
Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection


1
Department of Dental Hygiene, College of Bio-Health Convergence, Dongseo University, Busan 47011, Korea
2
Department of Physiology, College of Medicine, Yonsei University, Seoul 03722, Korea
3
Brain Korea 21 PLUS Project for Medical Science, College of Medicine, Yonsei University, Seoul 03722, Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(24), 13315; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413315
Submission received: 29 November 2021
/
Accepted: 3 December 2021
/
Published: 10 December 2021
(This article belongs to the Special Issue Neuroprotection: Rescue from Neuronal Death in the Brain 2.0)
{kind=link}
{kind=link}
{kind=link}
Abstract
:To counteract oxidative stress and associated brain diseases, antioxidant systems rescue neuronal cells from oxidative stress by neutralizing reactive oxygen species and preserving gene regulation. It is necessary to understand the communication and interactions between brain cells, including neurons, astrocytes and microglia, to understand oxidative stress and antioxidant mechanisms. Here, the role of glia in the protection of neurons against oxidative injury and glia–neuron crosstalk to maintain antioxidant defense mechanisms and brain protection are reviewed. The first part of this review focuses on the role of glia in the morphological and physiological changes required for brain homeostasis under oxidative stress and antioxidant defense mechanisms. The second part focuses on the essential crosstalk between neurons and glia for redox balance in the brain for protection against oxidative stress.
1. Introduction
The brain is highly susceptible to oxidative injury because of its high rate of oxidative metabolic activity, intense production of reactive oxygen metabolites, weak antioxidant capacity, relatively high lipid content, high energy requirements, non-replicating neuronal cells, and high membrane surface to cytoplasm ratio. Consequently, oxidative injury induces neurodegenerative disease [1,2]. Specifically, reactive oxygen species (ROS) increase vulnerability to brain cell damage and functional decline via a redox imbalance between pro-oxidant and antioxidant agents, which induce the formation of free radicals and other reactive molecules. There have been several studies on neuroprotection and the rescue of neurons after oxidative injury [3,4].
To develop new therapeutic interventions and diagnose the diseases that result from oxidative brain injury, it is necessary to understand the physiological functions of brain cells and the crosstalk between them. Astrocytes are important for brain homeostasis because they provide nutrition to neurons, maintain the integrity of the blood–brain barrier, regulate synapse activity, and process cell metabolites [5]. Microglia are crucial because they function as macrophages in the brain and rapidly respond to disturbances in the brain [6]. Targeting the interaction between astrocytes, microglia, and other brain cells may arrest or reverse oxidative injury, which results in neuroprotection. Specific glial cell-based diagnostic approaches that detect glial cell signaling pathways using biomarkers or neuroimaging may identify individuals at risk of neuronal dysfunction much earlier and more precisely, and these biomarkers may allow for the monitoring of oxidative disease progression and/or recovery. This review summarizes the current knowledge of the physiological roles and functions of astrocytes and microglia in response to oxidative stress, their interactions with neurons, and their neuroprotective capabilities.
2. Vulnerability of the Brain to Oxidative Stress
Oxidative stress, which can be induced by various mechanisms, plays a crucial role in neuronal death and brain dysfunction and induces neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), aging, and other neurodegenerative diseases [7,8,9]. The brain requires large amounts of adenosine triphosphate (ATP) and consumes over 25% of the circulating glucose and 20% of total basal oxygen (O2) to maintain neuronal activity [10,11]. However, glucose consumption may induce oxidative stress by inactivating proteins through the formation of advanced glycation end products, and oxygen utilization can produce ROS and reactive nitrogen species (RNS) via endogenous mechanisms during cellular respiration [12,13]. ROS/RNS production largely occurs during oxidative phosphorylation, and increased free radical production plays a crucial role in neuronal death. When ROS/RNS production exceeds the scavenging capacity of the antioxidant response system, extensive protein degradation, lipid oxidation, and DNA degeneration occur and subsequently induce an excessive and pathological loss of neurons [14,15].
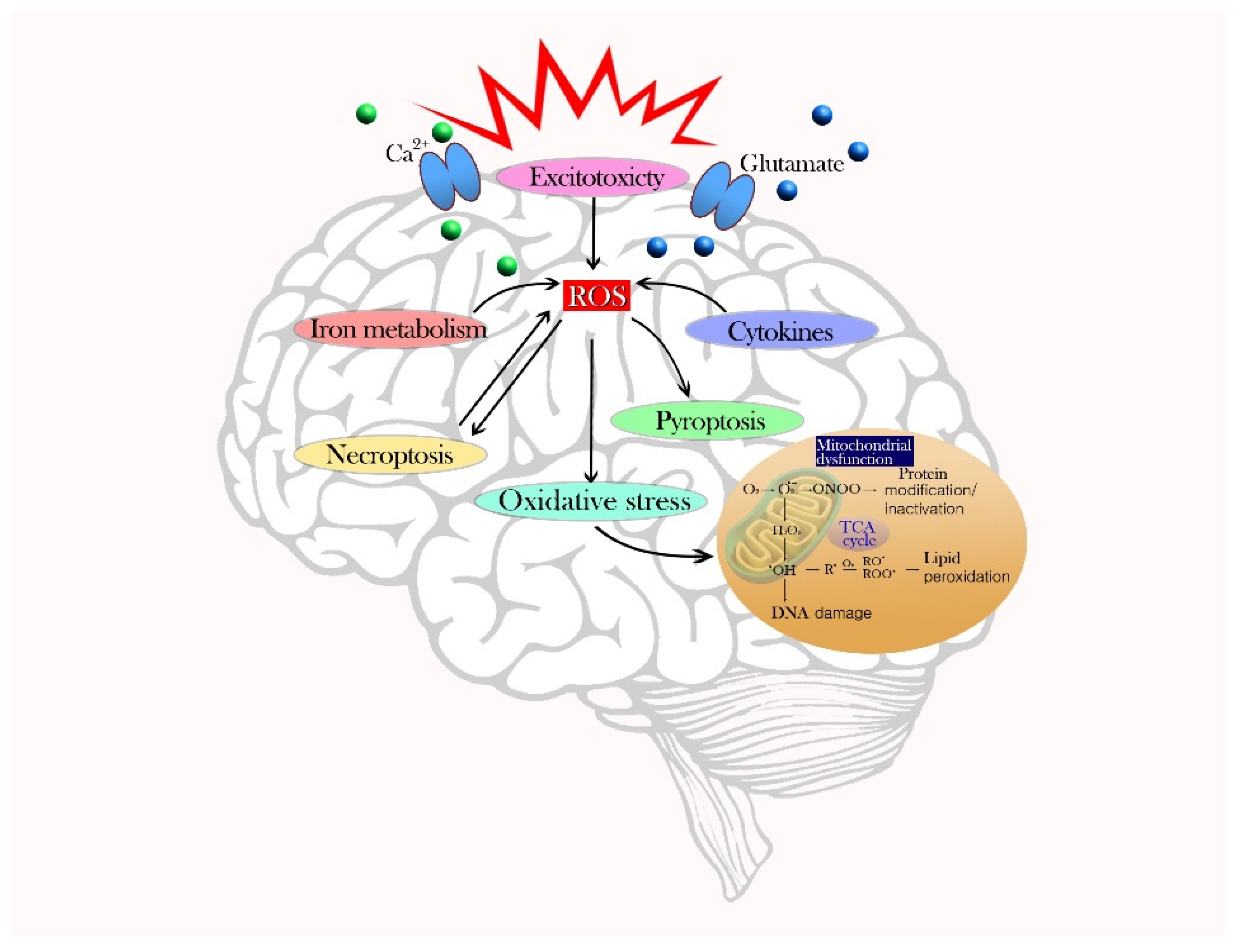
The primary mechanism of oxidative cell death is the formation of ROS and mitochondria dysfunction. Single-electron reactions produce reactive molecules as undesirable side-products of respiration or as a result of excess defense mechanisms. ROS/RNS include singlet oxygen, superoxide anion radicals, hydroxyl radicals, hydrogen peroxide, nitric oxide, and peroxynitrite anions [16]. These unstable molecules destroy cellular lipids and proteins, and consequently activate intracellular ROS production via nicotinamide adenine dinucleotide phosphate (NADPH) and the electron transport chain. Through the cell membrane, NADPH is used as an electron donor for electron transfers and, ultimately, molecular oxygen is reduced to ROS [17]. The mitochondria are the main sites of intracellular ROS production and the targets of ROS-induced injury. Slow electron transfer during the respiratory chain increases ROS production and seriously damages the antioxidant system [18]. Secondary mechanisms of cell death via ROS production are excitotoxicity, iron metabolism, cytokines, pyroptosis, and necroptosis. The excessive release of glutamate and an influx of Ca2+ causes calcium overload in neurons and a disturbance in intracellular Ca2+ homeostasis, which can intensify excitotoxicity by leading to ROS production [19]. Iron-dependent oxidative stress also causes brain function deterioration. When an overload of iron overwhelms a cell’s detoxification systems, iron content (especially Fe2+) increases and promotes the conversion of H2O2 to •OH through the Fenton reaction, thereby amplifying oxidative stress [20]. Inflammatory cells, immune factors, and chemokines can release harmful compounds and cytokines that exacerbate oxidative stress and impair neurons. Microglia, which are important for redox stability, activate NADPH oxidase (NOX) and nitric oxide synthase (NOS) enzymes, leading to an increased production of ROS and RNS [21,22]. Astrocytes stimulate the activation and proliferation of microglia, which produce many inflammatory mediators in the brain. Pyroptosis is another type of inflammatory programmed cell death. The leucine-rich-repeat (NLR) pyrin-domain-containing 3 (NLRP3) inflammasome signaling pathway that induces cell pyrolysis is triggered by ROS generation during brain injury [23]. NLRP3 inflammasome activation in astrocytes and microglia induce inflammatory responses and neuronal death [24]. Intracellular ROS accumulation can alter proteins, glucose, lipids, and nucleic acids to cause cell dysfunction and death. Tumor necrosis factor (TNF)-induced necroptosis (programmed necrosis) can also lead to ROS generation [25]. The pathophysiological mechanism of cell death due to oxidative stress is described in Figure 1.
