
Poly(aspartic acid) Biohydrogel as the Base of a New Hybrid Conducting Material

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
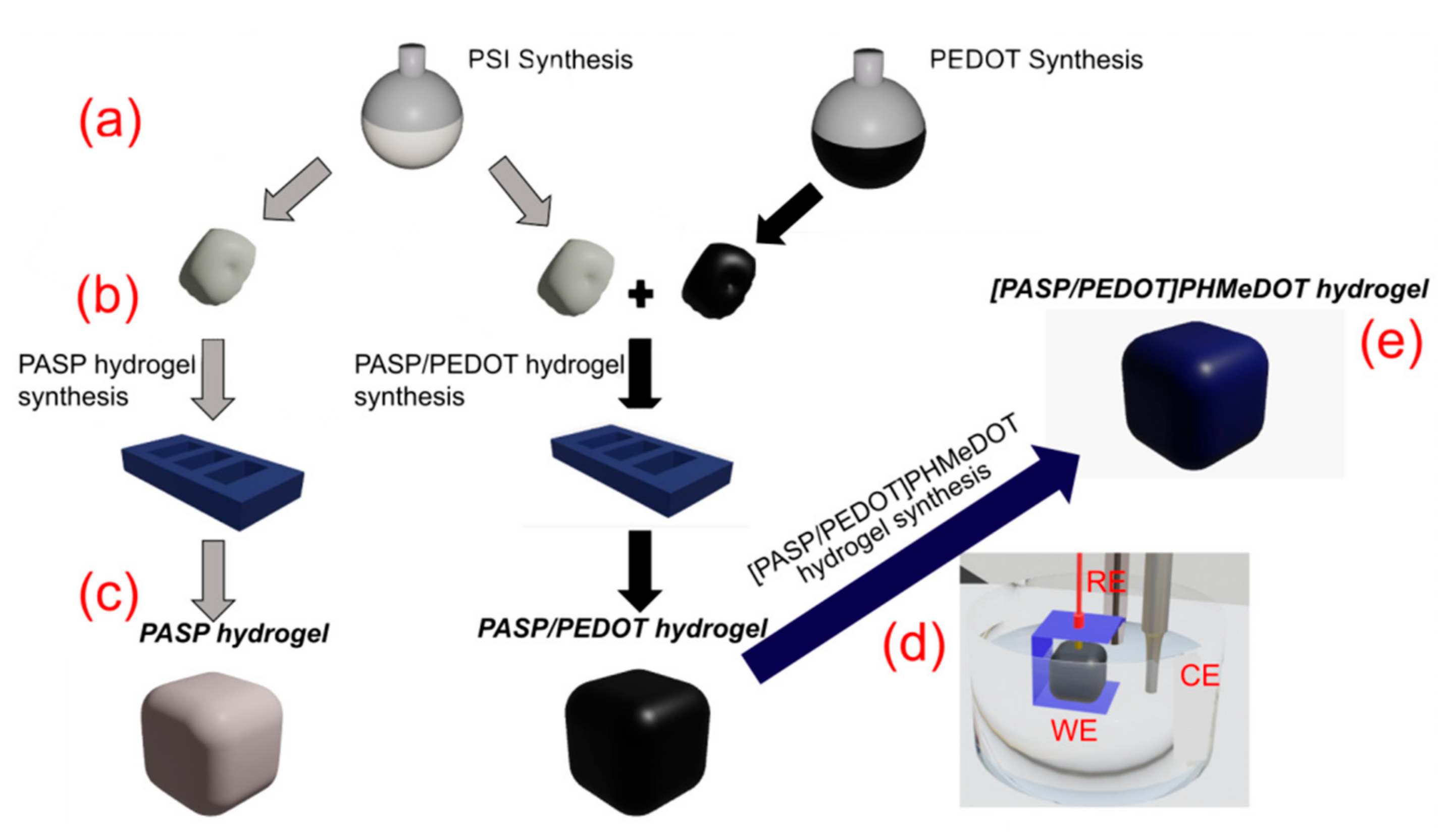
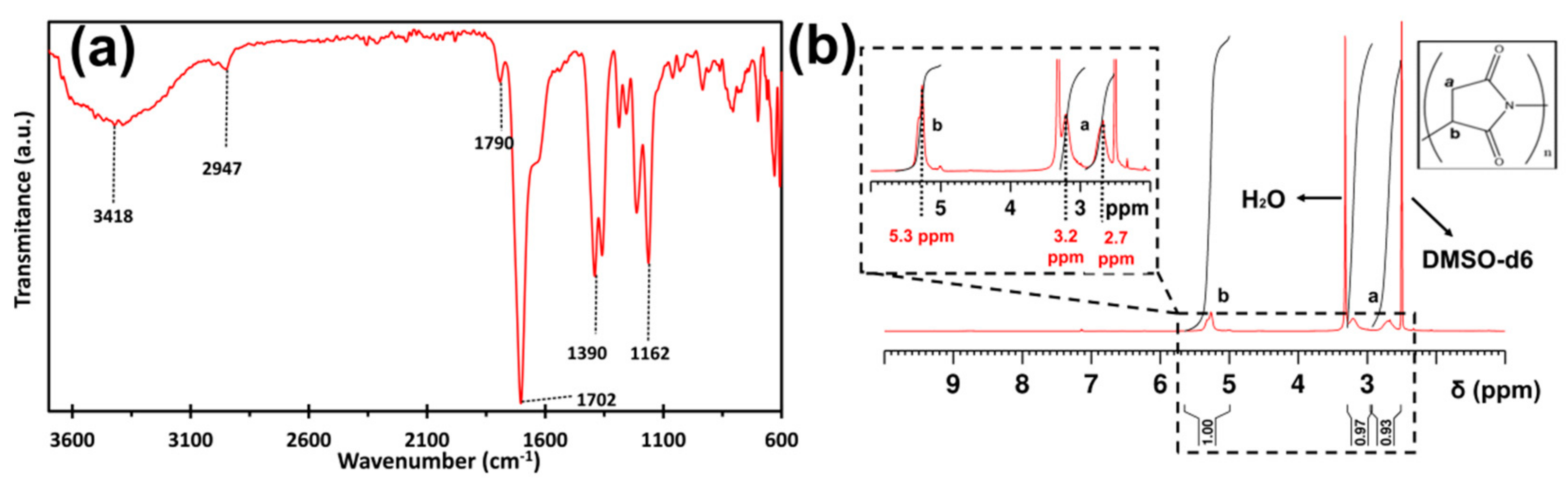
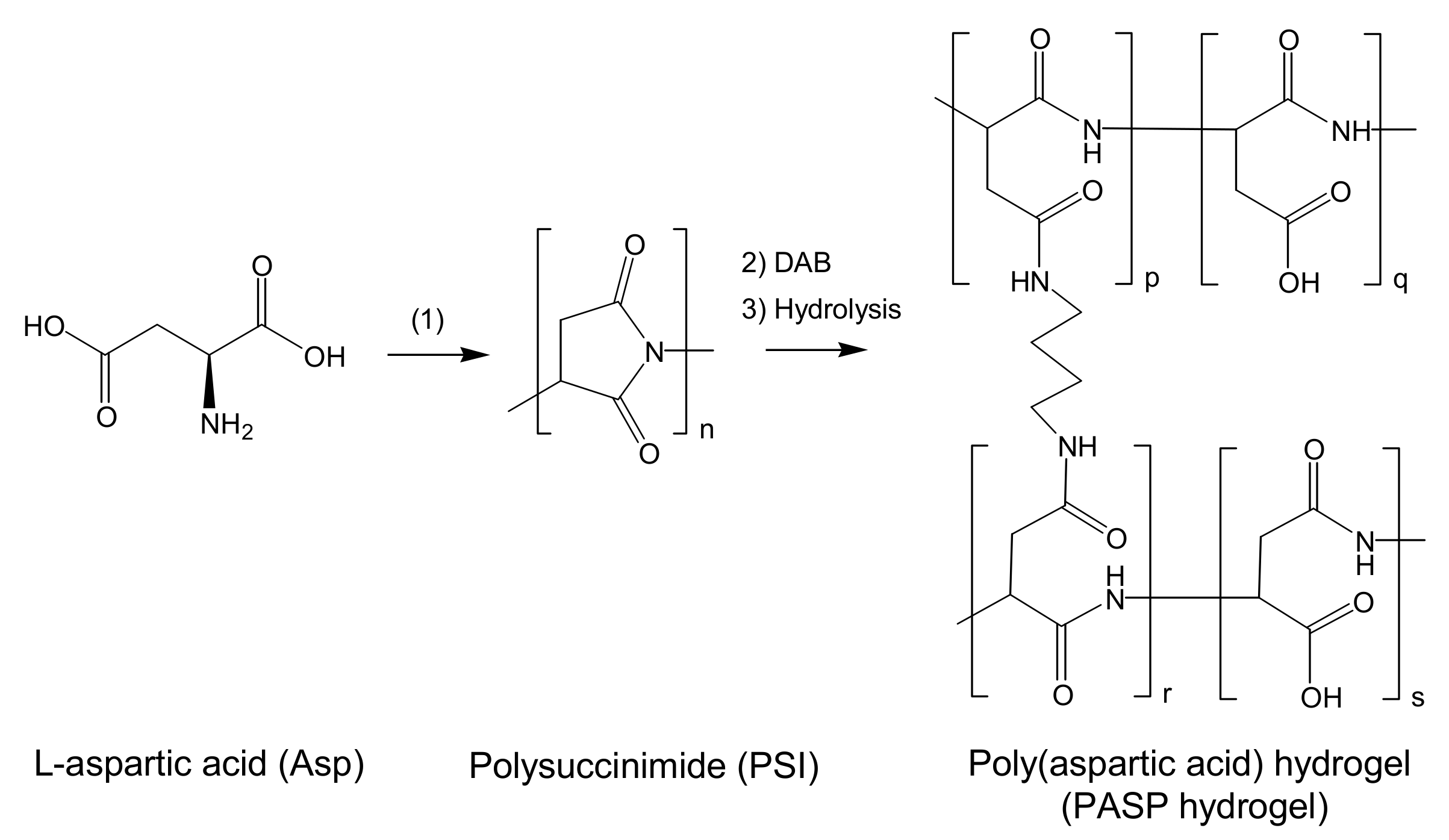
2.1. PSI: Synthesis and Characterization
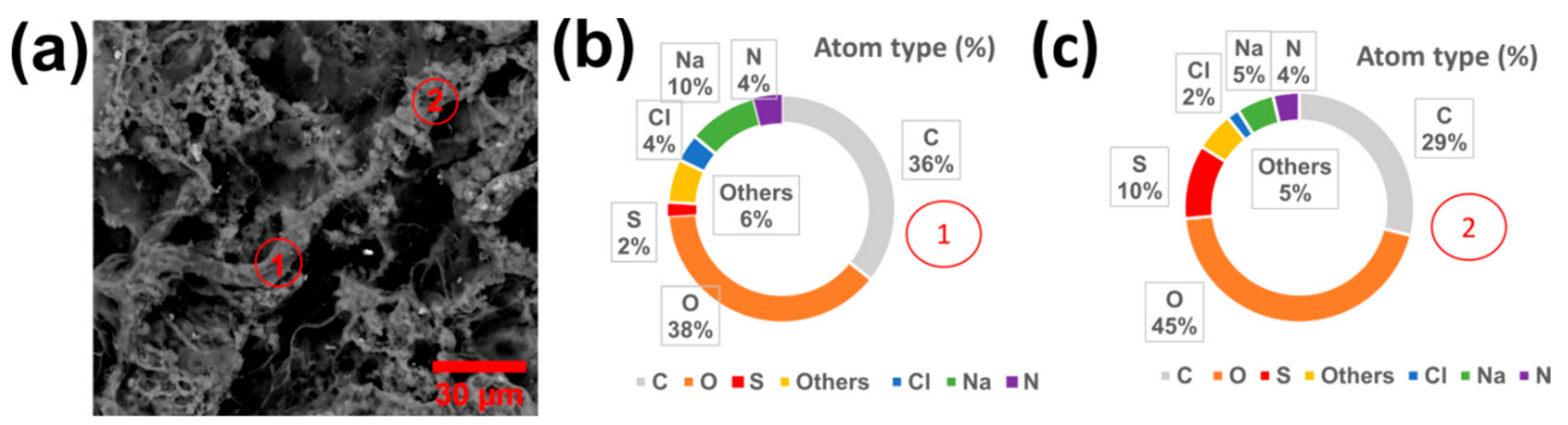
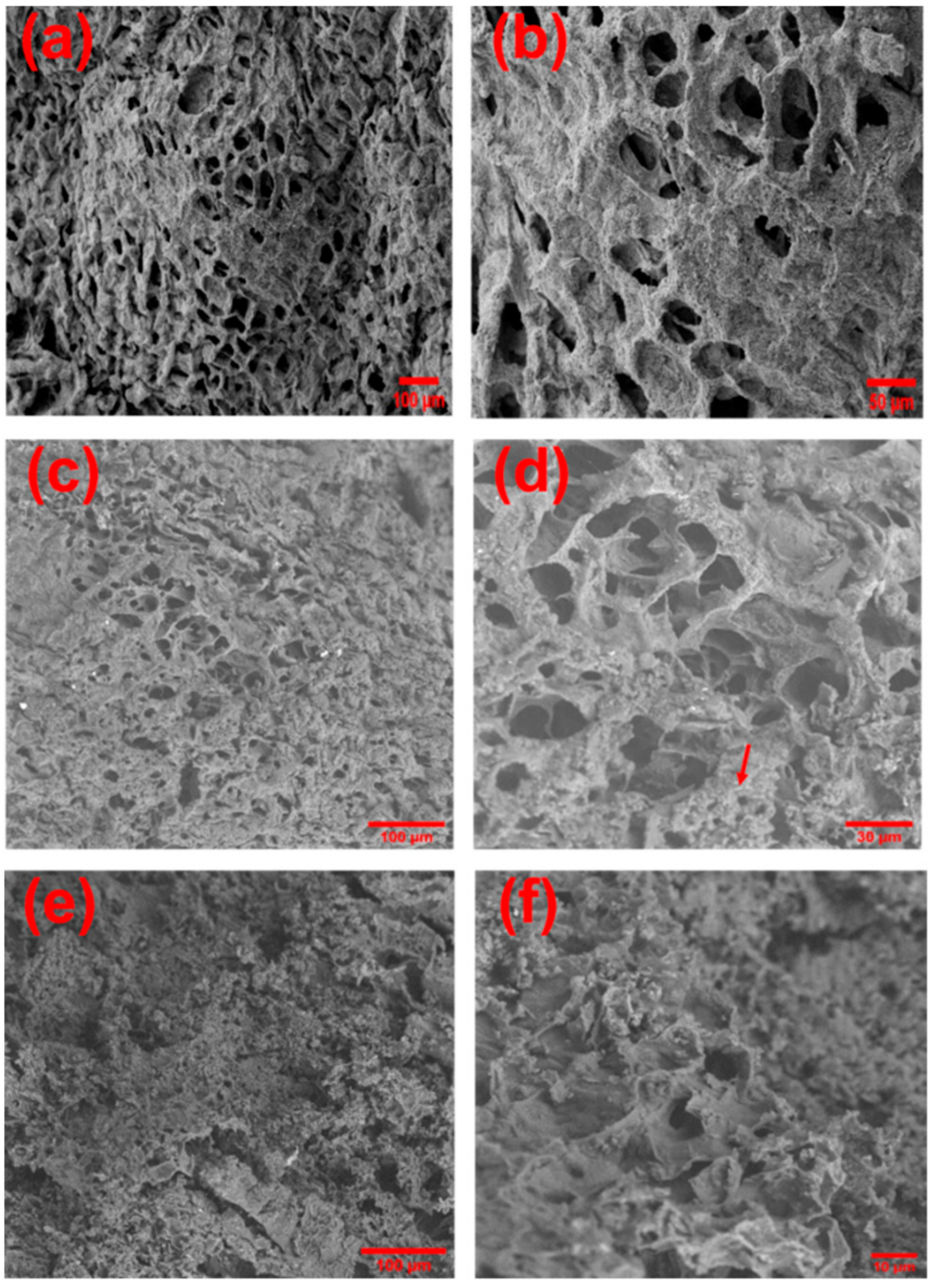
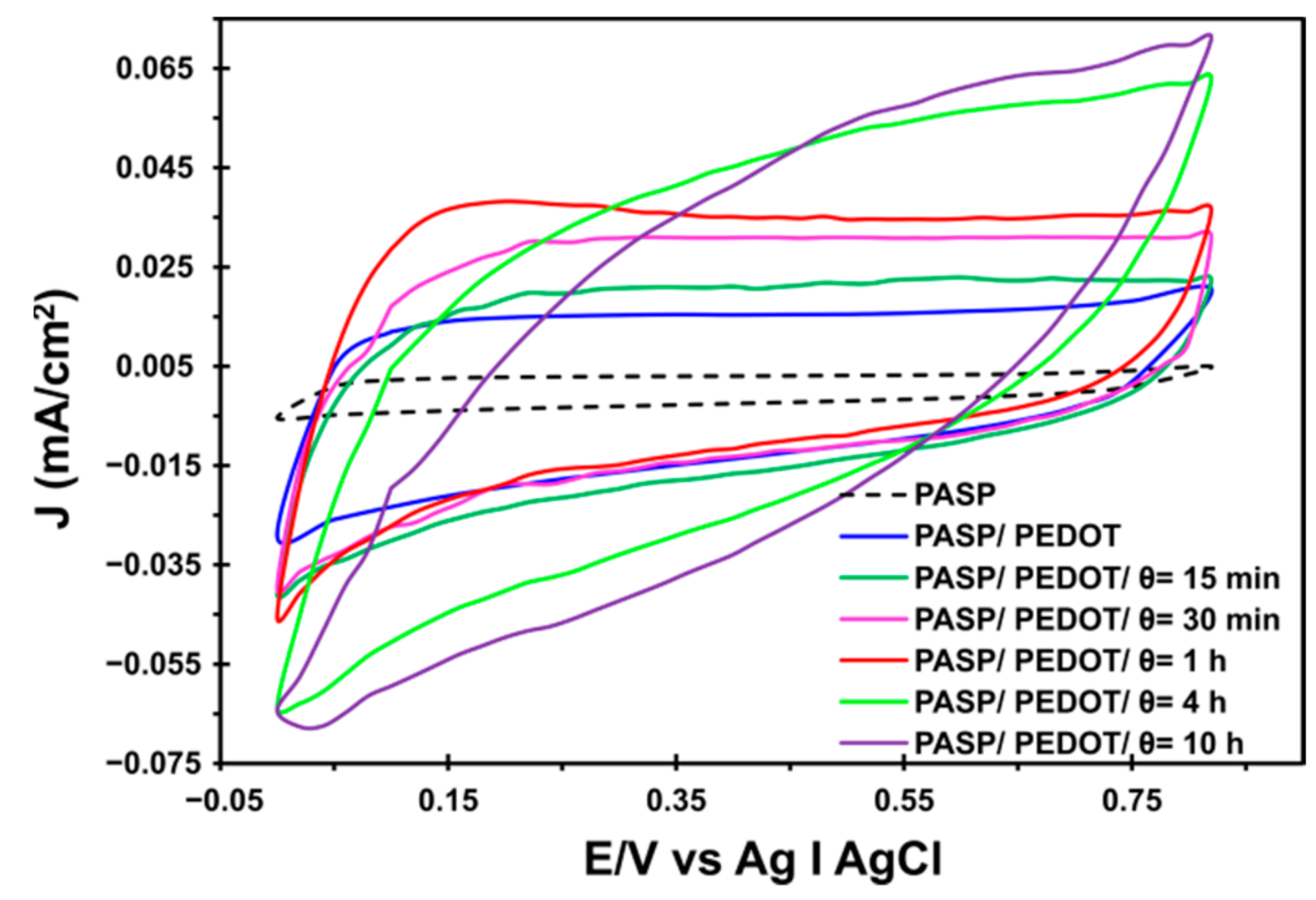
2.2. Synthesis of PASP/PEDOT Hydrogel
2.3. Preparation of [PASP/PEDOT]PHMeDOT Material
3. Material and Methods
3.1. Materials
3.2. Preparation of Poly(succinimide)
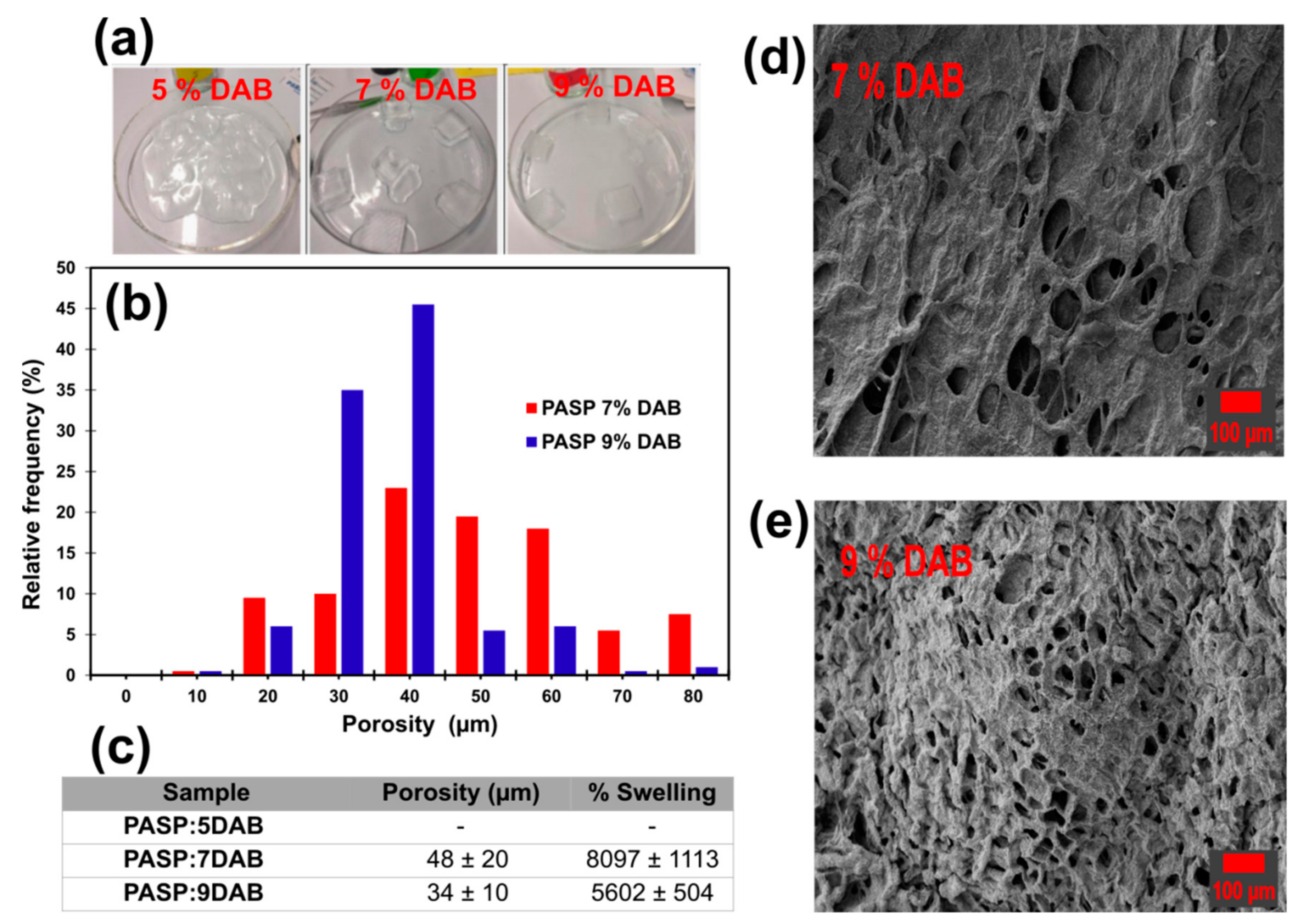
3.3. Preparation of Poly(aspartic acid) Hydrogels
3.4. Synthesis of PEDOT Particles
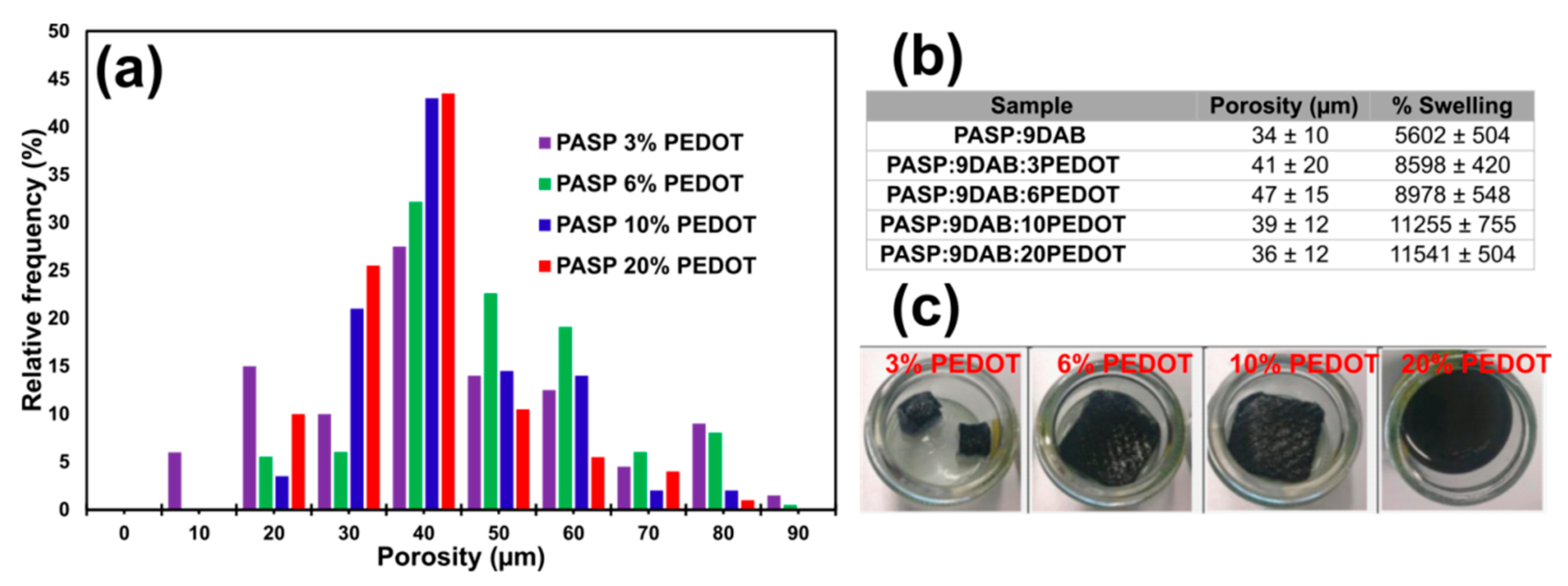
3.5. Preparation of PASP/PEDOT Hydrogels
3.6. Preparation of [PASP/PEDOT]PHMeDOT Hydrogels
3.7. Additive Manufacture
3.8. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ullah, F.; Othman, M.B.H.; Javed, F.; Ahmad, Z.; Akil, H.M. Classification, processing and application of hydrogels: A review. Mater. Sci. Eng. C 2015, 57, 414–433. [Google Scholar] [CrossRef]
- Van Vlierberghe, S.; Dubruel, P.; Schacht, E. Biopolymer-Based Hydrogels As Scaffolds for Tissue Engineering Applications: A Review. Biomacromolecules 2011, 12, 1387–1408. [Google Scholar] [CrossRef] [PubMed]
- Buenger, D.; Topuz, F.; Groll, J. Hydrogels in sensing applications. Prog. Polym. Sci. 2012, 37, 1678–1719. [Google Scholar] [CrossRef]
