeloma. Cancers 2019, 11, 731. [Google Scholar] [CrossRef] [Green Version]
- A Keane, N.; Glavey, S.V.; Krawczyk, J.; O’Dwyer, M. AKT as a therapeutic target in multiple myeloma. Expert Opin. Ther. Targets 2014, 18, 897–915. [Google Scholar] [CrossRef] [PubMed]
- Von Suskil, M.; Sultana, K.N.; Elbezanti, W.O.; Al-Odat, O.S.; Chitren, R.; Tiwari, A.K.; Challagundla, K.B.; Srivastava, S.K.; Jonnala-gadda, S.C.; Budak-Alpdogan, T.; et al. Bruton’s Tyrosine Kinase Targeting in Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 5707. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Phelan, J.D.; Wilson, W.H.; Staudt, L.M. Pathogenic B-cell receptor signaling in lymphoid malignancies: New insights to improve treatment. Immunol. Rev. 2019, 291, 190–213. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; David, M. Inhibition of B cell receptor-mediated apoptosis by IFN. J. Immunol. 1999, 162, 6317–6321. [Google Scholar] [PubMed]
- Mouhamad, S.; Besnault, L.; Auffredou, M.T.; Leprince, C.; Bourgeade, M.F.; Leca, G.; Vazquez, A. B cell receptor-mediated apoptosis of human lymphocytes is associated with a new regulatory pathway of Bim isoform expression. J. Immunol. 2004, 172, 2084–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiestner, A. The role of B-cell receptor inhibitors in the treatment of patients with chronic lymphocytic leukemia. Haematologica 2015, 100, 1495–1507. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Bowles, K.M.; Barrera, L.N.; Murray, M.Y.; Zaitseva, L.; MacEwan, D.J. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-kappaB. Cell Signal. 2013, 25, 106–112. [Google Scholar] [CrossRef]
- Takada, Y.; Aggarwal, B.B. TNF activates Syk protein tyrosine kinase leading to TNF-induced MAPK activation, NF-kappaB activation, and apoptosis. J. Immunol. 2004, 173, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ma, Y.; Wang, X.; Jiang, J.; Zhang, C.; Wang, X.; Jiang, Y.; Huang, H.; Hong, L. IL-17A Increases Multiple Myeloma Cell Viability by Positively Regulating Syk Expression. Transl. Oncol. 2019, 12, 1086–1091. [Google Scholar] [CrossRef]
- Xu, M.; Tao, J.; Yang, Y.; Tan, S.; Liu, H.; Jiang, J.; Zheng, F.; Wu, B. Ferroptosis involves in intestinal epithelial cell death in ul-cerative colitis. Cell Death Dis. 2020, 11, 86. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.; Fetterly, G.; Lugade, A.; Thanavala, Y. Sorafenib: A clinical and pharmacologic review. Expert Opin. Pharmacother. 2010, 11, 1943–1955. [Google Scholar] [CrossRef]
- Sun, X.; Niu, X.; Chen, R.; He, W.; Chen, D.; Kang, R.; Tang, D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016, 64, 488–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-L.; Hsu, L.-F.; Phyliky, R.L.; Li, C.-Y. Autocrine Expression of Platelet-Derived Growth Factor B in B Cell Chronic Lymphocytic Leukemia. Acta Haematol. 2005, 114, 133–140. [Google Scholar] [CrossRef]