3. Astrocytes
3.1. Astrocytes in the Brain
Astrocytes are the most dynamic and abundant cells in the human brain and are responsible for maintaining brain homeostasis. Astrocytes are called territorial cells and have several extended processes that communicate with adjacent cells; thus, they form organized anatomical domains with associated functional syncytia [26]. Astrocytes project vascular processes (astrocytic end-feet) onto intraparenchymal blood vessels and ensheath the vessel surfaces to control the movement of molecules and cells between the vascular compartment and the brain [27]. Human astrocytes are usually classified into four subdivisions based on their neuroanatomy [28]. First, interlaminar astrocytes have a round cell body and short processes and are located in layer I of the cortex. Second, protoplasmic astrocytes are found in gray matter and are located in layers II–VI of the cortex. They are the most abundant astrocytes and have numerous processes and a bushy morphology. Third, varicose projection astrocytes are located in layers V–VI and have short spiny processes with from one to five longer processes that may function in long-distance communication within the cortex. Fourth, fibrous astrocytes are located in white matter and are larger cells containing fewer processes. Fibrous astrocyte processes send numerous extensions to contact oligodendroglia that wrap myelinated axons [29]. Astrocytes are also classified into type I–III according to their morphological characteristics, such as cell body size, number of processes, thickness of processes, direction of processes, and length of processes. Type I astrocytes are characterized by a small cell body and numerous short processes. Type II astrocytes are characterized by a bipolar shape and long processes. Type III astrocytes are characterized by a star shape and long processes [30,31]. The function of astrocytes is to aid neurons by playing supportive roles in synaptic function and the modulation of neurotransmission. The processes of astrocytes ensheath synapses and contain a variety of receptors for neurotransmitters, cytokines, growth factors, and ion channels. Astrocytes are affected by intracellular Ca2+ release by extracellular glutamate, and maintain the ionic balance of synapses by increasing intracellular Ca2+ levels following the secretion of numerous gliotransmitters, such as glutamate, purines, GABA, and D-serine [32,33] Neurons are highly sensitive to small changes in the brain microenvironment, even though their metabolic consumption is high. The role of astrocytes in the normal brain is the maintenance of extracellular homeostasis through glutamate uptake and recycling, K+ buffering, supplying energy substrates, pH buffering, and defense against oxidative stress [28].
3.2. Astrocytes in Oxidative Injury
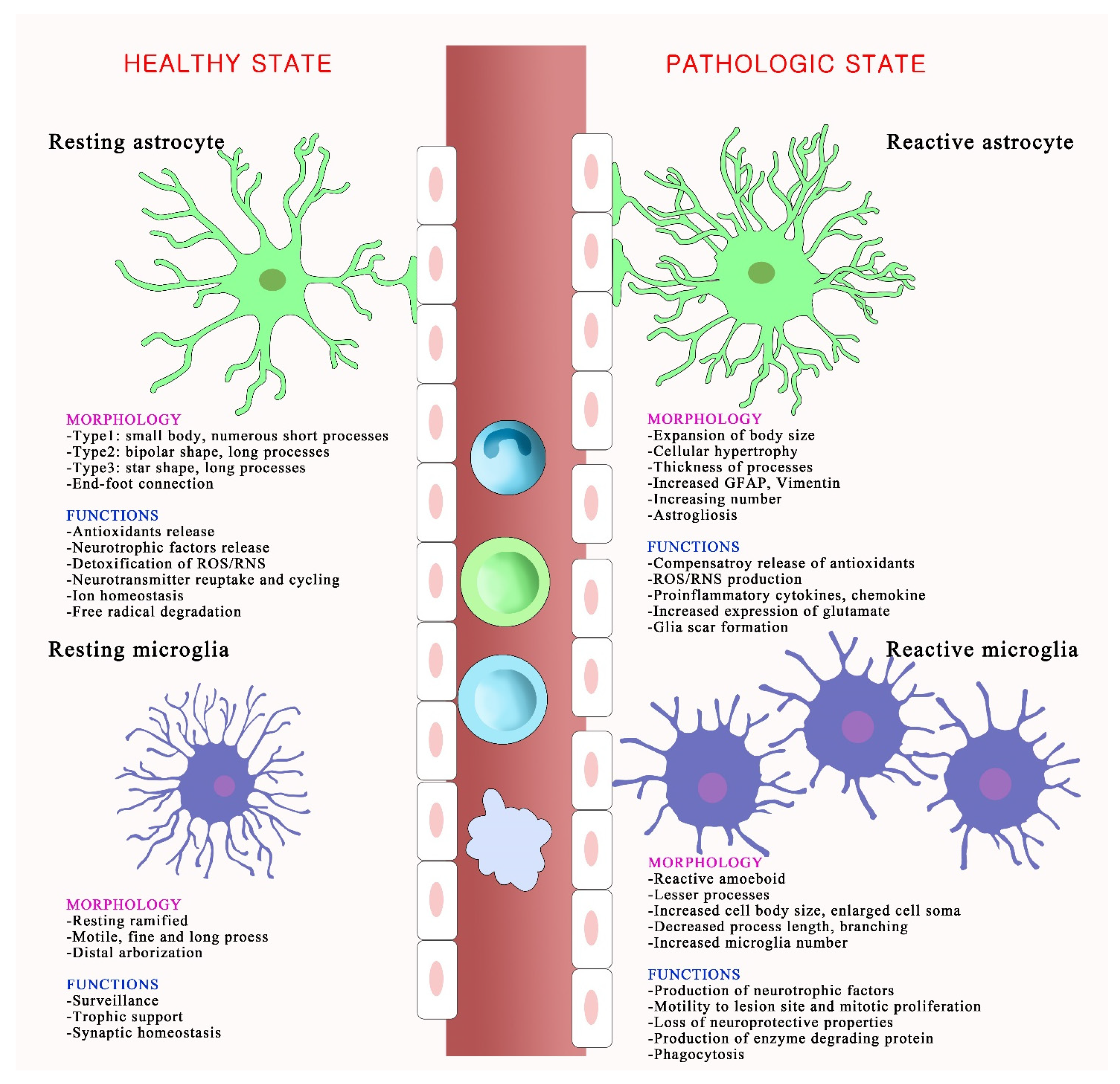
Astrocytes exist in a resting or reactive state in the brain, as shown in Figure 2. Reactive astrocytes release inflammatory cytokines including TNF and ROS, and form glial scars that impede axon regeneration and neurite outgrowth [34,35,36]. Activated astrocytes aid in the recovery of brain function after injury but can be neurotoxic. Reactive astrocytes release nitric oxide (NO) into the extracellular space; this can lead to neuronal injury and death by increasing lipid peroxidation, mitochondrial impairment, and inducing DNA strand breaks [37]. The astrocytic antioxidant system balances ROS (superoxides, hydroxyl radicals, and nitric monoxide) that are naturally produced during oxygen metabolism by the CNS [38]. Oxidative stress in reactive astrocytes leads to long-term effects on specific proteins, including connexins, glutamate transporters, and enzymes, which affect interactions between astrocytes and neurons [39]. The glutamate uptake by an astrocyte requires a high level of energy, needing more than one ATP molecule for one glutamate take-up. However, the lack of ATP is related to the mechanisms of ROS-induced glutamate uptake blockade in astrocytes [40,41]. Blocking astrocyte glutamate transporters increases neurotoxicity by potentiating neuronal excitability and excitatory neurotransmission [42]. Oxidative stress generated by astrocytes mainly occurs through mitochondria-derived oxidative stress, NADPH-derived oxidative stress, and RNS production. Mitochondria are distributed in the cell body and in the thin and long processes of astrocytes [43]. Disrupting mitochondrial function and increasing ROS in astrocytes lead to astrogliosis. NADPH-derived oxidative stress significantly affects the physiological function of astrocytes. Among the NOX family, NOX2 and NOX4 are the most abundantly expressed NOX isoforms in the CNS [43]. NOX4, but not NOX2, is expressed in astrocytes, and even a low expression of NOX4 regulates oxidative stress in astrocytes [44,45]. Astrocytic RNS production also affects astrocyte-derived oxidative stress. The main NOS isoforms, including Ca2+/calmodulin-dependent neuronal NOS, endothelial NOS, and Ca2+-independent inducible NOS, are observed in astrocytes [5,46]. Astrocytic NO leads to astrocyte-induced neuronal degeneration and Cu-Zn superoxide dismutase (SOD1) aggregation in astrocytes, which may induce ischemic/reperfusion CNS injury [47,48].