- Pinelli, F.; Magagnin, L.; Rossi, F. Progress in hydrogels for sensing applications: A review. Mater. Today Chem. 2020, 17, 100317. [Google Scholar] [CrossRef]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Kumar, V.; Kaith, B.S.; Kalia, S.; Swart, H.C. Conducting Polymer Hydrogels and Their Applications. In Conducting Polymer Hybrids; Kumar, V., Kalia, S., Swart, H.C., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 193–221. [Google Scholar]
- Gilmore, K.; Hodgson, A.J.; Luan, B.; Small, C.J.; Wallace, G.G. Preparation of hydrogel/conducting polymer composites. Polym. Gels Netw. 1994, 2, 135–143. [Google Scholar] [CrossRef]
- Yu, C.; Wang, C.; Liu, X.; Jia, X.; Naficy, S.; Shu, K.; Forsyth, M.; Wallace, G.G. A Cytocompatible Robust Hybrid Conducting Polymer Hydrogel for Use in a Magnesium Battery. Adv. Mater. 2016, 28, 9349–9355. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Dai, Z.; Sheng, X.; Xia, D.; Shao, P.; Yang, L.; Luo, X. Conducting polymer hydrogels as a sustainable platform for advanced energy, biomedical and environmental applications. Sci. Total Environ. 2021, 786, 147430. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, Y.; Hedenqvist, M.S.; Chen, C.; Cai, C.; Li, H.; Liu, H.; Fu, J. Multifunctional conductive hydrogels and their applications as smart wearable devices. J. Mater. Chem. B 2021, 9, 2561–2583. [Google Scholar] [CrossRef]