- Bilalis, A.; Pouliou, E.; Roussou, M.; Papanikolaou, A.; Tassidou, A.; Economopoulos, T.; Terpos, E. Increased expression of platelet derived growth factor receptor beta on trephine biopsies correlates with advanced myeloma. J. BUON. 2017, 22, 1032–1037. [Google Scholar] [PubMed]
- Lawrie, A.M.; Noble, M.E.; Tunnah, P.; Brown, N.R.; Johnson, L.N.; Endicott, J.A. Protein kinase inhibition by staurosporine revealed in details of the molecular interaction with CDK2. Nat. Struct Biol. 1997, 4, 796–801. [Google Scholar] [CrossRef]
- Shi, H.; Cheng, X.; Sze, S.K.; Yao, S.Q. Proteome profiling reveals potential cellular targets of staurosporine using a clickable cell-permeable probe. Chem. Commun. 2011, 47, 11306–11308. [Google Scholar] [CrossRef]
- Fischer, J.J.; Graebner Baessler, O.Y.; Dalhoff, C.; Michaelis, S.; Schrey, A.K.; Ungewiss, J.; Andrich, K.; Jeske, D.; Kroll, F.; Glinski, M.; et al. Comprehensive identification of staurosporine-binding kinases in the hepatocyte cell line HepG2 using Capture Compound Mass Spectrometry (CCMS). J. Proteome Res. 2010, 9, 806–817. [Google Scholar] [CrossRef]
- Wang, S.; Yi, X.; Wu, Z.; Guo, S.; Dai, W.; Wang, H.; Shi, Q.; Zeng, K.; Guo, W.; Li, C. CAMKK2 Defines Ferroptosis Sensitivity of Melanoma Cells by Regulating AMPK-NRF2 Pathway. J. Investig. Dermatol. 2021. [Google Scholar] [CrossRef]
- Kumar, N.; Perez-Novo, C.; Shaw, P.; Logie, E.; Privat-Maldonado, A.; Dewilde, S.; Smits, E.; Berghe, W.V.; Bogaerts, A. Physical plasma-derived oxidants sensitize pancreatic cancer cells to ferroptotic cell death. Free. Radic. Biol. Med. 2021, 166, 187–200. [Google Scholar] [CrossRef]
- Chirumamilla, C.S.; Pérez-Novo, C.; Van Ostade, X.; Berghe, W.V. Molecular insights into cancer therapeutic effects of the dietary medicinal phytochemical withaferin A. Proc. Nutr. Soc. 2017, 76, 96–105. [Google Scholar] [CrossRef]
- Mayola, E.; Gallerne, C.; Degli Esposti, D.; Martel, C.; Pervaiz, S.; Larue, L.; Debuire, B.; Lemoine, A.; Brenner, C.; Lemaire, C. Withaferin A induces apoptosis in human melanoma cells through generation of reactive oxygen species and down-regulation of Bcl-2. Apoptosis 2011, 16, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Stan, S.D.; Hahm, E.-R.; Warin, R.; Singh, S.V. Withaferin A Causes FOXO3a- and Bim-Dependent Apoptosis and Inhibits Growth of Human Breast Cancer Cells In vivo. Cancer Res. 2008, 68, 7661–7669. [Google Scholar] [CrossRef] [Green Version]