3.3. Astrocyte-Medicated Antioxidant Defense
Astrocytes are the main cells that maintain glutamate homeostasis, which indirectly affects the balance of oxidative stress, by regulating excitatory amino acids. Astrocytes also prevent excitotoxicity by releasing neurotrophic factors, such as glial-cell-line-derived neurotrophic factor (GDNF) and nerve growth factor (NGF), which support neuronal survival [39,49]. For neuroprotection during oxidative stress, astrocytes produce a variety of antioxidant molecules, including GSH, ascorbate, and vitamin E, and activate ROS-detoxifying enzymes, such as GSH S-transferase, GSH peroxidase, thioredoxin reductase, and catalase to improve neuronal survival [26,50,51]. Moreover, astrocytes participate in metal sequestration in the brain to prevent the generation of free radicals by redox-active metals. Astrocytes express high levels of metallothioneins and ceruloplasmin, which are involved in metal binding and ion trafficking [52].
Astrocytes can synthesize the GSH tripeptide with glutamate cysteine ligase and GSH synthetase. Astrocytes release GSH into the extracellular space and neurons take up the GSH directly or use extracellular neuronal aminopeptidase N to form glycine and cysteine [53]. A reduced neuronal protection against oxidative injury was observed in GSH-depleted astrocytes by limiting the substrate for GSH synthesis in neurons [54]. Astrocytes increase the capacity to synthesize GSH by increasing the capacity to uptake cysteine, thereby enhancing the neuroprotective effect of astrocytes against oxidative stress [5]. Another astrocyte antioxidant defense mechanism is the recycling of ascorbate, which can directly scavenge ROS and act as a cofactor for the recycling of oxidized vitamin E and GSH [2]. This recycled ascorbate is used intracellularly in astrocytes and/or released into the extracellular space for neurons to use for their own antioxidant defense mechanism. When ascorbic acid enters neurons, it inhibits glucose consumption and stimulates lactate transport. Ascorbic acid regulates the astrocyte-neuron lactate shuttle [55], and neurons produce glutamate, which stimulates ascorbic acid release from astrocytes during glutamatergic synaptic activity [56,57]. In the Nrf2-Keap1-ARE pathway, an important endogenous antioxidant system in the CNS, the ROS-inducible transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), regulates the GSH system, the thioredoxin system, and SOD [58]. Nrf2 is produced and ubiquitinated for degradation by binding to the Kelch-like ECH-associated protein 1 (Keap1) under basal conditions [59]. However, Keap1 binding to Nrf2 is inhibited by increased oxidative stress conditions, and this allows Nrf2 to escape degradation and interact with antioxidant response elements (AREs) in gene promoters [60,61]. Astrocytes show higher basal and stimulated levels of ARE binding by Nrf2 than neurons [62]. In addition, tertiary butylhydroquinone (tBHQ) activates Nrf2 and its downstream antioxidant enzymes, such as reduced coenzyme/quinone oxidoreductase 1 (NQO1), in astrocytes, but not in neurons [63]. Astrocytic Nrf2 is the main regulator of oxidative homeostasis as determined by the observation that Nrf2−/− astrocytes have more severe inflammatory responses. Further, astrocytic dopamine D2 receptor regulates GSH synthesis via Nrf2 transactivation in vivo [64,65].
4. Microglia
4.1. Microglia in the Brain
Microglia, which have numerous fine and motile processes that survey the parenchymal environment, represent approximately 10% of CNS cells. Each microglial cell has its own territory, which is approximately 50 µm in diameter [66]. Microglia, referred to as the resident macrophages in the CNS, are long-lived and self-renewing cells. In a healthy brain, microglia have a ramified morphology and are in a “quiescent” or “resting” state [67]. Microglial processes undergo continuous cycles of extension and withdrawal, scan their environment for disruptions in brain homeostasis, and systematically synapse to monitor and regulate neuronal activity via a specific signaling mechanism [68,69]. Microglia change their morphology from the resting state to the reactive amoeboid state during a pathological brain condition. Reactive microglia, which evolve into phagocytic or amoeboid microglia, have an increased cell body size, fewer processes, reduced process length and branching, and increased numbers and proliferation, indicating an intimate link between morphology and function [70,71,72,73] (Figure 2). Microglia are highly sensitive to environmental signals and respond to maintain their homeostatic phenotype in a disease-specific and brain-region-specific manner. White and gray matter microglia show a different immune regulation; cortex-associated microglia play a role in neurodegeneration and white-matter-associated microglia play a role in de-/remyelination [74].
Usually, activation of the neurotransmitter receptors inhibits the inflammatory activation of microglia and inhibits the production of abnormal molecules and abnormal concentrations of physiological molecules. Once activated upon brain injury or infection, microglia initiate immune responses and produce a number of cytokines, chemokines, and growth factors, and upregulate the expression of cell surface receptors, such as toll-like receptors (TLRs), phagocytic receptors, scavenger receptors, and various complement factors [75,76]. Microglia express several neurotransmitter receptors, including GABA, glutamate, dopamine, and noradrenaline [66,77].
4.2. Microglia in Oxidative Injury
During oxidative stress, activated microglia produce several inflammatory mediators, including NO and superoxide, which freely cross the cell membrane and act as signaling molecules. NO and superoxide can form peroxynitrite, which causes DNA fragmentation, lipid oxidation, and induces neuronal death [78,79]. In cultured microglia, superoxide production, which is catalyzed by nitrates/nitrites (NOx), is induced by phorbol ester, and NO production is stimulated by the induction of iNOS upon treatment with bacterial lipopolysaccharide (LPS) and interferon-γ (IFNγ) [80,81]. The expression of iNOS after intrahippocampal treatment with LPS was induced more rapidly in microglia than in astrocytes, and a lower concentration of LPS was required for iNOS induction in microglia than in astrocytes [82,83]. In addition, arginine is a well-known physiological substrate of NOS. Activated microglia with an insufficient amount of arginine leads to iNOS-mediated production of NO and superoxide, which form toxic peroxynitrite [84]. The induction of iNOS or activation of NOx alone does not cause substantial damage to microglia, but the simultaneous production of superoxide and NO by NOx and iNOS has the potential to harm microglia [85,86]. In activated microglia that generate superoxide upon NOx activation, the oxygen and H2O2 levels quickly become imbalanced and may affect microglial functions. ROS facilitates phagocytosis by amoeboid microglial cells and enhances vesicle formation, which was observed upon treatment of microglial cells with H2O2 [87]. Microglia-derived ROS can damage adjacent brain cells. Therefore, microglial proliferation and ROS production are potential therapeutic targets that may protect the brain from oxidative damage and neurodegenerative disease [88].
4.3. Microglia-Mediated Antioxidant Defense
To prevent oxidative stress by ROS, microglia contain a high cellular GSH concentration and express and upregulate diverse antioxidant enzymes, including SOD, GPx, GR, and catalase. Brain cell cultures labeled with fluorescence showed that microglia express a higher level of GSH than the other cell types in the rat brain [89]. This high concentration of intracellular GSH in microglia contributes to its antioxidant defense system against radical- and peroxide-mediated damage. Microglial cultures stimulated with TNFα showed twice as much GSH as unstimulated microglial cultures [90]. However, the cellular GSH content was lower in microglia treated with LPS/IFNγ, which induce iNOS production, but the mitochondrial GSH content was unaffected [91]. Thus, the microglial GSH content shows a binary effect, in which it increases upon improvements in GSH synthesis and decreases upon accelerated GSH consumption, depending on the type of stimulation. SOD, another antioxidant enzyme, was observed by immunocytochemical staining in activated microglia after quinolinic acid treatment, but was not detected in microglia under basal conditions [92,93]. The specific activity of MnSOD is 20 and 4 times higher in cultured microglia than in cultured astrocytes and oligodendrocytes, respectively [94]. In microglia treated with LPS/IFNγ or TNFα to induce oxidative stress, mitochondrial MnSOD expression was upregulated, which improved the ability of cells to decompose mitochondrial superoxide [90,95]. Elevated SOD activity in activated microglia reduces the risk of cell damage by superoxide-derived hydroxyl radicals and peroxynitrite. The upregulation of GSH peroxidases (GPx) in microglia is also a crucial mechanism against oxidative stress. The specific activity of GPx and GSH reductase (GR) is significantly higher in microglia than in neurons [96,97,98]. However, the specific activity of catalase was similar and/or a little lower in microglia than in other brain cell types, including neurons, astrocytes, and oligodendrocytes [97,99]. Although microglial GSH disulfide (GSSG) increases to almost 30% of total cellular GSH after exposure to H2O2, microglial GSSG is barely detectable under basal conditions [98,100].
