
Neuregulin 4 Downregulation Induces Insulin Resistance in 3T3-L1 Adipocytes through Inflammation and Autophagic Degradation of GLUT4 Vesicles

 ,
,  , ,
, ,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
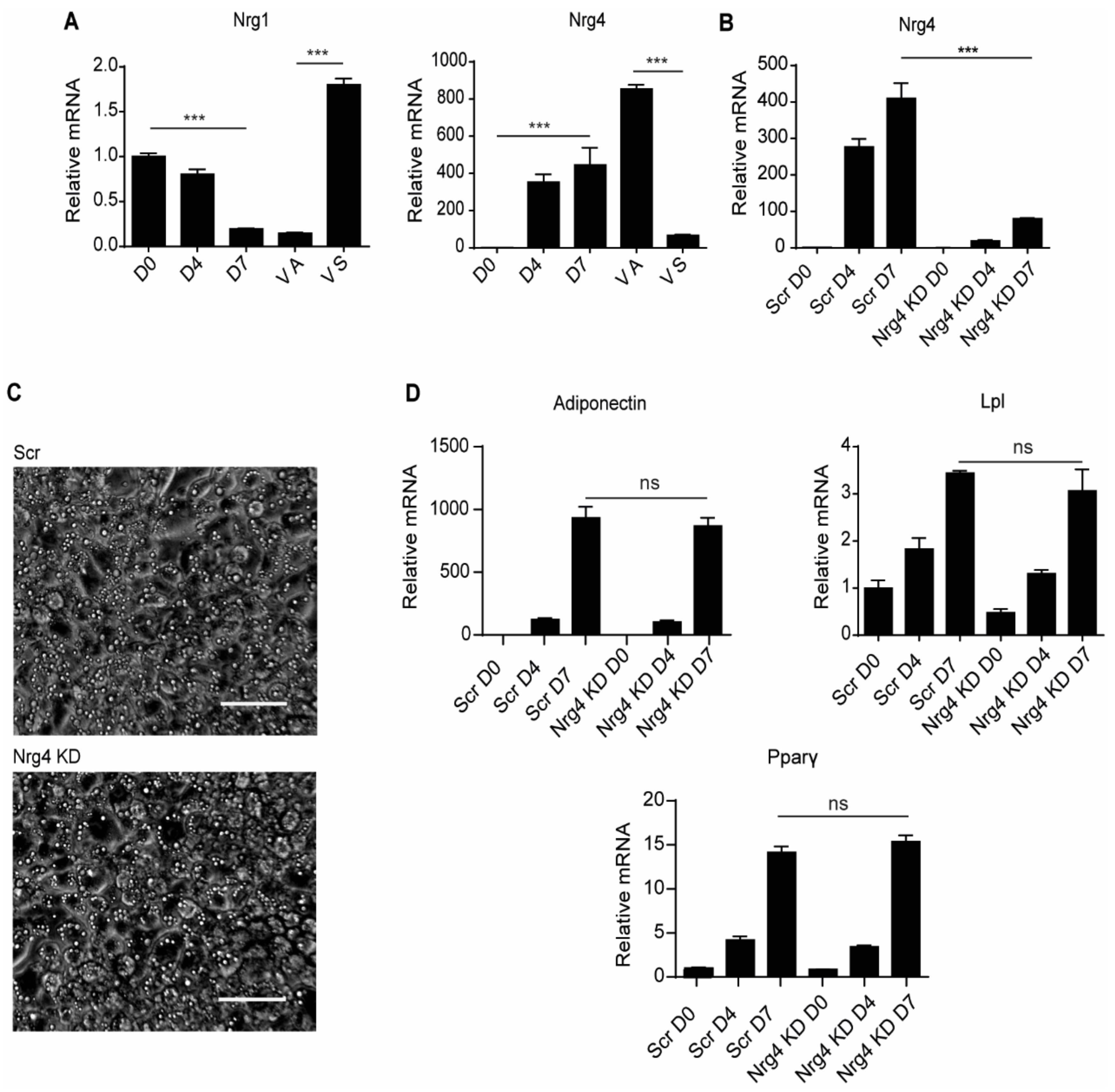
2.1. Nrg4 Deficiency Has No Effect on Differentiation of 3T3-L1 PRE-Adipocytes
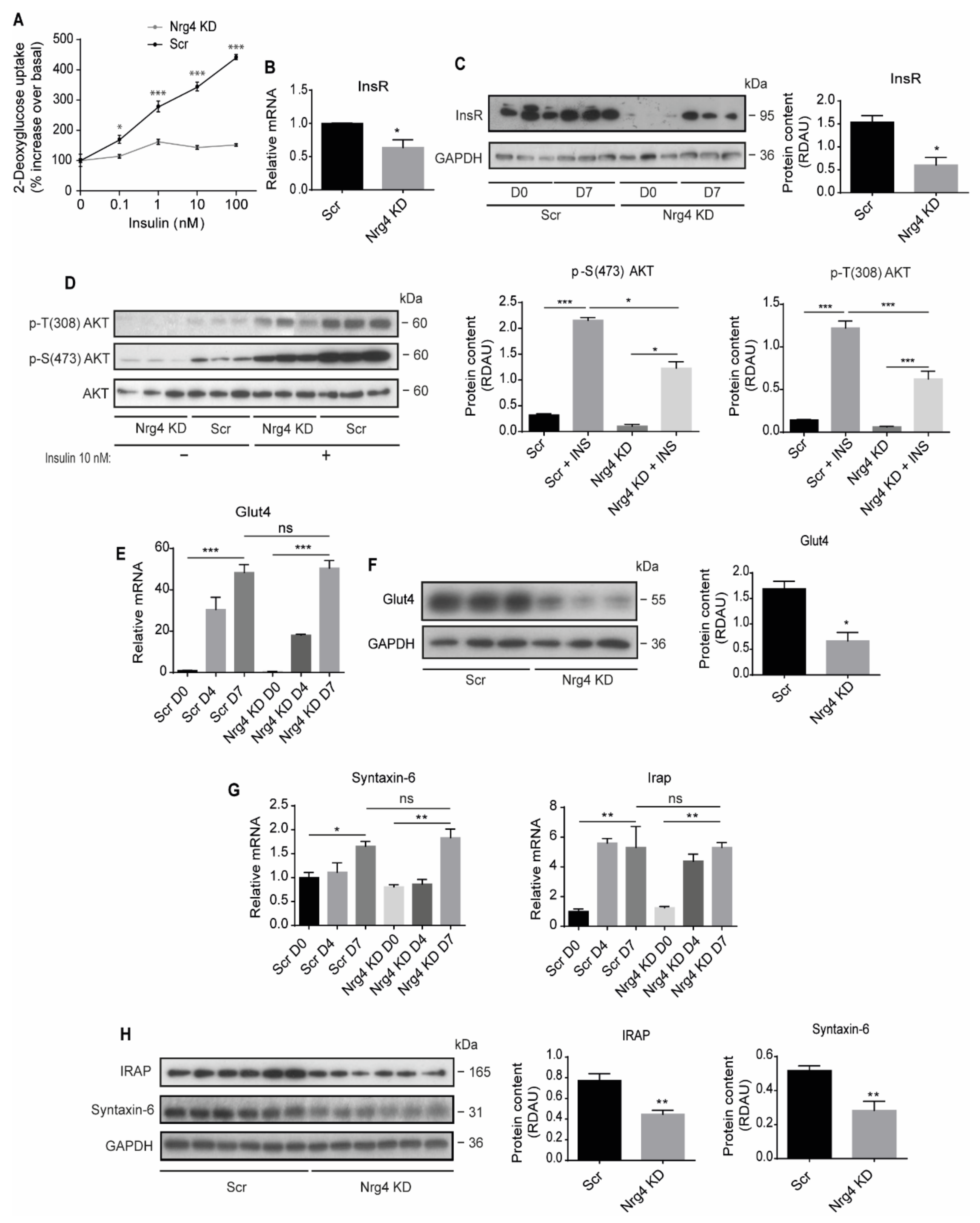
2.2. Nrg4-Deficient Adipocytes Have Impaired Insulin-Stimulated Glucose Transport
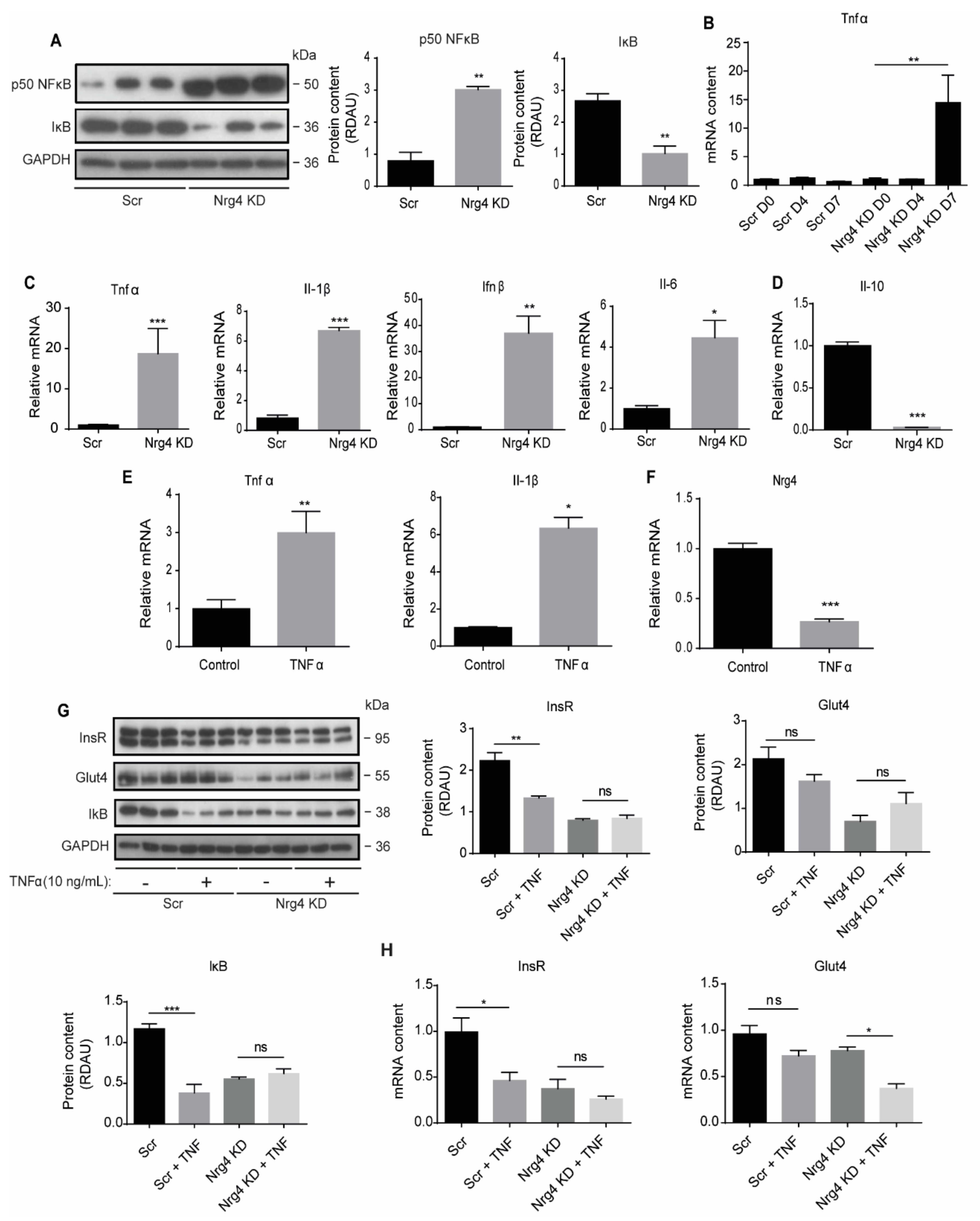
2.3. Nrg4 KD Adipocytes Show Enhanced Inflammation
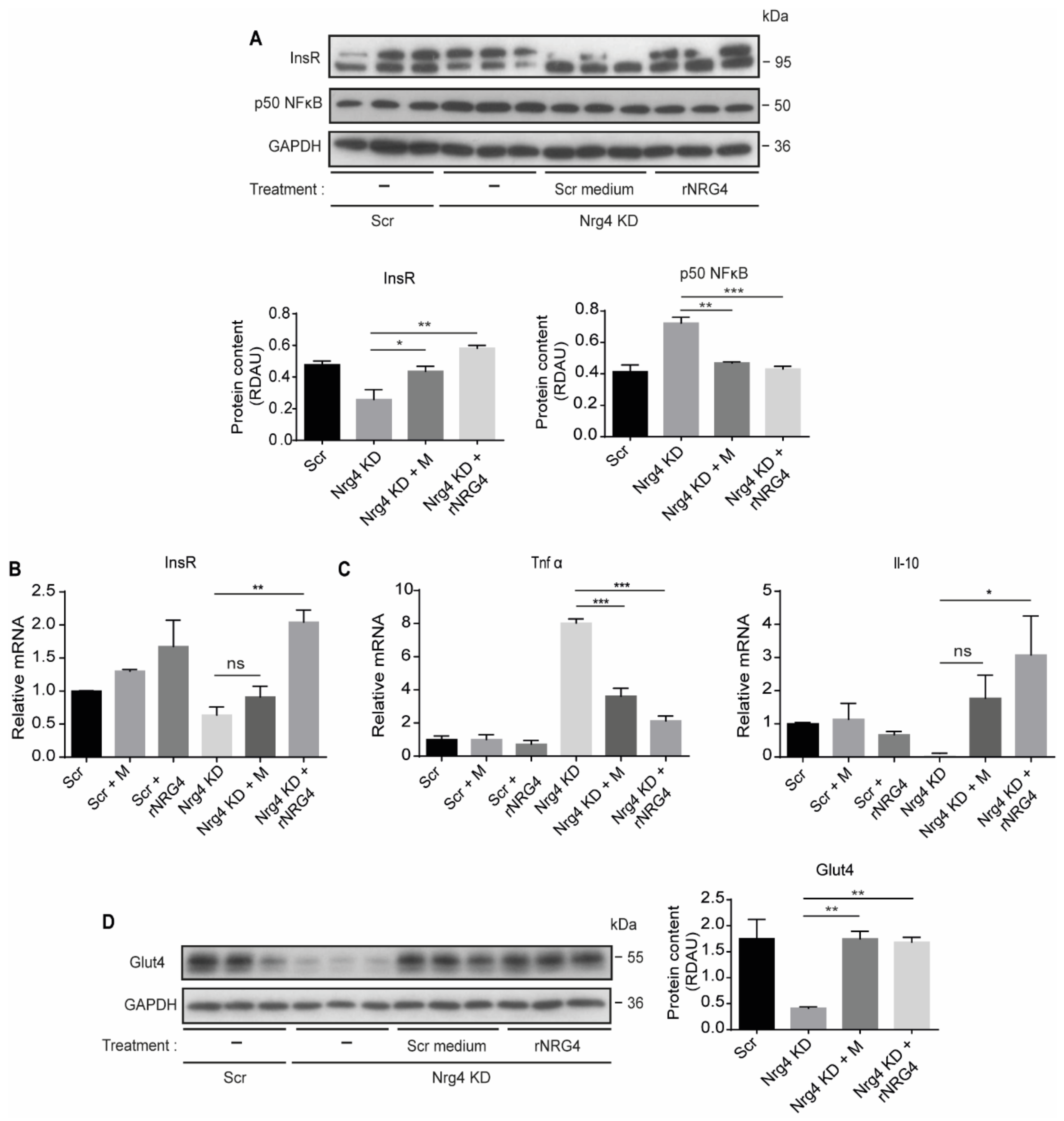
2.4. Treatment of Nrg4 KD Adipocytes with Conditioned Medium from Control Adipocytes or with Recombinant Nrg4 Reverses Inflammation and Restores Insulin Receptor and Glut4 Content
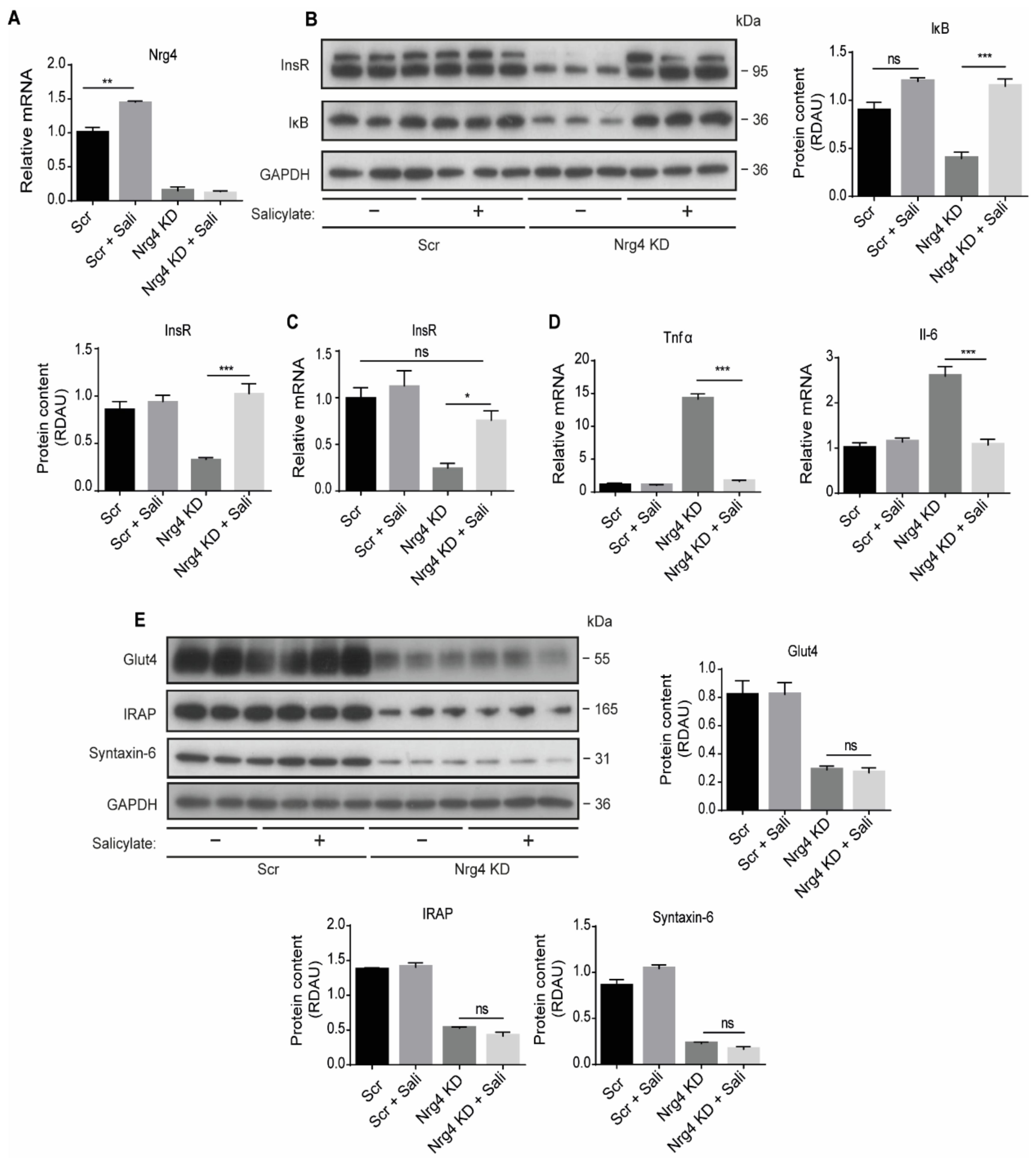
2.5. Sodium Salicylate and Dexamethasone Restore the Expression of the Insulin Receptor, but Not the Content of Glut4 Protein in Nrg4 KD Adipocytes
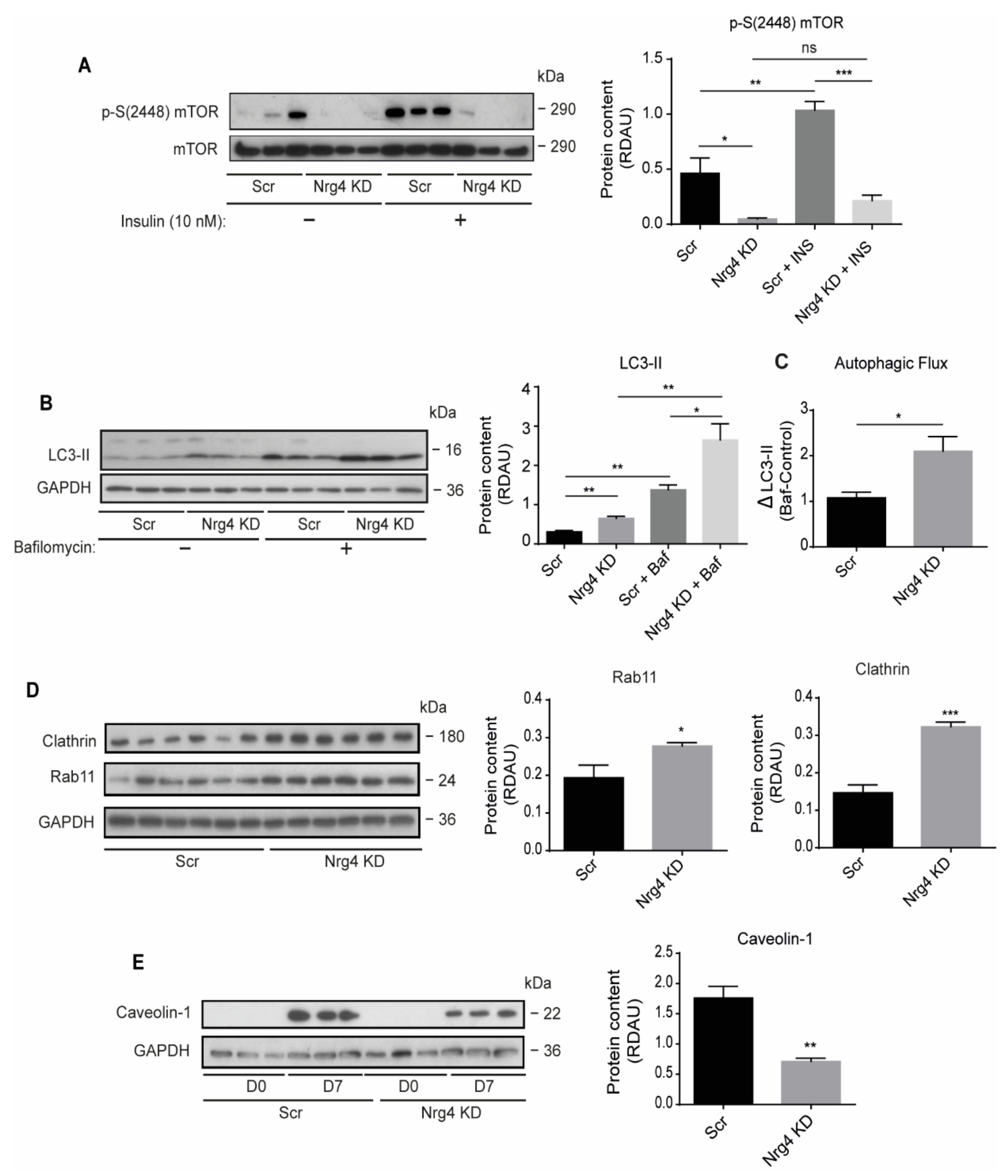
2.6. Autophagy Is Enhanced in Nrg4 KD Adipocytes
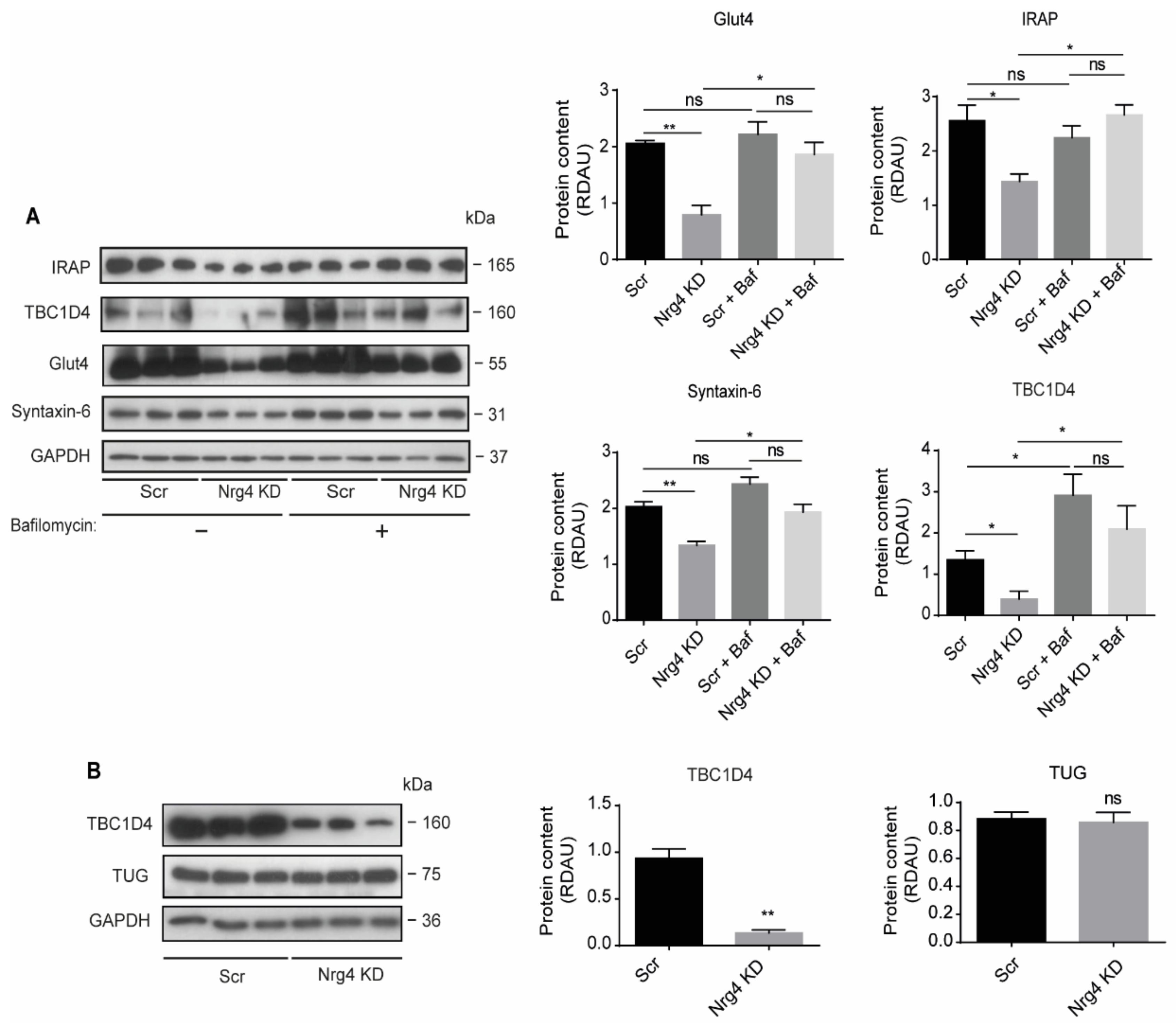
2.7. Blockade of Autophagy Restores Glut4 and GSV Proteins Content in Nrg4 KD Adipocytes
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. White Adipose Tissue Fractionation
4.3. T3-L1 Adipocyte Cell Culture and Differentiation
4.4. Lentiviral Production and Infection of 3T3-L1 Preadipocytes
4.5. Cell Treatments
4.6. Measurement of 2-d-Deoxyglucose Uptake
4.7. RNA Extraction and Quantitative PCR
4.8. Protein Extraction and Western Blotting