- Hahm, E.-R.; Moura, M.B.; Kelley, E.E.; Van Houten, B.; Shiva, S.; Singh, S.V. Withaferin A-Induced Apoptosis in Human Breast Cancer Cells Is Mediated by Reactive Oxygen Species. PLoS ONE 2011, 6, e23354. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, M.; Ambesi-Impiombato, A.; Qin, Y.; Herranz, D.; Bansal, M.; Girardi, T.; Paietta, E.; Tallman, M.S.; Rowe, J.M.; De Keersmaecker, K.; et al. Synergistic antileukemic therapies in NOTCH1-induced T-ALL. Proc. Natl. Acad. Sci. USA 2017, 114, 2006–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, N.; Banerjee, B.; Das, B.B.; Ganguly, A.; Sen, T.; Pramanik, S.; Mukhopadhyay, S.; Majumder, H.K. Apoptosis is induced in leishmanial cells by a novel protein kinase inhibitor withaferin A and is facilitated by apoptotic topoisomerase I–DNA complex. Cell Death Differ. 2007, 14, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Liu, J.; Wang, B.; Zhang, C.; Su, Z.; Zhao, M.; Qin, L.; Zhang, W.; Zheng, R. Withaferin A promotes white adipose browning and prevents obesity through sympathetic nerve-activated Prdm16-FATP1 axis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jackson, S.S.; Oberley, C.; Hooper, C.P.; Grindle, K.; Wuerzberger-Davis, S.; Wolff, J.; McCool, K.; Rui, L.; Miyamoto, S. Withaferin A disrupts ubiquitin-based NEMO reorganization induced by canonical NF-kappaB signaling. Exp. Cell Res. 2015, 331, 58–72. [Google Scholar] [CrossRef] [Green Version]
- Dom, M.; Offner, F.; Vanden Berghe, W.; Van Ostade, X. Proteomic characterization of Withaferin A-targeted protein networks for the treatment of monoclonal myeloma gammopathies. J. Proteomics. 2018, 179, 17–29. [Google Scholar] [CrossRef]
- Issa, M.E.; Cuendet, M. Withaferin A induces cell death and differentiation in multiple myeloma cancer stem cells. MedChemComm 2017, 8, 112–121. [Google Scholar] [CrossRef]
- Kemble, D.J.; Sun, G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc. Natl. Acad. Sci. USA 2009, 106, 5070–5075. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, C.; Carroll, K.S. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chem. Biol. 2010, 5, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Fowler, N.J.; Blanford, C.F.; de Visser, S.; Warwicker, J. Features of reactive cysteines discovered through computation: From kinase inhibition to enrichment around protein degrons. Sci. Rep. 2017, 7, 16338. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Cheng, C.K.; Nicolaides, T.P.; Hackett, C.S.; Knight, Z.A.; Shokat, K.M.; Weiss, W.A. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007, 67, 7960–7965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichhorn, P.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-Kinase Hyperactivation Results in Lapatinib Resistance that Is Reversed by the mTOR/Phosphatidylinositol 3-Kinase Inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [Google Scholar] [CrossRef] [Green Version]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Lig-and-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009, 15, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.