5. Neuron–Glia Crosstalk in the Antioxidant Defense Mechanism
Neurons depend on a continuous supply of glucose and oxygen from outside the brain via cerebral blood flow, even though they do not directly contact microvessels. However, 99% of the brain capillary surface is covered with astrocyte end-feet processes, indicating that neurons must interact with astrocytes to receive essential materials from the cerebral circulation [101]. In fact, crosstalk between astrocytes and neurons is essential for neuronal defense against ROS. Activated astrocytes exhibit ambidextrous properties such as A1 and A2 astrocytes. A1 astrocytes lead to neuronal loss by promoting inflammation via the NF-kB pathway, which loses the ability to protect neurons and control synaptogenesis [102,103]. A2 astrocytes promote neuronal survival via the Janus kinase/signal transducer and activator of transcription 3 (JAK-STAT3) signaling pathway by upregulating neurotrophic factors [104].
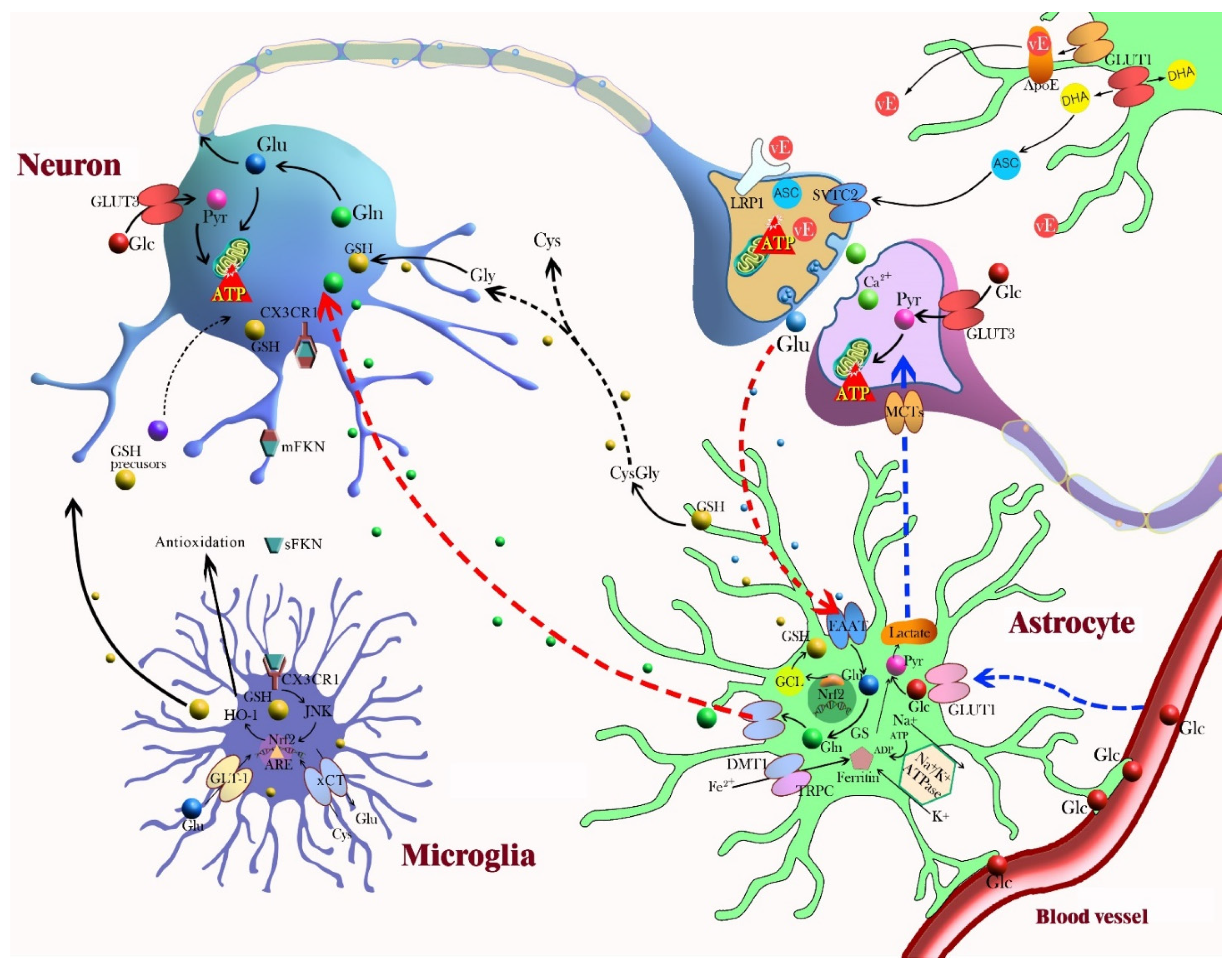
Neurons produce glutamate, which stimulates ascorbate release from astrocytes during glutamatergic synaptic activity, and then ascorbate enters neurons and inhibits glucose consumption and stimulates lactate transport. The antioxidant and metabolic interplay between neurons and astrocytes is described in Figure 3. Astrocytes are responsible for the maintenance and support of neurons by regulating oxidative stress via GSH production and glucose transformation into lactate, which ensures the energetic support of neurons [105]. The intrinsic antioxidant GSH, which is produced in both neurons and astrocytes, acts as an independent ROS scavenger and as a substrate for an antioxidant. Neuronal cells depend on astrocyte-derived GSH, for example, neurons depend on shuttling of the GSH precursor from astrocytes to neurons. Cysteine is the rate-limiting substrate for GSH synthesis, and extracellular cysteine is readily auto-oxidized to cystine [53]. Cystine uptake occurs via the cystine/glutamate exchange transporter in astrocytes, and then astrocytes reduce cystine back to cysteine for GSH synthesis. GSH directly reacts with ROS or acts as a substrate for GSH S-transferase or GSH peroxidase [50]. For the efficient use of extracellular cystine as a cysteine precursor, neurons depend on astrocytes to supply cysteine, even though neurons can synthesize GSH [54,106]. It has been shown that neuronal GSH levels are significantly higher when co-cultured with astrocytes [107]. Upon H2O2-induced oxidative stress, noradrenaline treatment protects neurons by increasing the supply of GSH from astrocytes to neurons via stimulation of the beta3-adenoreceptor in astrocytes [108]. The other interactions between neurons and astrocytes that are related to antioxidant activity include an astrocyte–neuron lactate shuttle and the recycling of ascorbate [55]. Astrocytes play a crucial role in coupling neuronal activity and brain glucose uptake through an astrocyte–neuron lactate shuttle [109]. Neuronal activity triggers glucose metabolism in astrocytes; glucose is converted to pyruvate by glycolysis and converted to lactate, which is released from astrocytes and taken up by neurons for oxidative phosphorylation. Ascorbate that is concentrated in the brain is released from glial reservoirs into the extracellular space and taken up by neurons. Highly activated neurons generate ROS, which oxidize ascorbate to dehydroascorbic acid (DHA), and scavenge ROS by taking up ascorbate [110,111].
In neurotransmitters, overstimulation with glutamate induces excitotoxicity, which is involved in the pathogenesis of many brain disorders. Astrocytes use two main transporters, excitatory amino acid transporter1 (EAAT1)/glutamate aspartate transporter (GLAST) and EAAT2/glutamate transporter-1 (GLT1), to take up glutamate and return glutamate to neurons via the well-established glutamate–glutamine cycle that involves the astrocyte-specific enzyme glutamine synthetase (GS), which converts glutamine into glutamate. If failure to convert glutamine back to glutamate occurs, the glutamate pool in presynaptic terminals would rapidly be depleted and excitatory neurotransmission would be disrupted [112,113]. An insufficient supply of glutamine to GABAergic neurons induces GABAergic dysfunction [114,115]. Glutamine in astrocytes is critical for GABA replenishment by glutamate decarboxylase, known as the GABA–glutamine cycle, in GABAergic neurons [116]. Neuronal activity and action potentials increase extracellular K+ in restricted spaces and lead to hyper-excitable membrane potentials when tight regulatory mechanisms are absent [117]. Astrocytes have a high number of membrane K+ channels and high K+ permeability [118,119]. Astrocytes capture and transport excess extracellular K+ to the astrocytic syncytium through Na+/K+ ATPase. Astrocytes also regulate the Ca2+ concentration within neurons via astrocytic calcium signaling and astrocyte-neuron crosstalk. Neuronal activation, which induces a reduction in extracellular Ca2+, evokes spatiotemporal changes via the Ca2+/Na+ exchanger in astrocytes and generates astrocytic Ca2+ waves that propagate from the cytoplasm into the extracellular space [120,121]. Astrocytes are also highly mechanosensitive, and a drop in extracellular Ca2+ due to synaptic activity leads to the release of ATP from astrocytes via the opening of connexin 43 hemichannels [122,123,124]. Neuronal activity can elicit metabolic changes in astrocytes via dual Na+ and Ca2+ signaling, which triggers glucose mobilization and glycolysis to support neuronal function. Astrocytic metabolism correlates with the high metabolic demands from neurons [125,126].