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Gumà, A.; Martínez-Redondo, V.; López-Soldado, I.; Cantó, C.; Zorzano, A. Emerging role of neuregulin as a modulator of muscle metabolism. Am. J. Physiol. Metab. 2010, 298, E742–E750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, D.; Birchmeier, C. Multiple essential functions of neuregulin in development. Nat. Cell Biol. 1995, 378, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-F.; Simon, H.; Chen, H.; Bates, B.; Hung, M.-C.; Hauser, C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nat. Cell Biol. 1995, 378, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, M.; Casagranda, F.; Orioli, D.; Simon, H.; Lai, C.; Klein, R.; Lemke, G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nat. Cell Biol. 1995, 378, 390–394. [Google Scholar] [CrossRef]
- Suárez, E.; Bach, D.; Cadefau, J.; Palacín, M.; Zorzano, A.; Gumà, A. A novel role of neuregulin in skeletal muscle: Neuregulin stimulates glucose uptake, glucose transporter translocation, and transporter expression in muscle cells. J. Biol. Chem. 2001, 276, 18257–18264. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Chibalin, A.; Barnes, B.R.; Glund, S.; Suárez, E.; Ryder, J.W.; Palacín, M.; Zierath, J.; Zorzano, A.; Gumà, A. Neuregulins Mediate Calcium-induced Glucose Transport during Muscle Contraction. J. Biol. Chem. 2006, 281, 21690–21697. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-X.; Zhao, X.-Y.; Meng, Z.-X.; Kern, M.; Dietrich, A.; Chen, Z.; Cozacov, Z.; Zhou, D.; Okunade, A.L.; Su, X.; et al. The brown fat–enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat. Med. 2014, 20, 1436–1443. [Google Scholar] [CrossRef]
- Harari, D.; Tzahar, E.; Romano, J.; Shelly, M.; Pierce, J.H.; Andrews, G.C.; Yarden, Y. Neuregulin-4: A novel growth factor that acts through the ErbB-4 receptor tyrosine kinase. Oncogene 1999, 18, 2681–2689. [Google Scholar] [CrossRef] [Green Version]