-W.; Knight, Z.; Goldenberg, D.D.; Yu, W.; Mostov, K.E.; Stokoe, D.; Shokat, K.M.; Weiss, W.A. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, E.A.; Ward, C.C.; Spradlin, J.; Bateman, L.A.; Huffman, T.; Miyamoto, D.K.; Kleinman, J.I.; Nomura, D.K. Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-cancer Natural Products. Cell Chem. Biol. 2017, 24, 1368–1376.e4. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Majumder, P.; Shioya, H.; Wu, F.; Kumar, S.; Weichselbaum, R.; Kharbanda, S.; Kufe, D. Activation of the Cytoplasmic c-Abl Tyrosine Kinase by Reactive Oxygen Species. J. Biol. Chem. 2000, 275, 17237–17240. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Pang, Q. Oxidative Stress-Associated Protein Tyrosine Kinases and Phosphatases in Fanconi Anemia. Antioxidants Redox Signal. 2014, 20, 2290–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmann, K.; Mayer, C.; Kehlbach, R.; Rothmund, M.-C.; Rodemann, H.P. Radiation-induced lipid peroxidation activates src kinase and triggers nuclear EGFR transport. Radiother. Oncol. 2009, 92, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Sousa, B.C.; Ahmed, T.; Dann, W.L.; Ashman, J.; Guy, A.; Durand, T.; Pitt, A.R.; Spickett, C.M. Short-chain lipid peroxidation products form covalent adducts with pyruvate kinase and inhibit its activity in vitro and in breast cancer cells. Free. Radic. Biol. Med. 2019, 144, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Palagani, A.; Op de Beeck, K.; Naulaerts, S.; Diddens, J.; Sekhar Chirumamilla, C.; Van Camp, G.; Laukens, K.; Heyninck, K.; Gerlo, S.; Mestdagh, P.; et al. Ectopic microRNA-150-5p transcription sensitizes glucocorticoid therapy response in MM1S multiple my-eloma cells but fails to overcome hormone therapy resistance in MM1R cells. PLoS ONE 2014, 9, e113842. [Google Scholar] [CrossRef] [PubMed]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; Dezonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]




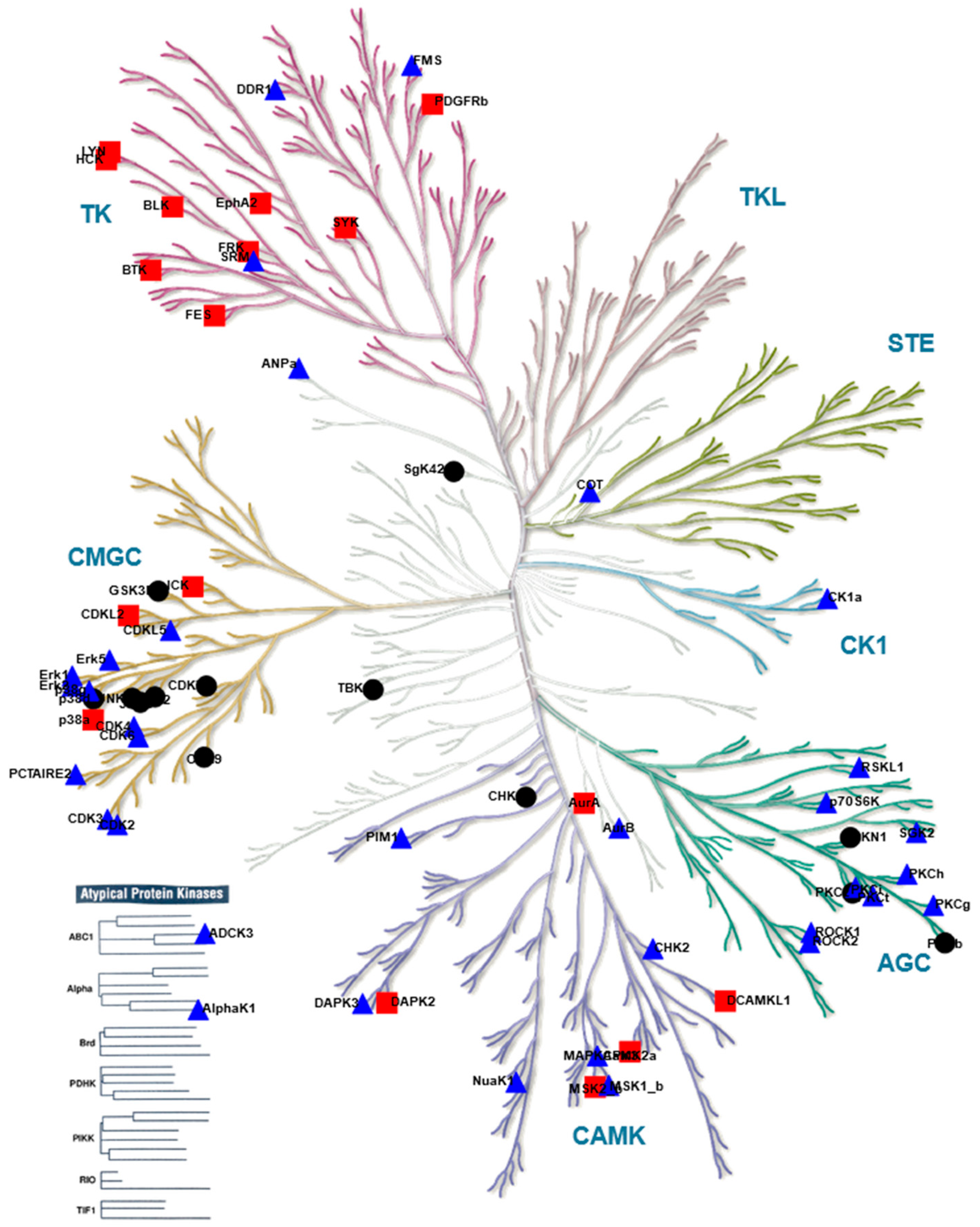
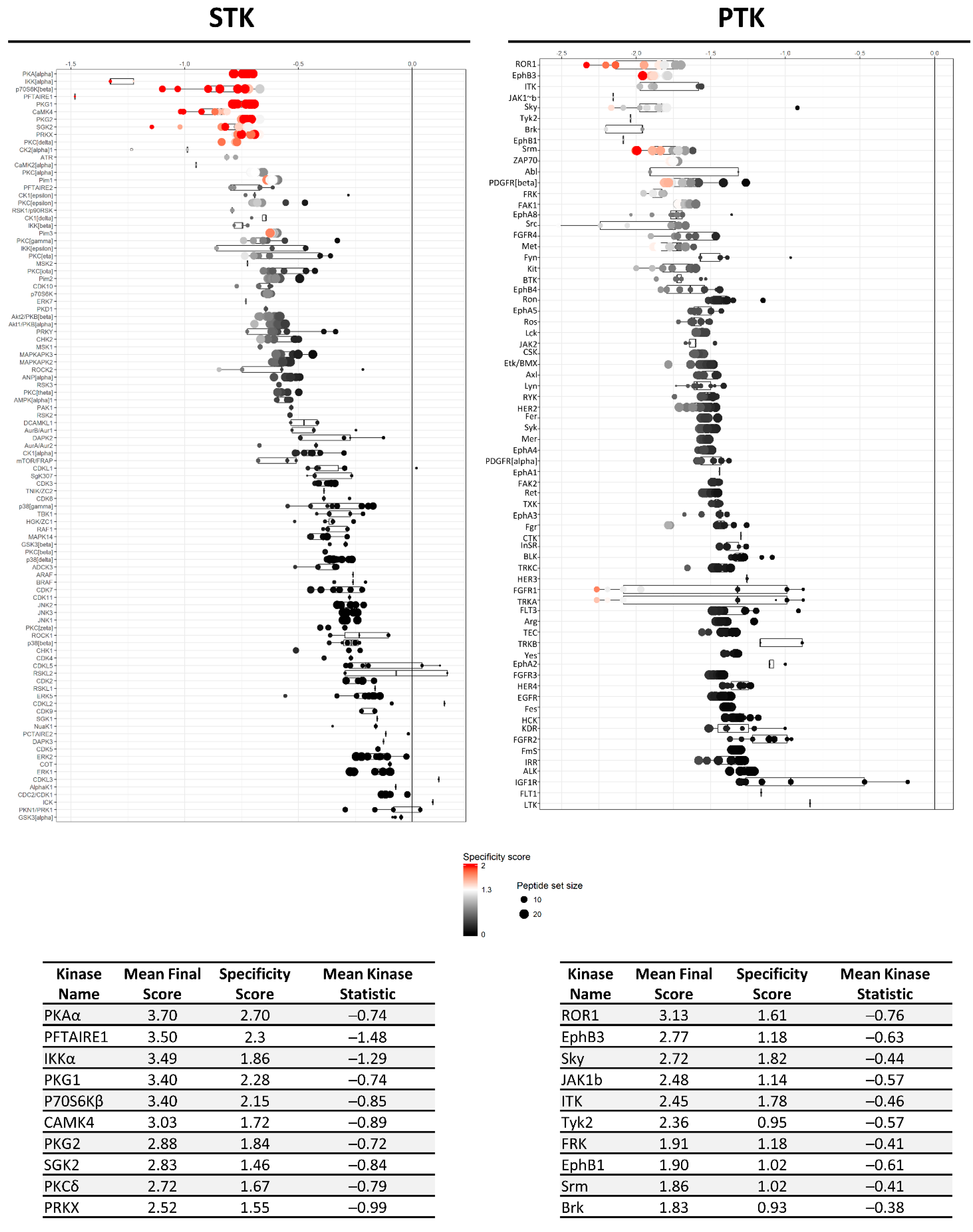
 ), kinases inhibited by both compounds are indicated as black circles (•). Only kinases with a mean final score ≥ 1.3 are included in the graph. Abbreviations: TK, tyrosine kinase group; TKL, tyrosine kinase-like group; STE, serine/threonine kinase group; CK1, casein kinase 1 group; AGC, protein kinase A, G, and C group; CAMK, Ca2+/calmodulin-dependent kinase group; CMGC, cyclin-dependent kinase, mitogen-activated protein kinase, glycogen synthase kinase, and CDC-like kinase group. llustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com, 27 September 2021).