The differential antioxidant response of neurons and astrocytes results from the preferential astrocytic expression of Nrf2, a redox-sensitive transcription factor. Nrf2-ARE is a critical pathway for the regulation of the antioxidant defense mechanism because it regulates the expression of phase II detoxifying enzymes and antioxidant genes [127]. The higher susceptibility of neurons to ROS is due to the continuous destabilization and degradation of the antioxidant transcriptional activator Nrf2, which regulates the GSH system, the thioredoxin system, and SOD [128,129]. Nrf2 is more stable in astrocytes; thus, they dispose of the ROS in the nervous system. Nrf2 induction of glutamate cysteine ligase (GCL) increases GSH synthesis in astrocytes, and GSH precursors are subsequently exported to the extracellular medium [130]. Moreover, Nrf2-induced GSH synthesis in astrocytes is used to replenish neuronal GSH through the astrocyte-neuron shuttle. Nrf2-induced molecules, such as GSH-related enzymes and metallothioneins, are more highly expressed in astrocytes than in neurons, indicating that Nrf2 activation in astrocytes protects neurons from oxidative stress [131,132].
Microglia exhibit a surveying phenotype via dynamic crosstalk between microglia and neurons in the healthy brain [133]. M1 microglia promote inflammation by producing proinflammatory cytokines and inducing NO synthase activity. M2 microglia regulate immune function and promote repair by secreting anti-inflammatory cytokines [134,135]. The function of redox regulators in microglia is unclear, but many antioxidant proteins are linked to inflammation via functional microglia. In the crosstalk between microglia and neurons described in Figure 3, the expression of classical antioxidant proteins is controlled by Nrf2 in microglia [6]. Nrf2 deficiency exacerbates cognitive impairment and reactive microgliosis upon LPS treatment in vivo [136]. Heme oxygenase-1 (HO-1), an antioxidant enzyme upregulated by Nrf2, inhibits NOX2 activation upon stimulation with LPS [137]. HO-1, which may facilitate the attenuation of TLR4 signaling by NOX inhibition, is responsible for the conversion of heme to biliverdin and carbon monoxide and functions as an antioxidant enzyme [138]. The overexpression of HO-1 in microglia reduced neurotoxic iron accumulation in aged mice [139]. The genetic deletion of microglial-specific proteins and mechanistic interruption of neuronal activity by microglia manipulation showed that microglia modulate neuronal activity. Fractalkine (FKN) is predominantly expressed in the CNS and localized on neuronal cells. The FKN receptor (CX3CR1) is exclusively expressed on microglia and neurons and is an interesting signaling axis for communication between microglia and neurons [69,140]. A CX3CR1 deficiency was linked to the disruption of neurogenesis and neural connectivity [141]. DAP12 is another microglia-specific protein which occurs as a result of alterations in glutamate receptor content at synapse through microglial BDNF [142]. In neurotransmission with microglia-specific manipulation, microglia-conditioned media enhanced excitatory postsynaptic potentials and current in dissociated cell cultures [143]. The inhibition of microglial activation by minocycline reduced neuronal cell death and spontaneous recurrent seizures in a rat lithium–pilocarpine model [144].
6. Conclusions
Neurons, which have high energy demands, engage in metabolic and redox crosstalk with surrounding cells for normal brain function. Glia play essential roles in the redox and metabolic needs of neurons for neurotransmission and survival. Several previous studies have demonstrated the molecular and cellular aspects of this glia–neuronal coupling and have used antioxidant therapies to slow down the progression of neurodegeneration [139,145,146,147]. We reviewed oxidant and antioxidant systems in activated due to paracrine redox signaling and the crucial role of neuron–glia crosstalk against oxidative stress in the CNS, where the extracellular space and distance to neighboring cells or cell structures is extremely limited. Glial cells show morphological and molecular alterations in response to oxidative injury and regulate neuronal activities under these conditions. This neuron–glia communication plays a critical role in oxidative conditions by delaying neurodegeneration and aberrant neurogenesis via redox-balancing mechanisms.
Author Contributions
All authors contributed substantially. K.H.L. designed and drafted the manuscript. M.C. assisted with drafting of the manuscript and preparation of the figures. B.H.L. oversaw the entire project and prepared the draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2016R1D1A3B2008194, NRF-2020R1A2C3008481).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
None.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AD | Alzheimer’s disease |
| ApoE | apolipoprotein E |
| ARE | antioxidant response element |
| ASC | ascorbate |
| ATP | adenosine triphosphate |
| CNS | central nervous system |
| CX3CR1 | fractalkine receptor |
| Cys | cysteine |
| DAP12 | DNAX activation protein of 12 kDa |
| DHA | dehydroascorbic acid |
| DMT1 | divalent metal transporter |
| DRD2 | dopamine d2 receptor |
| EAAT | excitatory amino acid transporter |
| FKN | fractalkine |
| mFKN | membrane-anchored fractalkine |
| sFKN | soluble fractalkine |
| GCL | glutamate cysteine ligase |
| GDNF | glial cell-line-derived neurotrophic factor |
| GLAST | glutamate aspartate transporter |
| Glc | glucose |
| Gln | glutamine |
| GLT | glutamate transporter |
| Glu | glutamate |
| GLUT | glucose transporter |
| Gly | glycine |
| GPx | GSH peroxidases |
| GR | GSH reductase |
| GS | glutamine synthetase |
| GSH | glutathione |
| GSSG | GSH disulfide |
| HO-1 | heme oxygenase-1 |
| IFNγ | interferon-γ |
| JAK-STAT3 | janus kinase/signal transducer and activator of transcription 3 |
| Keap1 | kelch-like ech-associated protein |
| LRP | lipoprotein receptor-related protein |
| LPS | lipopolysaccharide |
| MCT | monocarboxylate transporter |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NGF | nerve growth factor |
| NLR | leucine-rich-repeat |
| NLRP3 | leucine-rich-repeat pyrin domain-containing 3 |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| NOx | nitrates/nitrites |
| NOX | NADPH oxidase |
| NQO1 | quinone oxidoreductase 1 |
| Nrf2 | nuclear erythroid-related factor 2 |
| O2 | oxygen |
| PD | parkinson’s disease |
| Pry | pyruvate |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| SVTC-2 | sodium-dependent transporter |
| tBHQ | tertiary butylhydroquinone |
| TLRs | toll-like receptors |
| TNF | tumor necrosis factor |
| TRPC | transient receptor potential canonical |
| vE | vitamin E |
| xCT | cysteine–glutamate exchanger |
References
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective effect of antioxidants in the brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Lee, B.H. Neuroprotection: Rescue from neuronal death in the brain. Int. J. Mol. Sci. 2021, 22, 5525. [Google Scholar] [CrossRef] [PubMed]
- Tanioka, M.; Park, W.K.; Park, J.; Lee, J.E.; Lee, B.H. Lipid emulsion improves functional recovery in an animal model of stroke. Int. J. Mol. Sci. 2020, 21, 7373. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [Green Version]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Li, J.; Wuliji, O.; Li, W.; Jiang, Z.-G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef] [Green Version]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, A.; Balachandran, B. Role of oxidative stress and antioxidants in neurodegenerative diseases. Nutr. Neurosci. 2002, 5, 291–309. [Google Scholar] [CrossRef]
- Bauernfeind, A.L.; Barks, S.K.; Duka, T.; Grossman, L.I.; Hof, P.R.; Sherwood, C.C. Aerobic glycolysis in the primate brain: Reconsidering the implications for growth and maintenance. Brain Struct. Funct. 2013, 219, 1149–1167. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Abate, G.; Vezzoli, M.; Sandri, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Mitochondria and cellular redox state on the route from ageing to Alzheimer’s disease. Mech. Ageing Dev. 2020, 192, 111385. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The good, the bad, and the ugly of ros: New insights on aging and aging-related diseases from eukaryotic and prokaryotic model organisms. Oxid. Med. Cell. Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Heikkila, R.E.; MacNamee, D. The generation of hydrogen peroxide, superoxide radical, and hydroxyl radical by 6-hydroxydopamine, dialuric acid, and related cytotoxic agents. J. Biol. Chem. 1974, 249, 2447–2452. [Google Scholar] [CrossRef]