- Zhao, F.; Yao, D.; Guo, R.; Deng, L.; Dong, A.; Zhang, J. Composites of Polymer Hydrogels and Nanoparticulate Systems for Biomedical and Pharmaceutical Applications. Nanomaterials 2015, 5, 2054–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stejskal, J. Conducting polymer hydrogels. Chem. Pap. 2017, 71, 269–291. [Google Scholar] [CrossRef]
- Wolfram, J.; Zhu, M.; Yang, Y.; Shen, J.; Gentile, E.; Paolino, D.; Fresta, M.; Nie, G.; Chen, C.; Shen, H.; et al. Safety of Nanoparticles in Medicine. Curr. Drug Targets 2015, 16, 1671–1681. [Google Scholar] [CrossRef] [Green Version]
- Adabi, M.; Naghibzadeh, M.; Adabi, M.; Zarrinfard, M.A.; Esnaashari, S.S.; Seifalian, A.M.; Faridi-Majidi, R.; Aiyelabegan, H.T.; Ghanbari, H. Biocompatibility and nanostructured materials: Applications in nanomedicine. Artif. Cells Nanomed. Biotechnol. 2017, 45, 833–842. [Google Scholar] [CrossRef]
- Aoki, K.; Saito, N. Biocompatibility and Carcinogenicity of Carbon Nanotubes as Biomaterials. Nanomaterials 2020, 10, 264. [Google Scholar] [CrossRef]
- Goding, J.; Gilmour, A.; Martens, P.; Poole-Warren, L.; Green, R. Interpenetrating Conducting Hydrogel Materials for Neural Interfacing Electrodes. Adv. Healthc. Mater. 2017, 6, 1601177. [Google Scholar] [CrossRef]
