
Bifunctional Inhibitors of Influenza Virus Neuraminidase: Molecular Design of a Sulfonamide Linker


Abstract
:1. Introduction
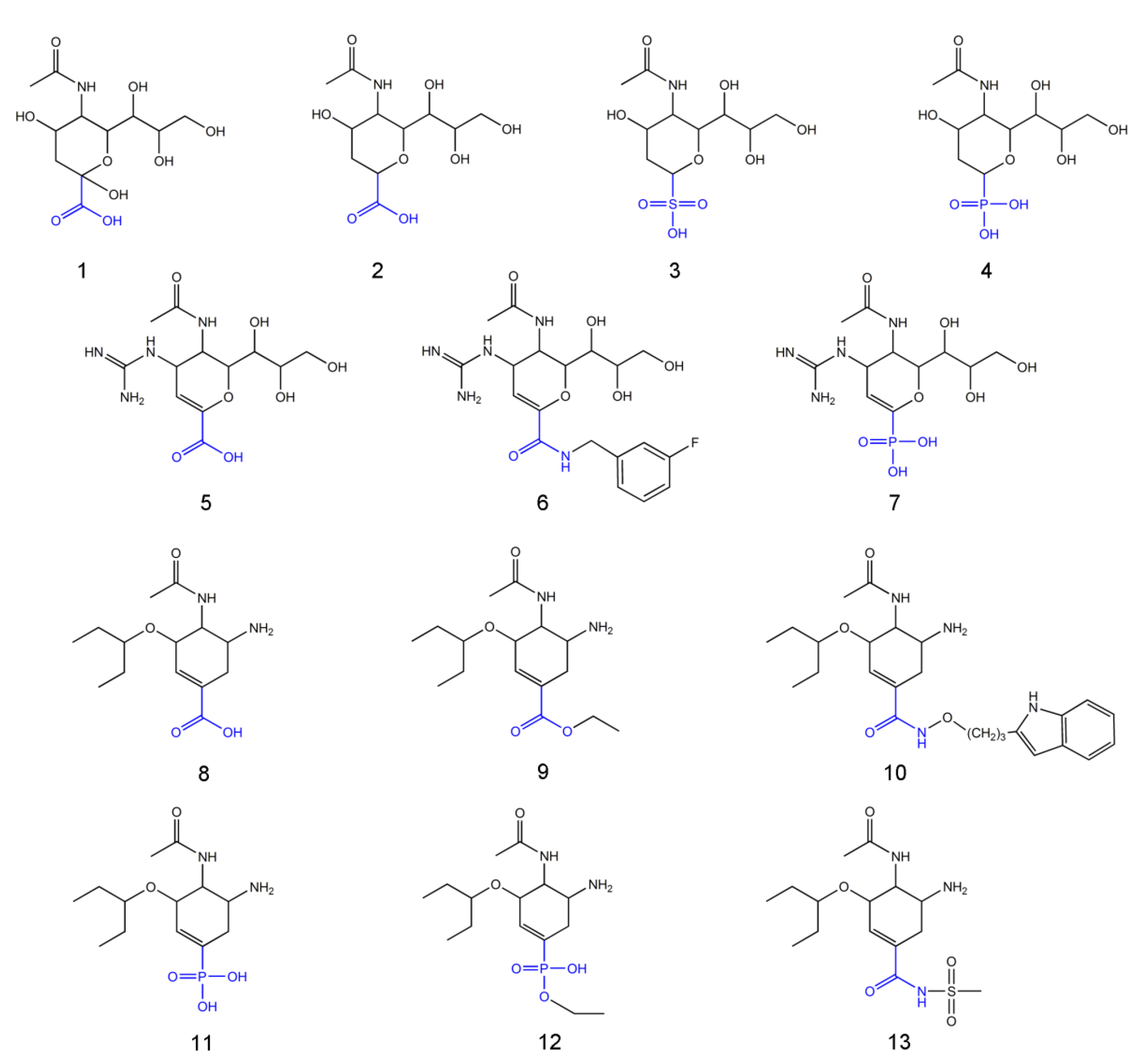
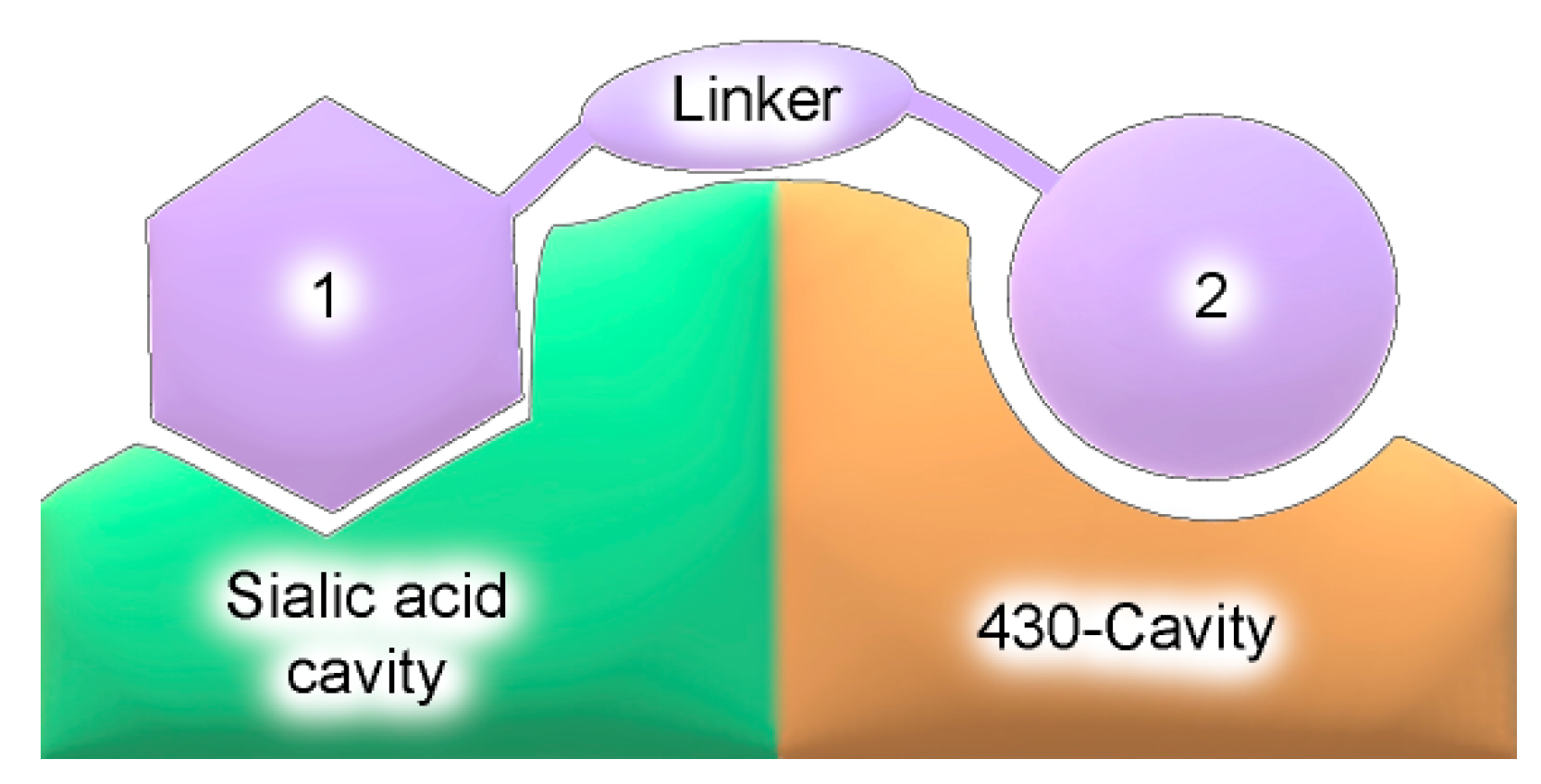
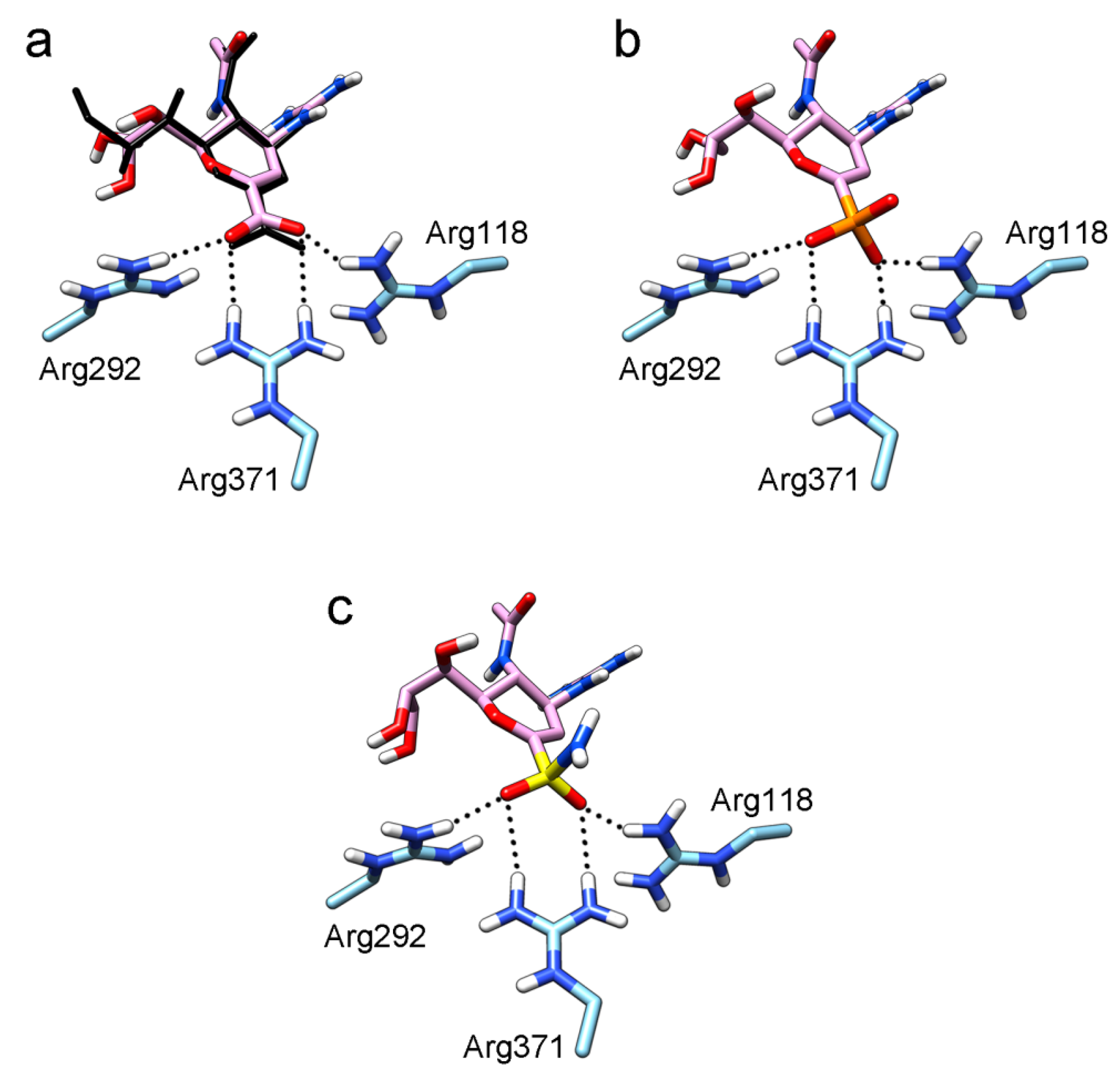
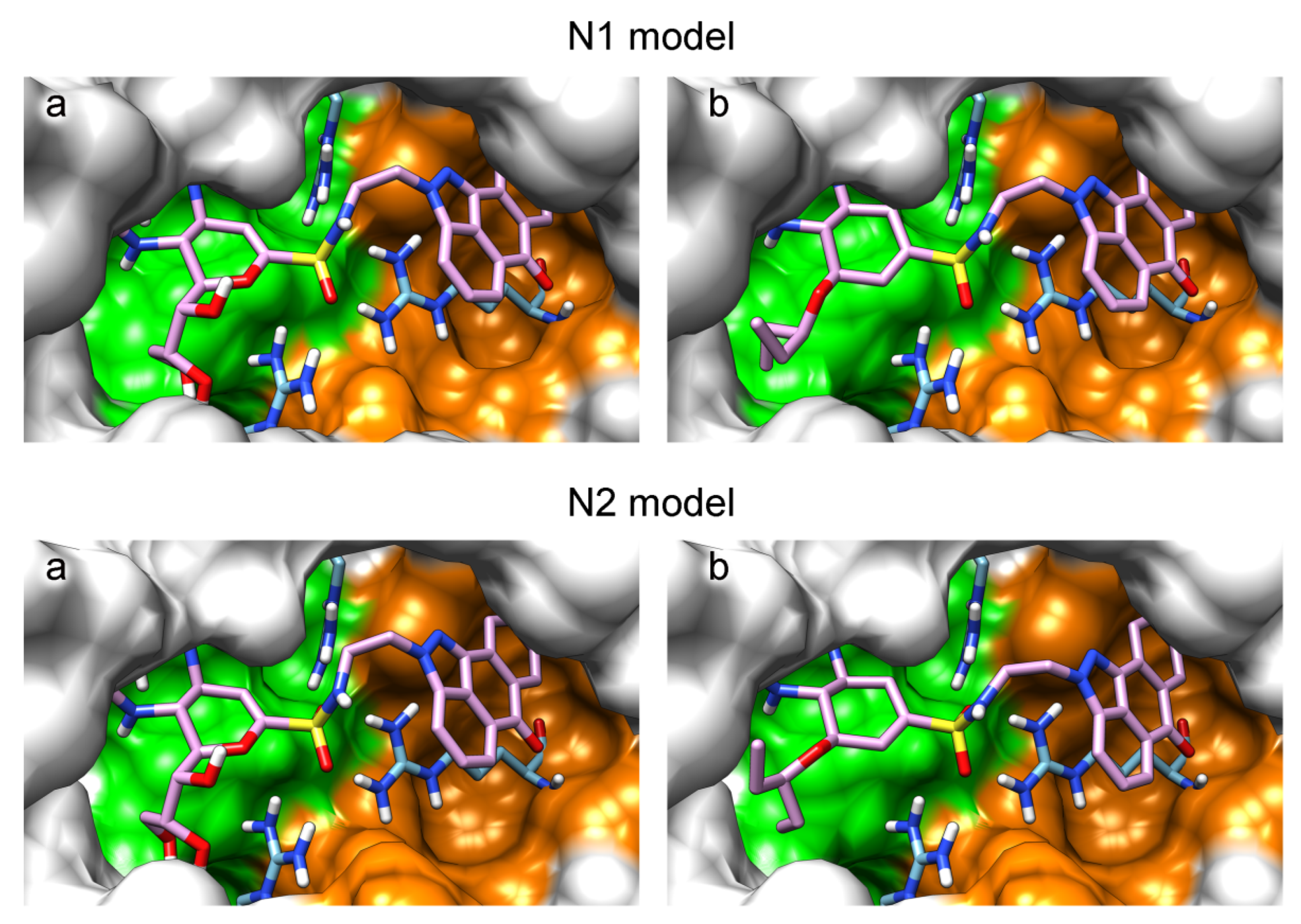
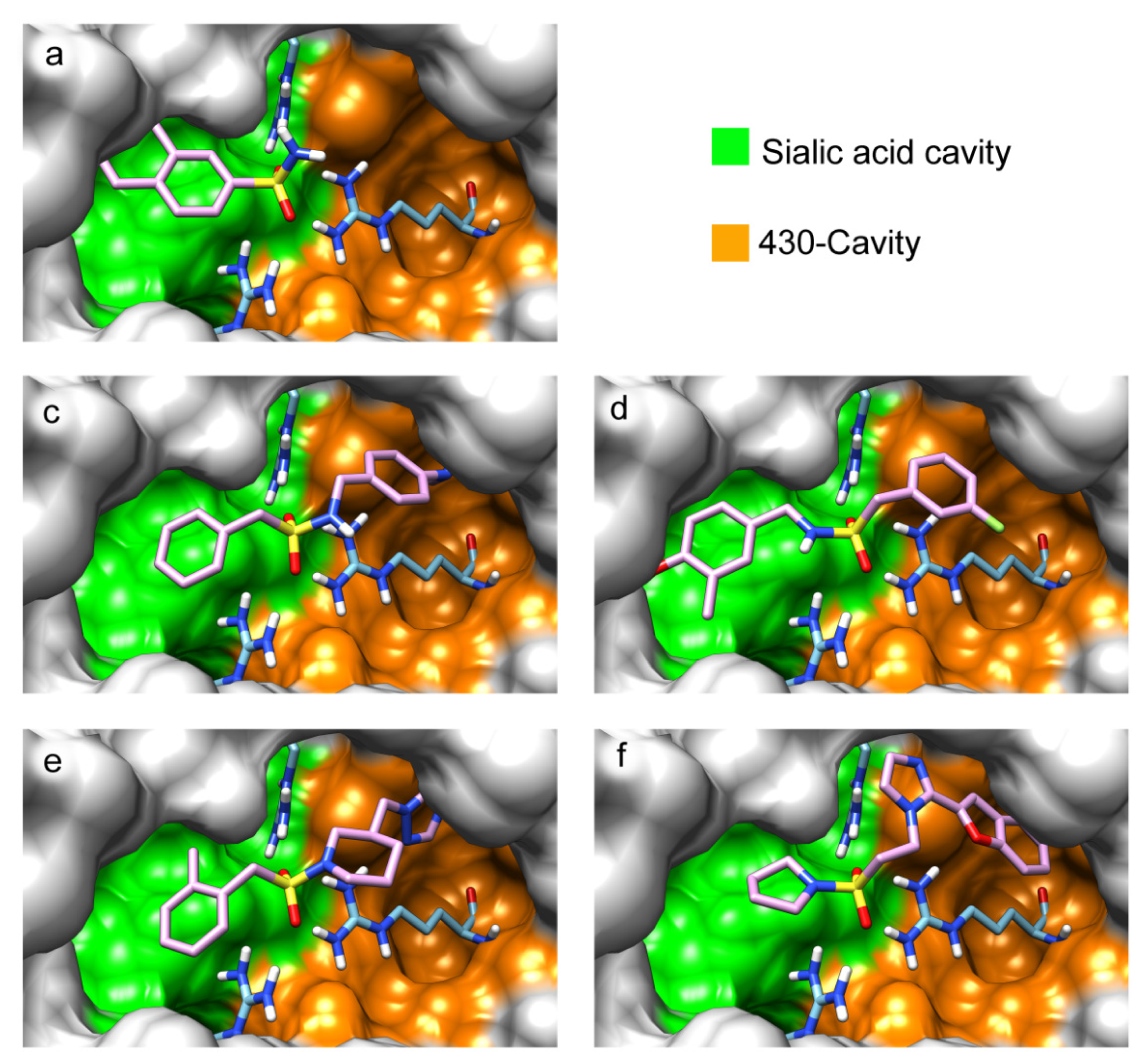
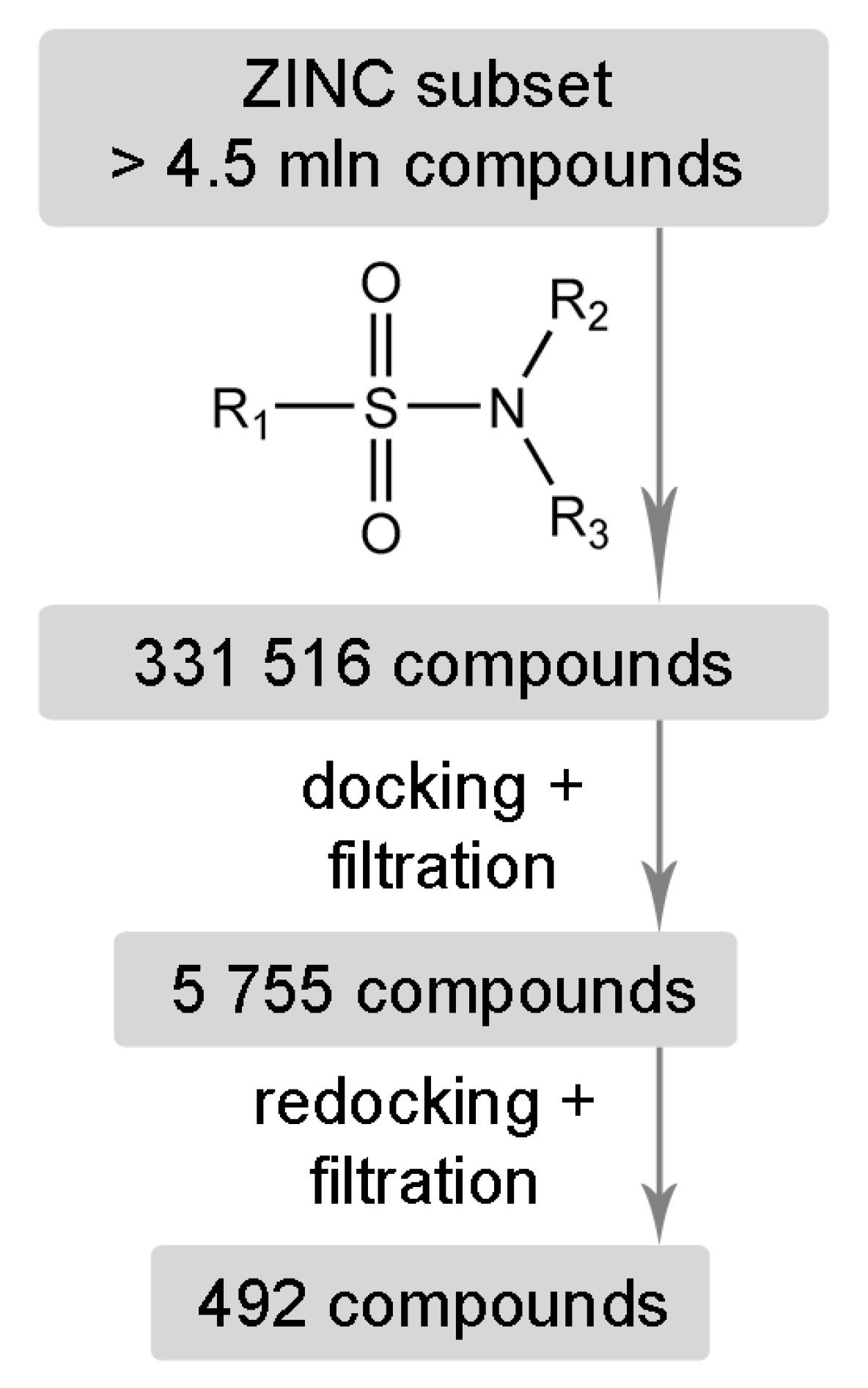
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zambon, M.C. Epidemiology and pathogenesis of influenza. J. Antimicrob. Chemother. 1999, 44, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Postnikova, Y.; Treshchalina, A.; Boravleva, E.; Gambaryan, A.; Ishmukhametov, A.; Matrosovich, M.; Fouchier, R.A.M.; Sadykova, G.; Prilipov, A.; Lomakina, N. Diversity and reassortment rate of influenza A viruses in wild ducks and gulls. Viruses 2021, 13, 1010. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Morens, D.M. Pandemic influenza--including a risk assessment of H5N1. Rev. Sci. Tech. 2009, 28, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef] [Green Version]
- Herold, S.; Becker, C.; Ridge, K.M.; Budinger, G.R. Influenza virus-induced lung injury: Pathogenesis and implications for treatment. Eur. Respir. J. 2015, 45, 1463–1478. [Google Scholar] [CrossRef] [Green Version]
- Von Itzstein, M.; Wu, W.Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef]
- Von Itzstein, M. The war against influenza: Discovery and development of sialidase inhibitors. Nat. Rev. Drug Discov. 2007, 6, 967–974. [Google Scholar] [CrossRef]