- Goncalves, R.L.; Rothschild, D.E.; Quinlan, C.L.; Scott, G.K.; Benz, C.C.; Brand, M.D. Sources of superoxide/H2O2 during mitochondrial proline oxidation. Redox Biol. 2014, 2, 901–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A.-M. ROS and brain diseases: The good, the bad, and the ugly. Oxidative Med. Cell. Longev. 2013, 2013, 963520. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Kumar Dabla, P. Oxidative stress and antioxidants in hypertension—A current review. Curr. Hypertens. Rev. 2015, 11, 132–142. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Yudkoff, M. Interactions in the metabolism of glutamate and the branched-chain amino acids and ketoacids in the CNS. Neurochem. Res. 2017, 42, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garza-Lombó, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity linked to dysfunctional metal ion homeostasis and xenobiotic metal exposure: Redox signaling and oxidative stress. Antioxid. Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef] [Green Version]
- Morgan, L.; Shah, B.; Rivers, L.; Barden, L.; Groom, A.; Chung, R.; Higazi, D.; Desmond, H.; Smith, T.; Staddon, J. Inflammation and dephosphorylation of the tight junction protein occludin in an experimental model of multiple sclerosis. Neuroscience 2007, 147, 664–673. [Google Scholar] [CrossRef]
- Qin, L.; Crews, F.T. NADP oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflammation 2012, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.R.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid. Med. Cell. Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Gajavelli, S.; Spurlock, M.S.; Andreoni, C.; de Rivero Vaccari, J.P.; Bullock, M.R.; Keane, R.W.; Dietrich, W.D. Microglial inflammasome activation in penetrating ballistic-like brain injury. J. Neurotrauma 2018, 35, 1681–1693. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar]
- Hawkins, B.; Davis, T. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Mulica, P.; Grünewald, A.; Pereira, S.L. Astrocyte-neuron metabolic crosstalk in neurodegeneration: A mitochondrial perspective. Front. Endocrinol. 2021, 12, 668517. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Ahn, S.; Yang, E.-J.; Kim, H.; Chong, Y.H.; Kim, H.-S. Hippocampus-based contextual memory alters the morphological characteristics of astrocytes in the dentate gyrus. Mol. Brain 2016, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Gulbransen, B.; Sharkey, K. Novel functional roles for enteric glia in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Sasaki, T.; Ishikawa, T.; Abe, R.; Nakayama, R.; Asada, A.; Matsuki, N.; Ikegaya, Y. Astrocyte calcium signalling orchestrates neuronal synchronization in organotypic hippocampal slices. J. Physiol. 2014, 592, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, R.; Baez-Jurado, E.; Hidalgo-Lanussa, O.; Echeverria, V.; Ashrad, G.M.; Sahebkar, A.; Barreto, G.E. Growth factors and neuroglobin in astrocyte protection against neurodegeneration and oxidative stress. Mol. Neurobiol. 2019, 56, 2339–2351. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K.; Nedergaard, M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics 2010, 7, 338–353. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Reactive astrocytes in neural repair and protection. Neuroscientist 2005, 11, 400–407. [Google Scholar] [CrossRef]
- Teismann, P.; Schulz, J.B. Cellular pathology of parkinson’s disease: Astrocytes, microglia and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef]
- Tsang, A.H.; Chung, K.K. Oxidative and nitrosative stress in parkinson’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, I.; Lukomska, B. The role of astrocytes in the physiology and pathology of the central nervous system. Acta Neurobiol. Exp. 2006, 66, 343–358. [Google Scholar]
- Lu, M.; Hu, L.-F.; Hu, G.; Bian, J.-S. Hydrogen sulfide protects astrocytes against H2O2-induced neural injury via enhancing glutamate uptake. Free. Radic. Biol. Med. 2008, 45, 1705–1713. [Google Scholar] [CrossRef]
- Sun, X.-L.; Zeng, X.-N.; Zhou, F.; Dai, C.-P.; Ding, J.-H.; Hu, G. Katp channel openers facilitate glutamate uptake by gluts in rat primary cultured astrocytes. Neuropsychopharmacology 2008, 33, 1336–1342. [Google Scholar] [CrossRef] [Green Version]
- Barbeito, L.H.; Pehar, M.; Cassina, P.; Vargas, M.; Peluffo, H.; Viera, L.; Estévez, A.G.; Beckman, J.S. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res. Brain Res. Rev. 2004, 47, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Shih, E.K.; Robinson, M.B. Role of astrocytic mitochondria in limiting ischemic brain injury? Physiology 2018, 33, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Nayernia, Z.; Jaquet, V.; Krause, K.-H. New insights on nox enzymes in the central nervous system. Antioxid. Redox Signal. 2014, 20, 2815–2837. [Google Scholar] [CrossRef] [Green Version]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.S. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in alzheimer’s diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef]
- Galea, E.; Feinstein, D.L.; Reis, D.J. Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proc. Natl. Acad. Sci. USA 1992, 89, 10945–10949. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Excitotoxicity and nitric oxide in parkinson’s disease pathogenesis. Ann. Neurol. 1998, 44, S110–S114. [Google Scholar] [CrossRef]
- Chen, X.; Guan, T.; Li, C.; Shang, H.; Cui, L.; Li, X.-M.; Kong, J. SOD1 aggregation in astrocytes following ischemia/reperfusion injury: A role of NO-mediated S-nitrosylation of protein disulfide isomerase (PDI). J. Neuroinflammation 2012, 9, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberto, C.M.; Albrecht, P.J.; Herx, L.M.; Yong, V.W.; Levison, S. Pro-regenerative properties of cytokine-activated astrocytes. J. Neurochem. 2004, 89, 1092–1100. [Google Scholar] [CrossRef]
- Dringen, R.; Kussmaul, L.; Gutterer, J.M.; Hirrlinger, J.; Hamprecht, B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 1999, 72, 2523–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.X. Antioxidant defense of the brain: A role for astrocytes. Can. J. Physiol. Pharmacol. 1997, 75, 1149–1163. [Google Scholar] [CrossRef]
- Dringen, R.; Bishop, G.; Koeppe, M.; Dang, T.N.; Robinson, S. The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem. Res. 2007, 32, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Vartiainen, N.E.; Ying, W.; Chan, P.H.; Koistinaho, J.; Swanson, R.A. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J. Neurochem. 2001, 77, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Beltrán, F.A.; Brauchi, S.; Concha, I.I. A metabolic switch in brain: Glucose and lactate metabolism modulation by ascorbic acid. J. Neurochem. 2009, 110, 423–440. [Google Scholar] [CrossRef]
- Castro, M.A.; Pozo, M.; Cortés, C.; García Mde, L.; Concha, I.I.; Nualart, F. Intracellular ascorbic acid inhibits transport of glucose by neurons, but not by astrocytes. J. Neurochem. 2007, 102, 773–782. [Google Scholar] [CrossRef]
- Salazar, K.; Espinoza, F.; Cerda-Gallardo, G.; Ferrada, L.; Magdalena, R.; Ramírez, E.; Ulloa, V.; Saldivia, N.; Troncoso, N.; Oviedo, M.J.; et al. SVCT2 overexpression and ascorbic acid uptake increase cortical neuron differentiation, which is dependent on vitamin c recycling between neurons and astrocytes. Antioxidants 2021, 10, 1413. [Google Scholar] [CrossRef]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the nrf2–keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxidants Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.F.; Al-Mubarak, B.; Martel, M.-A.; McKay, S.; Wheelan, N.; Hasel, P.; Márkus, N.M.; Baxter, P.; Deighton, R.F.; Serio, A.; et al. Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat. Commun. 2015, 6, 7066. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. keap1 is a redox-regulated substrate adaptor protein for a cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, D.; Tambone, S.; Cerretani, M.; Fezzardi, P.; Missineo, A.; Sherman, L.-T.; Munoz-Sajuan, I.; Harper, S.; Dominquez, C.; Pacifici, R.; et al. Nrf2 activation by reversible Keap1 binding induces the antioxidant response in primary neurons and astrocytes of a huntington’s disease mouse model. Free Radical Biol. Med. 2021, 162, 243–254. [Google Scholar] [CrossRef]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.-C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Wang, H.; Zhu, L.; Mao, L.; Qiao, L.; Su, X. Depletion of Nrf2 Enhances Inflammation Induced by Oxyhemoglobin in Cultured Mice Astrocytes. Neurochem. Res. 2011, 36, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lu, M.; Mei, M.; Wang, H.; Han, Z.; Chen, M.; Yao, H.; Song, N.; Ding, X.; Ding, J.; et al. Pyridoxine induces glutathione synthesis via PKM2-mediated Nrf2 transactivation and confers neuroprotection. Nat. Commun. 2020, 11, 941. [Google Scholar] [CrossRef] [Green Version]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Dissing-Olesen, L.; LeDue, J.M.; Rungta, R.; Hefendehl, J.; Choi, H.B.; MacVicar, B.A. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J. Neurosci. 2014, 34, 10511–10527. [Google Scholar] [CrossRef] [Green Version]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional microglia–neuron communication in health and disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Eyo, U.; Murugan, M.; Wu, L.-J. Microglia-neuron communication in epilepsy. Glia 2017, 65, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Stence, N.; Waite, M.; Dailey, M.E. Dynamics of microglial activation: A confocal time-lapse analysis in hippocampal slices. Glia 2001, 33, 256–266. [Google Scholar] [CrossRef]