- Rosell, M.; Kaforou, M.; Frontini, A.; Okolo, A.; Chan, Y.-W.; Nikolopoulou, E.; Millership, S.; Fenech, M.E.; MacIntyre, D.; Turner, J.O.; et al. Brown and white adipose tissues: Intrinsic differences in gene expression and response to cold exposure in mice. Am. J. Physiol. Metab. 2014, 306, E945–E964. [Google Scholar] [CrossRef] [Green Version]
- Comas, F.; Martínez, C.; Sabater, M.; Ortega, F.; Latorre, J.; Díaz-Sáez, F.; Aragonés, J.; Camps, M.; Gumà, A.; Ricart, W.; et al. Neuregulin 4 Is a Novel Marker of Beige Adipocyte Precursor Cells in Human Adipose Tissue. Front. Physiol. 2019, 10, 39. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, G.-X.; Ma, S.L.; Jung, D.Y.; Ha, H.; Altamimi, T.; Zhao, X.-Y.; Guo, L.; Zhang, P.; Hu, C.-R.; et al. Nrg4 promotes fuel oxidation and a healthy adipokine profile to ameliorate diet-induced metabolic disorders. Mol. Metab. 2017, 6, 863–872. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, P.; Chen, Z.; Xia, H.; Li, S.; Zhang, Y.; Kobberup, S.; Zou, W.; Lin, J.D. Hepatic neuregulin 4 signaling defines an endocrine checkpoint for steatosis-to-NASH progression. J. Clin. Investig. 2017, 127, 4449–4461. [Google Scholar] [CrossRef] [Green Version]
- Nugroho, D.B.; Ikeda, K.; Barinda, A.J.; Wardhana, D.A.; Yagi, K.; Miyata, K.; Oike, Y.; Hirata, K.-I.; Emoto, N. Neuregulin-4 is an angiogenic factor that is critically involved in the maintenance of adipose tissue vasculature. Biochem. Biophys. Res. Commun. 2018, 503, 378–384. [Google Scholar] [CrossRef]