- Dhand, A.P.; Galarraga, J.H.; Burdick, J.A. Enhancing Biopolymer Hydrogel Functionality through Interpenetrating Networks. Trends Biotechnol. 2021, 39, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.; Das, P.; Itzhaki, E.; Hadad, E.; Gedanken, A.; Margel, S. Microwave-Synthesized Polysaccharide-Derived Carbon Dots as Therapeutic Cargoes and Toughening Agents for Elastomeric Gels. ACS Appl. Mater. Interfaces 2020, 12, 51940–51951. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.; Das, P.; Das, T.K.; Ghosh, S.; Das, S.; Bose, M.; Mondal, M.; Das, A.K.; Das, N.C. Acoustic cavitation assisted destratified clay tactoid reinforced in situ elastomer-mimetic semi-IPN hydrogel for catalytic and bactericidal application. Ultrason. Sonochem. 2020, 60, 104797. [Google Scholar] [CrossRef] [PubMed]
- Saborío, M.C.G.; Lanzalaco, S.; Fabregat, G.; Puiggalí, J.; Estrany, F.; Alemán, C. Flexible Electrodes for Supercapacitors Based on the Supramolecular Assembly of Biohydrogel and Conducting Polymer. J. Phys. Chem. C 2018, 122, 1078–1090. [Google Scholar] [CrossRef] [Green Version]
- Saborío, M.G.; Svelic, P.; Casanovas, J.; Ruano, G.; Pérez-Madrigal, M.M.; Franco, L.; Torras, J.; Estrany, F.; Alemán, C. Hydrogels for flexible and compressible free standing cellulose supercapacitors. Eur. Polym. J. 2019, 118, 347–357. [Google Scholar] [CrossRef]
- Molina, B.G.; Llampayas, A.; Fabregat, G.; Estrany, F.; Alemán, C.; Torras, J. Electroactive interpenetrated biohydrogels as hybrid materials based on conducting polymers. J. Appl. Polym. Sci. 2021, 138, 50062. [Google Scholar] [CrossRef]