- Priller, J.; Prinz, M. Targeting microglia in brain disorders. Science 2019, 365, 32–33. [Google Scholar] [CrossRef]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflammation 2012, 9, 155. [Google Scholar] [CrossRef] [Green Version]
- von Bernhardi, R.; Heredia, F.; Salgado, N.; Muñoz, P. Microglia function in the normal brain. Glial Cells Health Dis. CNS 2016, 949, 67–92. [Google Scholar]
- Pocock, J.M.; Kettenmann, H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007, 30, 527–535. [Google Scholar] [CrossRef]
- Minghetti, L.; Levi, G. Microglia as effector cells in brain damage and repair: Focus on prostanoids and nitric oxide. Prog. Neurobiol. 1998, 54, 99–125. [Google Scholar] [CrossRef]
- Nakanishi, H. Microglial Functions and Proteases. Mol. Neurobiol. 2003, 27, 163–176. [Google Scholar] [CrossRef]
- Colton, C.A.; Gilbert, D.L. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987, 223, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Dodd, C.A.; Filipov, N.M. Manganese potentiates LPS-induced heme-oxygenase 1 in microglia but not dopaminergic cells: Role in controlling microglial hydrogen peroxide and inflammatory cytokine output. NeuroToxicology 2011, 32, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Herber, D.L.; Maloney, J.L.; Roth, L.M.; Freeman, M.J.; Morgan, D.; Gordon, M.N. Diverse microglial responses after intrahippocampal administration of lipopolysaccharide. Glia 2006, 53, 382–391. [Google Scholar] [CrossRef]
- Liu, B.; Gao, H.-M.; Wang, J.-Y.; Jeohn, G.-H.; Cooper, C.L.; Hong, J.-S. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann. N. Y. Acad. Sci. 2002, 962, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, H. Arginine metabolism and the synthesis of nitric oxide in the nervous system. Prog. Neurobiol. 2001, 64, 365–391. [Google Scholar] [CrossRef]
- Bal-Price, A.; Matthias, A.; Brown, G.C. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J. Neurochem. 2002, 80, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Possel, H.; Noack, H.; Keilhoff, G.; Wolf, G. Life imaging of peroxynitrite in rat microglial and astroglial cells: Role of superoxide and antioxidants. Glia 2002, 38, 339–350. [Google Scholar] [CrossRef]
- Takeda, H.; Tomita, M.; Tanahashi, N.; Kobari, M.; Yokoyama, M.; Takao, M.; Ito, D.; Fukuuchi, Y. Hydrogen peroxide enhances phagocytic activity of ameboid microglia. Neurosci. Lett. 1998, 240, 5–8. [Google Scholar] [CrossRef]
- Dringen, R. Oxidative and Antioxidative Potential of Brain Microglial Cells. Antioxid. Redox Signal. 2005, 7, 1223–1233. [Google Scholar] [CrossRef]
- Chatterjee, S.; Noack, H.; Possel, H.; Keilhoff, G.; Wolf, G. Glutathione levels in primary glial cultures: Monochlorobimane provides evidence of cell type-specific distribution. Glia 1999, 27, 152–161. [Google Scholar] [CrossRef]
- Dopp, J.M.; Sarafian, T.A.; Spinella, F.M.; Kahn, M.A.; Shau, H.; de Vellis, J. Expression of the p75 TNF receptor is linked to TNF-induced NFκB translocation and oxyradical neutralization in glial cells. Neurochem. Res. 2002, 27, 1535–1542. [Google Scholar] [CrossRef]
- Chatterjee, S.; Noack, H.; Possel, H.; Wolf, G. Induction of nitric oxide synthesis lowers intracellular glutathione in microglia of primary glial cultures. Glia 2000, 29, 98–101. [Google Scholar] [CrossRef]
- Lindenau, J.; Noack, H.; Possel, H.; Asayama, K.; Wolf, G. Cellular distribution of superoxide dismutases in the rat CNS. Glia 2000, 29, 25–34. [Google Scholar] [CrossRef]
- Noack, H.; Lindenau, J.; Rothe, F.; Asayama, K.; Wolf, G. Differential expression of superoxide dismutase isoforms in neuronal and glial compartments in the course of excitotoxically mediated neurodegeneration: Relation to oxidative and nitrergic stress. Glia 1998, 23, 285–297. [Google Scholar] [CrossRef]
- Hollensworth, S.B.; Shen, C.-C.; Sim, J.E.; Spitz, D.R.; Wilson, G.L.; LeDoux, S.P. Glial cell type-specific responses to menadione-induced oxidative stress. Free. Radic. Biol. Med. 2000, 28, 1161–1174. [Google Scholar] [CrossRef]
- Noack, H.; Possel, H.; Rethfeldt, C.; Keilhoff, G.; Wolf, G. Peroxynitrite mediated damage and lowered superoxide tolerance in primary cortical glial cultures after induction of the inducible isoform of NOS. Glia 1999, 28, 13–24. [Google Scholar] [CrossRef]
- Chan, T.E.; Grossman, Y.S.; Bloss, E.B.; Janssen, W.G.; Lou, W.; McEwen, B.S.; Dumitriu, D.; Morrison, J.H. Cell-type specific changes in glial morphology and glucocorticoid expression during stress and aging in the medial prefrontal cortex. Front. Aging Neurosci. 2018, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, N.; Kato, H.; Araki, T. Age-related changes of astorocytes, oligodendrocytes and microglia in the mouse hippocampal CA1 sector. Mech. Ageing Dev. 2007, 128, 311–316. [Google Scholar] [CrossRef]
- Long, J.M.; Kalehua, A.N.; Muth, N.J.; Calhoun, M.E.; Jucker, M.; Hengemihle, J.M.; Ingram, D.K.; Mouton, P.R. Stereological analysis of astrocyte and microglia in aging mouse hippocampus. Neurobiol. Aging 1998, 19, 497–503. [Google Scholar] [CrossRef]
- Sharaf, A.; Krieglstein, K.; Spittau, B. Distribution of microglia in the postnatal murine nigrostriatal system. Cell Tissue Res. 2012, 351, 373–382. [Google Scholar] [CrossRef]
- Edler, M.K.; Mhatre-Winters, I.; Richardson, J. Microglia in aging and Alzheimer’s disease: A comparative species review. Cells 2021, 10, 1138. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L. NFκB -activated astroglial release of complement C3 compromises neuronal morphology and function associated with alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhang, A.; Zhu, Y.; He, W.; Di, W.; Fang, Y.; Shi, X. MFG-E8 reverses microglial-induced neurotoxic astrocyte (A1) via NFκB and PI3K-Akt pathways. J. Cell. Physiol. 2018, 234, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive astrocytes in neurodegenerative diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Sidoryk-Wegrzynowicz, M.; Węgrzynowicz, M.; Lee, E.; Bowman, A.B.; Aschner, M. Role of astrocytes in brain function and disease. Toxicol. Pathol. 2011, 39, 115–123. [Google Scholar] [CrossRef]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/Glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Mercer, J.F.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116, 33–57. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Negoro, R.; Kadoi, H.; Motegi, T.; Shibagaki, F.; Yamamuro, A.; Ishimaru, Y.; Maeda, S. Noradrenaline protects neurons against H2O2-induced death by increasing the supply of glutathione from astrocytes via β 3 -adrenoceptor stimulation. J. Neurosci. Res. 2021, 99, 621–637. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias-Pinto, A.; Acuña, A.I.; Beltrán, F.A.; Torres-Díaz, L.; Castro, M.A. Old things new view: Ascorbic acid protects the brain in neurodegenerative disorders. Int. J. Mol. Sci. 2015, 16, 28194–28217. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kim, U.J.; Cha, M.; Lee, B.H. Chronic treatment of ascorbic acid leads to age-dependent neuroprotection against oxidative injury in hippocampal slice cultures. Int. J. Mol. Sci. 2021, 22, 1608. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Dringen, R.; Schousboe, A.; Robinson, S.R. Astrocytes: Glutamate producers for neurons. J. Neurosci. Res. 1999, 57, 417–428. [Google Scholar] [CrossRef]
- Tani, H.; Dulla, C.G.; Farzampour, Z.; Taylor-Weiner, A.; Huguenard, J.R.; Reimer, R.J. A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron 2014, 81, 888–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajcy, K.; Lochynski, S.; Librowski, T. A role of GABA analogues in the treatment of neurological diseases. Curr. Med. Chem. 2010, 17, 2338–2347. [Google Scholar] [CrossRef] [PubMed]
- Ting Wong, C.G.; Bottiglieri, T.; Snead, O.C., III. GABA, gamma-hydroxybutyric acid, and neurological disease. Ann. Neurol. 2003, 54, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.B.; Waagepetersen, H.S.; Bak, L.K.; Schousboe, A.; Sonnewald, U. The glutamine–glutamate/GABA cycle: Function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem. Res. 2015, 40, 402–409. [Google Scholar] [CrossRef]
- Heinemann, U.; Lux, H.D. Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res. 1977, 120, 231–249. [Google Scholar] [CrossRef]
- Barres, B.A. Glial ion channels. Curr. Opin. Neurobiol. 1991, 1, 354–359. [Google Scholar] [CrossRef]