- Nugroho, D.B.; Ikeda, K.; Kajimoto, K.; Hirata, K.-I.; Emoto, N. Activation of neuregulin-4 in adipocytes improves metabolic health by enhancing adipose tissue angiogenesis. Biochem. Biophys. Res. Commun. 2018, 504, 427–433. [Google Scholar] [CrossRef]
- Zeng, F.; Wang, Y.; Kloepfer, L.A.; Wang, S.; Harris, R.C. ErbB4 deletion predisposes to development of metabolic syndrome in mice. Am. J. Physiol. Metab. 2018, 315, E583–E593. [Google Scholar] [CrossRef]
- Bogan, J.S.; Kandror, K.V. Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr. Opin. Cell Biol. 2010, 22, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Perera, H.K.I.; Clarke, M.; Morris, N.; Hong, W.; Chamberlain, L.H.; Gould, G.W. Syntaxin 6 Regulates Glut4 Trafficking in 3T3-L1 Adipocytes. Mol. Biol. Cell 2003, 14, 2946–2958. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Miles, P.D.G.; Ladd, C.M.; Ross, K.; Golub, T.R.; Olefsky, J.M.; Lodish, H.F. Profiling Gene Transcription In Vivo Reveals Adipose Tissue as an Immediate Target of Tumor Necrosis Factor-α: Implications for Insulin Resistance. Diabetes 2002, 51, 3176–3188. [Google Scholar] [CrossRef] [Green Version]
- Pellegrinelli, V.; Peirce, V.J.; Howard, L.; Virtue, S.; Türei, D.; Senzacqua, M.; Frontini, A.; Dalley, J.W.; Horton, A.R.; Bidault, G.; et al. Adipocyte-secreted BMP8b mediates adrenergic-induced remodeling of the neuro-vascular network in adipose tissue. Nat. Commun. 2018, 9, 4974. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Chiang, G.G.; Abraham, R.T. Phosphorylation of Mammalian Target of Rapamycin (mTOR) at Ser-2448 IsMediated by p70S6 Kinase. J. Biol. Chem. 2005, 280, 25485–25490. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Longatti, A.; Lamb, C.; Razi, M.; Yoshimura, S.-I.; Barr, F.; Tooze, S.A. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J. Cell Biol. 2012, 197, 659–675. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.; Liu, M.; Ma, L.; Du, W.; Zhang, H.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.; et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef]