- Nakato, T.; Yoshitake, M.; Matsubara, K.; Tomida, M.; Kakuchi, T. Relationships between Structure and Properties of Poly(aspartic acid)s. Macromolecules 1998, 31, 2107–2113. [Google Scholar] [CrossRef]
- Tabata, K.; Abe, H.; Doi, Y. Microbial Degradation of Poly(aspartic acid) by Two Isolated Strains of Pedobacter sp. and Sphingomonas sp. Biomacromolecules 2000, 1, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-M.; Oh, B.C.; Kim, J.H.; Ahn, T.; Nam, H.-S.; Park, C.W.; Kim, J.-D. Multifunctional poly(aspartic acid) nanoparticles containing iron oxide nanocrystals and doxorubicin for simultaneous cancer diagnosis and therapy. Colloids Surf. A 2011, 391, 208–215. [Google Scholar] [CrossRef]
- Gandhimathi, C.; Venugopal, J.; Ravichandran, R.; Sundarrajan, S.; Suganya, S.; Ramakrishna, S. Mimicking Nanofibrous Hybrid Bone Substitute for Mesenchymal Stem Cells Differentiation into Osteogenesis. Macromol. Biosci. 2013, 13, 696–706. [Google Scholar] [CrossRef]
- Sattari, S.; Tehrani, A.D.; Adeli, M.; Azarbani, F. Development of new nanostructure based on poly(aspartic acid)-g-amylose for targeted curcumin delivery using helical inclusion complex. J. Mol. Liq. 2018, 258, 18–26. [Google Scholar] [CrossRef]
- Adelnia, H.; Blakey, I.; Little, P.J.; Ta, H.T. Hydrogels Based on Poly(aspartic acid): Synthesis and Applications. Front. Chem. 2019, 7, 755. [Google Scholar] [CrossRef] [PubMed]