- Lehnert, R.; Pletz, M.; Reuss, A.; Schaberg, T. Antiviral medications in seasonal and pandemic influenza. Dtsch. Arztebl. Int. 2016, 113, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colman, P.M. Influenza virus neuraminidase: Structure, antibodies, and inhibitors. Protein Sci. 1994, 3, 1687–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtyrya, Y.A.; Mochalova, L.V.; Bovin, N.V. Influenza virus neuraminidase: Structure and function. Acta Nat. 2009, 1, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Thorlund, K.; Awad, T.; Boivin, G.; Thabane, L. Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect. Dis. 2011, 11, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharan, N.J.; Gubareva, L.V.; Meyer, J.J.; Okomo-Adhiambo, M.; McClinton, R.C.; Marshall, S.A.; St. George, K.; Epperson, S.; Brammer, L.; Klimov, A.I.; et al. Infections with oseltamivir-resistant influenza A(H1N1) virus in the United States. JAMA 2009, 301, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Ilyushina, N.A.; Seiler, J.P.; Rehg, J.E.; Webster, R.G.; Govorkova, E.A. Effect of neuraminidase inhibitor-resistant mutations on pathogenicity of clade 2.2 A/Turkey/15/06 (H5N1) influenza virus in ferrets. PLoS Pathog. 2010, 6, e1000933. [Google Scholar]
- Meijer, A.; Rebelo-de-Andrade, H.; Correia, V.; Besselaar, T.; Drager-Dayal, R.; Fry, A.; Gregory, V.; Gubareva, L.; Kageyama, T.; Lackenby, A.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2012–2013. Antiviral Res. 2014, 110, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Hurt, A.C.; Besselaar, T.G.; Daniels, R.S.; Ermetal, B.; Fry, A.; Gubareva, L.; Huang, W.; Lackenby, A.; Lee, R.T.; Lo, J.