- Le Lay, S.; Briand, N.; Blouin, C.M.; Chateau, D.; Prado, C.; Lasnier, F.; Liepvre, X.; Hajduch, E.; Dugail, I. The lipoatrophic caveolin-1 deficient mouse model reveals autophagy in mature adipocytes. Autophagy 2010, 6, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 Is a Critical Determinant of Autophagy, Metabolic Switching, and Oxidative Stress in Vascular Endothelium. PLoS ONE 2014, 9, e87871. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Tan, S.-H.; Ng, S.; Zhou, J.; Yang, N.-D.; Koo, G.-B.; McMahon, K.-A.; Parton, R.G.; Hill, M.M.; Del Pozo, M.A.; et al. Critical role of CAV1/caveolin-1 in cell stress responses in human breast cancer cells via modulation of lysosomal function and autophagy. Autophagy 2015, 11, 769–784. [Google Scholar] [CrossRef] [Green Version]
- Sano, H.; Kane, S.; Sano, E.; Mîinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-stimulated Phosphorylation of a Rab GTPase-activating Protein Regulates GLUT4 Translocation. J. Biol. Chem. 2003, 278, 14599–14602. [Google Scholar] [CrossRef] [Green Version]
- Larance, M.; Ramm, G.; Stöckli, J.; van Dam, E.M.; Winata, S.; Wasinger, V.; Simpson, F.; Graham, M.; Junutula, J.R.; Guilhaus, M.; et al. Characterization of the Role of the Rab GTPase-activating Protein AS160 in Insulin-regulated GLUT4 Trafficking. J. Biol. Chem. 2005, 280, 37803–37813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadt, A.; Immisch, A.; de Wendt, C.; Springer, C.; Zhou, Z.; Stermann, T.; Holman, G.D.; Loffing-Cueni, D.; Loffing, J.; Joost, H.