- Patwadkar, M.V.; Gopinath, C.S.; Badiger, M.V. An efficient Ag-nanoparticle embedded semi-IPN hydrogel for catalytic applications. RSC Adv. 2015, 5, 7567–7574. [Google Scholar] [CrossRef]
- Jv, X.; Zhao, X.; Ge, H.; Sun, J.; Li, H.; Wang, Q.; Lu, H. Fabrication of a Magnetic Poly(aspartic acid)-Poly(acrylic acid) Hydrogel: Application for the Adsorptive Removal of Organic Dyes from Aqueous Solution. J. Chem. Eng. Data 2019, 64, 1228–1236. [Google Scholar] [CrossRef]
- Tomida, M.; Nakato, T.; Kuramochi, M.; Shibata, M.; Matsunami, S.; Kakuchi, T. Novel method of synthesizing poly(succinimide) and its copolymeric derivatives by acid-catalysed polycondensation of l-aspartic acid. Polymer 1996, 37, 4435–4437. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, M.; Zhou, C. Thermal Stability and Decomposition Kinetics of Polysuccinimide. Am. J. Anal. Chem. 2013, 4, 749–755. [Google Scholar] [CrossRef] [Green Version]
- Yeh, J.-C.; Hsu, Y.-T.; Su, C.-M.; Wang, M.-C.; Lee, T.-H.; Lou, S.-L. Preparation and characterization of biocompatible and thermoresponsive micelles based on poly(N-isopropylacrylamide-co-N,N-dimethylacrylamide) grafted on polysuccinimide for drug delivery. J. Biomater. Appl. 2014, 29, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Jalalvandi, E.; Shavandi, A. Polysuccinimide and its derivatives: Degradable and water soluble polymers (review). Eur. Polym. J. 2018, 109, 43–54. [Google Scholar] [CrossRef]