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2014–2015. Antiviral Res. 2016, 132, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.T.; Tawa, P.; Auger, A.; Wang, D.; Su, H.P.; Yan, Y.; Hazuda, D.J.; Miller, M.D.; Asante-Appiah, E.; Melnyk, R.A. Identification of novel bifunctional HIV-1 reverse transcriptase inhibitors. J. Antimicrob. Chemother. 2018, 73, 109–117. [Google Scholar] [CrossRef]
- Ciubotaru, M.; Musat, M.G.; Surleac, M.; Ionita, E.; Petrescu, A.J.; Abele, E.; Abele, R. The design of new HIV-IN tethered bifunctional inhibitors using multiple microdomain targeted docking. Curr. Med. Chem. 2019, 26, 2574–2600. [Google Scholar] [CrossRef]
- Amaro, R.E.; Minh, D.D.; Cheng, L.S.; Lindstrom, W.M., Jr.; Olson, A.J.; Lin, J.H.; Li, W.W.; McCammon, J.A. Remarkable loop flexibility in avian influenza N1 and its implications for antiviral drug design. J. Am. Chem. Soc. 2007, 129, 7764–7765. [Google Scholar] [CrossRef]
- Landon, M.R.; Amaro, R.E.; Baron, R.; Ngan, C.H.; Ozonoff, D.; McCammon, J.A.; Vajda, S. Novel druggable hot spots in avian influenza neuraminidase H5N1 revealed by computational solvent mapping of a reduced and representative receptor ensemble. Chem. Biol. Drug Des. 2008, 71, 106–116. [Google Scholar] [CrossRef]
- Cheng, L.S.; Amaro, R.E.; Xu, D.; Li, W.W.; Arzberger, P.W.; McCammon, J.A. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. J. Med. Chem. 2008, 51, 3878–3894. [Google Scholar] [CrossRef] [Green Version]