), kinases inhibited by both compounds are indicated as black circles (•). Only kinases with a mean final score ≥ 1.3 are included in the graph. Abbreviations: TK, tyrosine kinase group; TKL, tyrosine kinase-like group; STE, serine/threonine kinase group; CK1, casein kinase 1 group; AGC, protein kinase A, G, and C group; CAMK, Ca2+/calmodulin-dependent kinase group; CMGC, cyclin-dependent kinase, mitogen-activated protein kinase, glycogen synthase kinase, and CDC-like kinase group. llustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com, 27 September 2021).
), kinases inhibited by both compounds are indicated as black circles (•). Only kinases with a mean final score ≥ 1.3 are included in the graph. Abbreviations: TK, tyrosine kinase group; TKL, tyrosine kinase-like group; STE, serine/threonine kinase group; CK1, casein kinase 1 group; AGC, protein kinase A, G, and C group; CAMK, Ca2+/calmodulin-dependent kinase group; CMGC, cyclin-dependent kinase, mitogen-activated protein kinase, glycogen synthase kinase, and CDC-like kinase group. llustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com, 27 September 2021).
), kinases inhibited by both compounds are indicated as black circles (•). Only kinases with a mean final score ≥ 1.3 are included in the graph. Abbreviations: TK, tyrosine kinase group; TKL, tyrosine kinase-like group; STE, serine/threonine kinase group; CK1, casein kinase 1 group; AGC, protein kinase A, G, and C group; CAMK, Ca2+/calmodulin-dependent kinase group; CMGC, cyclin-dependent kinase, mitogen-activated protein kinase, glycogen synthase kinase, and CDC-like kinase group. llustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com, 27 September 2021).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Logie, E.; Novo, C.P.; Driesen, A.; Van Vlierberghe, P.; Vanden Berghe, W. Phosphocatalytic Kinome Activity Profiling of Apoptotic and Ferroptotic Agents in Multiple Myeloma Cells. Int. J. Mol. Sci. 2021, 22, 12731. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312731
Logie E, Novo CP, Driesen A, Van Vlierberghe P, Vanden Berghe W. Phosphocatalytic Kinome Activity Profiling of Apoptotic and Ferroptotic Agents in Multiple Myeloma Cells. International Journal of Molecular Sciences. 2021; 22(23):12731. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312731
Chicago/Turabian StyleLogie, Emilie, Claudina Perez Novo, Amber Driesen, Pieter Van Vlierberghe, and Wim Vanden Berghe. 2021. "Phosphocatalytic Kinome Activity Profiling of Apoptotic and Ferroptotic Agents in Multiple Myeloma Cells" International Journal of Molecular Sciences 22, no. 23: 12731. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312731