- Nakato, T.; Kusuno, A.; Kakuchi, T. Synthesis of poly(succinimide) by bulk polycondensation of L-aspartic acid with an acid catalyst. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 117–122. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Gyarmati, B.; Mészár, E.Z.; Kiss, L.; Deli, M.A.; László, K.; Szilágyi, A. Supermacroporous chemically cross-linked poly(aspartic acid) hydrogels. Acta Biomater. 2015, 22, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Aradilla, D.; Estrany, F.; Alemán, C. Symmetric Supercapacitors Based on Multilayers of Conducting Polymers. J. Phys. Chem. C 2011, 115, 8430–8438. [Google Scholar] [CrossRef]
- Lee, T.H.; Do, K.; Lee, Y.W.; Jeon, S.S.; Kim, C.; Ko, J.; Im, S.S. High-performance dye-sensitized solar cells based on PEDOT nanofibers as an efficient catalytic counter electrode. J. Mater. Chem. 2012, 22, 21624–21629. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Swelling (%) |
|---|---|
| PASP | 5602 ± 504 |
| PASP/PEDOT | 11,255 ± 755 |
| [PASP/PEDOT] PHMeDOT θ = 15 min | 11,842 ± 438 |
| [PASP/PEDOT] PHMeDOT θ = 30 min | 10,659 ± 672 |
| [PASP/PEDOT] PHMeDOT θ = 1 h | 13,375 ± 780 |
| [PASP/PEDOT] PHMeDOT θ = 4 h | 13,598 ± 526 |
| [PASP/PEDOT] PHMeDOT θ = 10 h | 13,915 ± 589 |
| System | LEA (%) | SC (mF cm−2) (×10−5) | |
|---|---|---|---|
| Cycle 30 | Cycle 2 | Cycle 30 | |