- Amaro, R.E.; Cheng, X.; Ivanov, I.; Xu, D.; McCammon, J.A. Characterizing loop dynamics and ligand recognition in human- and avian-type influenza neuraminidases via generalized born molecular dynamics and end-point free energy calculations. J. Am. Chem. Soc. 2009, 131, 4702–4709. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, K.; Dyason, J.C.; Maggioni, A.; von Itzstein, M.; Downard, K.M. Binding of a natural anthocyanin inhibitor to influenza neuraminidase by mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 6563–6572. [Google Scholar] [CrossRef]
- Varghese, J.N.; McKimm-Breschkin, J.L.; Caldwell, J.B.; Kortt, A.A.; Colman, P.M. The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins 1992, 14, 327–332. [Google Scholar] [CrossRef]
- Taylor, N.R.; von Itzstein, M. Molecular modeling studies on ligand binding to sialidase from influenza virus and the mechanism of catalysis. J. Med. Chem. 1994, 37, 616–624. [Google Scholar] [CrossRef]
- Cheng, T.J.; Weinheimer, S.; Tarbet, E.B.; Jan, J.T.; Cheng, Y.S.; Shie, J.J.; Chen, C.L.; Chen, C.A.; Hsieh, W.C.; Huang, P.W.; et al. Development of oseltamivir phosphonate congeners as anti-influenza agents. J. Med. Chem. 2012, 55, 8657–8670. [Google Scholar] [CrossRef] [Green Version]
- Feng, E.; Shin, W.J.; Zhu, X.; Li, J.; Ye, D.; Wang, J.; Zheng, M.; Zuo, J.P.; No, K.T.; Liu, X.; et al. Structure-based design and synthesis of C-1- and C-4-modified analogs of zanamivir as neuraminidase inhibitors. J. Med. Chem. 2013, 56, 671–684. [Google Scholar] [CrossRef]