-G.; et al. Deletion of Both Rab-GTPase–Activating Proteins TBC1D1 and TBC1D4 in Mice Eliminates Insulin- and AICAR-Stimulated Glucose Transport. Diabetes 2014, 64, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klip, A.; McGraw, T.E.; James, D.E. Thirty sweet years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belman, J.; Bian, R.R.; Habtemichael, E.N.; Li, D.; Jurczak, M.; Alcazar-Roman, A.R.; McNally, L.J.; Shulman, G.; Bogan, J. Acetylation of TUG Protein Promotes the Accumulation of GLUT4 Glucose Transporters in an Insulin-responsive Intracellular Compartment. J. Biol. Chem. 2015, 290, 4447–4463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, E.; Calvo, J.C. ErbB2 and EGFR are downmodulated during the differentiation of 3T3-L1 preadipocytes. J. Cell. Biochem. 2003, 90, 561–572. [Google Scholar] [CrossRef]
- Kim, D.; Chi, S.; Lee, K.H.; Rhee, S.; Kwon, Y.K.; Chung, C.H.; Kwon, H.; Kang, M.-S. Neuregulin Stimulates Myogenic Differentiation in an Autocrine Manner. J. Biol. Chem. 1999, 274, 15395–15400. [Google Scholar] [CrossRef] [Green Version]
- Bernard, J.K.; McCann, S.P.; Bhardwaj, V.; Washington, M.K.; Frey, M.R. Neuregulin-4 Is a Survival Factor for Colon Epithelial Cells both in Culture and in Vivo. J. Biol. Chem. 2012, 287, 39850–39858. [Google Scholar] [CrossRef] [Green Version]
- McElroy, S.; Castle, S.L.; Bernard, J.K.; Almohazey, D.; Hunter, C.J.; Bell, B.A.; Al Alam, D.; Wang, L.; Ford, H.R.; Frey, M.R. The ErbB4 Ligand Neuregulin-4 Protects against Experimental Necrotizing Enterocolitis. Am. J. Pathol. 2014, 184, 2768–2778. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Hedl, M.; Abraham, C.; Bernard, J.K.; Lozano, P.R.; Hsieh, J.J.; Almohazey, D.; Bucar, E.B.; Punit, S.; Dempsey, P.J.; et al. ErbB4 signaling stimulates pro-inflammatory macrophage apoptosis and limits colonic inflammation. Cell Death Dis. 2017, 8, e2622. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [Green Version]
- Stephens, J.M.; Lee, J.; Pilch, P.F. Tumor Necrosis Factor-α-induced Insulin Resistance in 3T3-L1 Adipocytes Is Accompanied by a Loss of Insulin Receptor Substrate-1 and GLUT4 Expression without a Loss of Insulin Receptor-mediated Signal Transduction. J. Biol. Chem. 1997, 272, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Rotter, V.; Nagaev, I.; Smith, U. Interleukin-6 (IL-6) Induces Insulin Resistance in 3T3-L1 Adipocytes and Is, Like IL-8 and Tumor Necrosis Factor-α, Overexpressed in Human Fat Cells from Insulin-resistant Subjects. J. Biol. Chem. 2003, 278, 45777–45784. [Google Scholar] [CrossRef] [Green Version]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Ros-Baró, A.; López-Iglesias, C.; Peiró, S.; Bellido, D.; Palacín, M.; Zorzano, A.; Camps, M. Lipid rafts are required for GLUT4 internalization in adipose cells. Proc. Natl. Acad. Sci. USA 2001, 98, 12050–12055. [Google Scholar] [CrossRef] [Green Version]
- Shi, A.; Sun, L.; Banerjee, R.; Tobin, M.; Zhang, Y.; Grant, B.D. Regulation of endosomal clathrin and retromer-mediated endosome to Golgi retrograde transport by the J-domain protein RME-8. EMBO J. 2009, 28, 3290–3302. [Google Scholar] [CrossRef] [Green Version]
- Gonzaález-Munñoz, E.; Loópez-Iglesias, C.; Calvo, M.; Palaciín, M.; Zorzano, A.; Camps, M. Caveolin-1 Loss of Function Accelerates Glucose Transporter 4 and Insulin Receptor Degradation in 3T3-L1 Adipocytes. Endocrinology 2009, 150, 3493–3502. [Google Scholar] [CrossRef] [Green Version]