| PASP | 39 ± 1 | 0.19 ±0.00 | 0.12 ± 0.00 |
| PASP/PEDOT | 62 ± 1 | 19.7 ± 0.4 | 7.49 ± 0.06 |
| [PASP/PEDOT] PHMeDOT θ = 15 min | 57 ± 4 | 16.7 ± 0.9 | 7.07 ± 0.29 |
| [PASP/PEDOT] PHMeDOT θ = 30 min | 13 ± 3 | 20.3 ± 0.2 | 22.9 ± 0.3 |
| [PASP/PEDOT] PHMeDOT θ = 1 h | 12 ± 3 | 45.9 ± 1.1 | 40.3 ± 1.2 |
| [PASP/PEDOT] PHMeDOT θ = 4 h | 14 ± 3 | 141.0 ± 1.6 | 122.0 ± 3.7 |
| [PASP/PEDOT] PHMeDOT θ = 10 h | 61 ± 2 | 228.0 ± 7.6 | 88.0 ± 1.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana-Escartín, A.; Ruano, G.; Silva, F.M.; Estrany, F.; Puiggalí, J.; Alemán, C.; Torras, J. Poly(aspartic acid) Biohydrogel as the Base of a New Hybrid Conducting Material. Int. J. Mol. Sci. 2021, 22, 13165. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313165
Fontana-Escartín A, Ruano G, Silva FM, Estrany F, Puiggalí J, Alemán C, Torras J. Poly(aspartic acid) Biohydrogel as the Base of a New Hybrid Conducting Material. International Journal of Molecular Sciences. 2021; 22(23):13165. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313165
Chicago/Turabian StyleFontana-Escartín, Adrián, Guillem Ruano, Fiorella M. Silva, Francesc Estrany, Jordi Puiggalí, Carlos Alemán, and Juan Torras. 2021. "Poly(aspartic acid) Biohydrogel as the Base of a New Hybrid Conducting Material" International Journal of Molecular Sciences 22, no. 23: 13165. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313165