- Hadházi, Á.; Pascolutti, M.; Bailly, B.; Dyason, J.C.; Borbás, A.; Thomson, R.J.; von Itzstein, M. A sialosyl sulfonate as a potent inhibitor of influenza virus replication. Org. Biomol. Chem. 2017, 15, 5249–5253. [Google Scholar] [CrossRef] [Green Version]
- Shie, J.J.; Fang, J.M. Development of effective anti-influenza drugs: Congeners and conjugates—A review. J. Biomed. Sci. 2019, 26, 84. [Google Scholar] [CrossRef] [Green Version]
- Ballatore, C.; Huryn, D.M.; Smith, A.B., 3rd. Carboxylic acid (bio)isosteres in drug design. ChemMedChem 2013, 8, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Lassalas, P.; Gay, B.; Lasfargeas, C.; James, M.J.; Tran, V.; Vijayendran, K.G.; Brunden, K.R.; Kozlowski, M.C.; Thomas, C.J.; Smith, A.B., 3rd; et al. Structure property relationships of carboxylic acid isosteres. J. Med. Chem. 2016, 59, 3183–3203. [Google Scholar] [CrossRef]
- Lemke, T.L. Review of Organic Functional Groups: Introduction to Medicinal Organic Chemistry; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2012; pp. 86–87. [Google Scholar]
- White, C.L.; Janakiraman, M.N.; Laver, W.G.; Philippon, C.; Vasella, A.; Air, G.M.; Luo, M. A sialic acid-derived phosphonate analog inhibits different strains of influenza virus neuraminidase with different efficiencies. J. Mol. Biol. 1995, 245, 623–634. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M.; Lai, P.T.; Wen, W.H.; Wang, S.Y.; Cheng, Y.S.; Tsai, K.C.; Yang, A.S.; Wong, C.H. A practical synthesis of zanamivir phosphonate congeners with potent anti-influenza activity. J. Am. Chem. Soc. 2011, 133, 17959–17965. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.T.; Chen, C.L.; Fang, J.M.; Tsai, K.C.; Wang, S.Y.; Huang, W.I.; Cheng, Y.E.; Wong, C.H. Oseltamivir hydroxamate and acyl sulfonamide derivatives as influenza neuraminidase inhibitors. Bioorg. Med. Chem. 2014, 22, 6647–6654. [Google Scholar] [CrossRef]
- Vavricka, C.J.; Muto, C.; Hasunuma, T.; Kimura, Y.; Araki, M.; Wu, Y.; Gao, G.F.; Ohrui, H.; Izumi, M.; Kiyota, H. Synthesis of sulfo-sialic acid analogues: Potent neuraminidase inhibitors in regards to anomeric functionality. Sci. Rep. 2017, 7, 8239. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Escarpe, P.A.; Eisenberg, E.J.; Cundy, K.C.; Sweet, C.; Jakeman, K.J.; Merson, J.; Lew, W.; Williams, M.; Zhang, L.; et al. Identification of GS 4104 as an orally bioavailable prodrug of the influenza virus neuraminidase inhibitor GS 4071. Antimicrob. Agents Chemother. 1998, 42, 647–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.E. Pharmacokinetics of oseltamivir: An oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J. Antimicrob. Chemother. 2010, 65, ii5–ii10. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur containing scaffolds in drugs: Synthesis and application in medicinal chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef]
- Apaydın, S.; Török, M. Sulfonamide derivatives as multi-target agents for complex diseases. Bioorg. Med. Chem. Lett. 2019, 29, 2042–2050. [Google Scholar] [CrossRef]