- Gumà, A.; Mora, C.; Santalucía, T.; Viñals, F.; Testar, X.; Palacín, M.; Zorzano, A. System A transport activity is stimulated in skeletal muscle in response to diabetes. FEBS Lett. 1992, 310, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Sáez, F.; Blanco-Sinfreu, C.; Archilla-Ortega, A.; Sebastian, D.; Romero, M.; Hernández-Alvarez, M.I.; Mora, S.; Testar, X.; Ricart, W.; Fernández-Real, J.M.; et al. Neuregulin 4 Downregulation Induces Insulin Resistance in 3T3-L1 Adipocytes through Inflammation and Autophagic Degradation of GLUT4 Vesicles. Int. J. Mol. Sci. 2021, 22, 12960. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312960
Díaz-Sáez F, Blanco-Sinfreu C, Archilla-Ortega A, Sebastian D, Romero M, Hernández-Alvarez MI, Mora S, Testar X, Ricart W, Fernández-Real JM, et al. Neuregulin 4 Downregulation Induces Insulin Resistance in 3T3-L1 Adipocytes through Inflammation and Autophagic Degradation of GLUT4 Vesicles. International Journal of Molecular Sciences. 2021; 22(23):12960. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312960
Chicago/Turabian StyleDíaz-Sáez, Francisco, Carla Blanco-Sinfreu, Adrià Archilla-Ortega, David Sebastian, Montserrat Romero, Maria Isabel Hernández-Alvarez, Sílvia Mora, Xavier Testar, Wifredo Ricart, José Manuel Fernández-Real, and et al. 2021. "Neuregulin 4 Downregulation Induces Insulin Resistance in 3T3-L1 Adipocytes through Inflammation and Autophagic Degradation of GLUT4 Vesicles" International Journal of Molecular Sciences 22, no. 23: 12960. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312960