- Vega-Hissi, E.G.; Andrada, M.F.; Zamarbide, G.N.; Estrada, M.R.; Tomás-Vert, F. Theoretical studies on sulfanilamide and derivatives with antibacterial activity: Conformational and electronic analysis. J. Mol. Model. 2011, 17, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., 3rd; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Xu, X.; Zhu, X.; Dwek, R.A.; Stevens, J.; Wilson, I.A. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol. 2008, 82, 10493–10501. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Vavricka, C.J.; Li, Q.; Wu, Y.; Qi, J.; Wang, M.; Liu, Y.; Gao, F.; Liu, J.; Feng, E.; He, J.; et al. Structural and functional analysis of laninamivir and its octanoate prodrug reveals group specific mechanisms for influenza NA inhibition. PLoS Pathog. 2011, 7, e1002249. [Google Scholar] [CrossRef]
- Schwab, C.H. Conformations and 3D pharmacophore searching. Drug Discov. Today Technol. 2010, 7, e245–e253. [Google Scholar] [CrossRef] [PubMed]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. Lead finder: An approach to improve accuracy of protein-ligand docking, binding energy estimation, and virtual screening. J. Chem. Inf. Model. 2008, 48, 2371–2385. [Google Scholar] [CrossRef] [PubMed]
- Novikov, F.N.; Stroylov, V.S.; Zeifman, A.A.; Stroganov, O.V.; Kulkov, V.; Chilov, G.G. Lead Finder docking and virtual screening evaluation with Astex and DUD test sets. J. Comput. Aided Mol. Des. 2012, 26, 725–735. [Google Scholar] [CrossRef]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. Engl. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Gushchina, I.V.; Polenova, A.M.; Suplatov, D.A.; Švedas, V.K.; Nilov, D.K. vsFilt: A tool to improve virtual screening by structural filtration of docking poses. J. Chem. Inf. Model. 2020, 60, 3692–3696. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Group | Linker Structure | Optimal Interaction 1 | Resistance to Hydrolysis |
|---|---|---|---|
| Carboxyl |  | + | − |
| Amide |  | − | + |
| Hydroxamic |  | − | +− |
| Sulfo |  | + | − |
| Phosphono |  | + | − |
| Sulfonamide |  | ??? | + |
| a |  | 31 | b |  | 0 |
| c |  | 214 | d |  | 59 |
| e |  | 122 (46) | f |  | 66 (63) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evteev, S.; Nilov, D.; Polenova, A.; Švedas, V. Bifunctional Inhibitors of Influenza Virus Neuraminidase: Molecular Design of a Sulfonamide Linker. Int. J. Mol. Sci. 2021, 22, 13112. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313112
Evteev S, Nilov D, Polenova A, Švedas V. Bifunctional Inhibitors of Influenza Virus Neuraminidase: Molecular Design of a Sulfonamide Linker. International Journal of Molecular Sciences. 2021; 22(23):13112. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313112
Chicago/Turabian StyleEvteev, Sergei, Dmitry Nilov, Aleksandra Polenova, and Vytas Švedas. 2021. "Bifunctional Inhibitors of Influenza Virus Neuraminidase: Molecular Design of a Sulfonamide Linker" International Journal of Molecular Sciences 22, no. 23: 13112. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313112