- Orkand, R.K.; Nicholls, J.G.; Kuffler, S.W. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 1966, 29, 788–806. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.P.; Zorec, R. Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotti, S.; Charles, A. Extracellular calcium sensing by glial cells: Low extracellular calcium induces intracellular calcium release and intercellular signaling. J. Neurochem. 1997, 69, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Teschemacher, A.G.; Kasparov, S. Neuroprotective potential of astroglia. J. Neurosci. Res. 2017, 95, 2126–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maneshi, M.M.; Maki, B.; Gnanasambandam, R.; Belin, S.; Popescu, G.K.; Sachs, F.; Hua, S.Z. Mechanical stress activates NMDA receptors in the absence of agonists. Sci. Rep. 2017, 7, 39610. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.; Wang, F.; Xu, Q.; Fujita, T.; Dobrowolski, R.; Willecke, K.; Takano, T.; Nedergaard, M. Extracellular Ca2+ acts as a mediator of communication from neurons to glia. Sci. Signal. 2012, 5, ra8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvat, A.; Muhič, M.; Smolič, T.; Begić, E.; Zorec, R.; Kreft, M.; Vardjan, N. Ca2+ as the prime trigger of aerobic glycolysis in astrocytes. Cell Calcium 2021, 95, 102368. [Google Scholar] [CrossRef]
- Porras, O.H.; Ruminot, I.; Loaiza, A.; Barros, L.F. Na+-Ca2+cosignaling in the stimulation of the glucose transporter GLUT1 in cultured astrocytes. Glia 2008, 56, 59–68. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, J.A. The Nrf2–ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 2009, 11, e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of endogenous neuroprotective effects of astrocytes in brain injury. Oxid. Med. Cell. Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef]
- Zhang, R.; Chae, S.; Lee, J.H.; Hyun, J.W. The cytoprotective effect of butin against oxidative stress is mediated by the up-regulation of manganese superoxide dismutase expression through a PI3K/Akt/Nrf2-dependent pathway. J. Cell. Biochem. 2012, 113, 1987–1997. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Neuron-astrocyte interactions in Parkinson’s disease. Cells 2020, 9, 2623. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Kikkawa, Y.; Takeshima, M.; Murakami, S.; Miyoshi, K.; Sogawa, N.; Kita, T. Astrocyte-derived metallothionein protects dopaminergic neurons from dopamine quinone toxicity. Glia 2011, 59, 435–451. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Microglia in steady state. J. Clin. Investig. 2017, 127, 3201–3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michell-Robinson, M.A.; Touil, H.; Healy, L.M.; Owen, D.R.; Durafourt, B.A.; Bar-Or, A.; Antel, J.P.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, D.G.; Lue, L.-F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimer’s Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Kelly, M.G.; Yang, X.R.; Fernandez, T.G.; Wierzbicki, E.L.; Skrobach, A.; Doré, S. Nrf2 deficiency exacerbates cognitive impairment and reactive microgliosis in a lipopolysaccharide-induced neuroinflammatory mouse model. Cell. Mol. Neurobiol. 2020, 40, 1185–1197. [Google Scholar] [CrossRef]
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taillé, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases NOS2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. 2005, 19, 1890–1892. [Google Scholar] [CrossRef] [Green Version]
- Prawan, A.; Kundu, J.K.; Surh, Y.-J. Molecular basis of heme oxygenase-1 induction: Implications for chemoprevention and chemoprotection. Antioxid. Redox Signal. 2005, 7, 1688–1703. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Mendívil, C.; Luengo, E.; Trigo-Alonso, P.; García-Magro, N.; Negredo, P.; López, M.G. Protective role of microglial HO-1 blockade in aging: Implication of iron metabolism. Redox Biol. 2021, 38, 101789. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Chu, S.-F.; Zhang, Z.; Xia, C.-Y.; Chen, N.-H. Fractalkine/CX3CR1 is involved in the cross-talk between neuron and glia in neurological diseases. Brain Res. Bull. 2019, 146, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Morganti, J.M.; Bachstetter, A.D.; Hudson, C.E.; Peters, M.M.; Grimmig, B.A.; Weeber, E.J.; Bickford, P.C.; Gemma, C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 2011, 31, 16241–16250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates III, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Ishibashi, H.; Hashimoto, K.; Nakanishi, H. Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors. Glia 2006, 53, 660–668. [Google Scholar] [CrossRef]
- Wang, N.; Mi, X.; Gao, B.; Gu, J.; Wang, W.; Zhang, Y.; Wang, X. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience 2015, 287, 144–156. [Google Scholar] [CrossRef]
- Fracassi, A.; Marcatti, M.; Zolochevska, O.; Tabor, N.; Woltjer, R.; Moreno, S.; Taglialatela, G. Oxidative damage and antioxidant response in frontal cortex of demented and nondemented individuals with Alzheimer’s neuropathology. J. Neurosci. 2021, 41, 538–554. [Google Scholar] [CrossRef]
- Kosa, P.; Wu, T.; Phillips, J.; Leinonen, M.; Masvekar, R.; Komori, M.; Wichman, A.; Sandford, M.; Bielekova, B. Idebenone does not inhibit disability progression in primary progressive MS. Mult. Scler. Relat. Disord. 2020, 45, 102434. [Google Scholar] [CrossRef] [PubMed]
- Turati, J.; Ramírez, D.; Carniglia, L.; Saba, J.; Caruso, C.; Quarleri, J.; Durand, D.; Lasaga, M. Antioxidant and neuroprotective effects of mGlu3 receptor activation on astrocytes aged in vitro. Neurochem. Int. 2020, 140, 104837. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic of the main pathophysiological mechanism of cell death due to oxidative stress. The presence of ROS due to an imbalance of pro-oxidants and antioxidants can damage a variety of cells in the brain. Formation of ROS and mitochondria dysfunction occurs during the primary mechanism of oxidative stress. Secondary mechanisms of cell death by ROS production include excitotoxicity, iron metabolism, cytokines, pyroptosis, and necroptosis, which amplify cell death.
Figure 1.
Schematic of the main pathophysiological mechanism of cell death due to oxidative stress. The presence of ROS due to an imbalance of pro-oxidants and antioxidants can damage a variety of cells in the brain. Formation of ROS and mitochondria dysfunction occurs during the primary mechanism of oxidative stress. Secondary mechanisms of cell death by ROS production include excitotoxicity, iron metabolism, cytokines, pyroptosis, and necroptosis, which amplify cell death.

Figure 2.
Schematic depicting morphological and functional changes in activated glial cells following oxidative stress. The resting and reactive states of astrocytes and microglia have different morphologies and functions. In a healthy brain, astrocytes are called territorial cells and they maintain extracellular homeostasis via numerous cellular processes. Microglia use their defense mechanisms to rapidly respond to disturbances in the brain environment, and assist in specific immune functions. However, under oxidative stress, reactive astrocytes undergo astrogliosis, which associates with cellular hypertrophy, astrocyte proliferation, increasing numbers and thickness of processes, and expanded cell body size. Reactive microglia, also called amoeboid microglia, exhibit morphological modifications and proliferation and produce several inflammatory mediators, including nitric oxide and superoxide. The disruption of vascular integrity is observed and increases the permeability of immune cells in pathological conditions.
Figure 2.
Schematic depicting morphological and functional changes in activated glial cells following oxidative stress. The resting and reactive states of astrocytes and microglia have different morphologies and functions. In a healthy brain, astrocytes are called territorial cells and they maintain extracellular homeostasis via numerous cellular processes. Microglia use their defense mechanisms to rapidly respond to disturbances in the brain environment, and assist in specific immune functions. However, under oxidative stress, reactive astrocytes undergo astrogliosis, which associates with cellular hypertrophy, astrocyte proliferation, increasing numbers and thickness of processes, and expanded cell body size. Reactive microglia, also called amoeboid microglia, exhibit morphological modifications and proliferation and produce several inflammatory mediators, including nitric oxide and superoxide. The disruption of vascular integrity is observed and increases the permeability of immune cells in pathological conditions.

Figure 3.
This diagram represents neuron–glia crosstalk involved in neuroprotection and the antioxidant defense mechanism. Astrocyte-neuron: Astrocytes contain a variety of antioxidant molecules, including glutathione (GSH), ascorbate, and vitamin E (vE), and ROS-detoxifying enzymes, such as GSH S-transferase, GSH peroxidase, thioredoxin reductase, and catalase. Astrocytes project the end-feet processes onto brain capillary surface so that astrocytes control the movement of molecules and cells between the vascular compartments and the brain. In the lactate shuttle, astrocytes support neurons by regulating glucose transformation into lactate, which ensures the energetic support of neurons. Neuronal activity triggers glucose metabolism in astrocytes. Glucose is converted to pyruvate by glycolysis and to lactate, which is released from astrocytes and taken up by neurons (blue arrow). Astrocytes can synthesize GSH via activation of Nrf2 and can shuttle GSH precursors to neurons for GSH synthesis. Astrocytes release GSH into the extracellular space and neurons take up the GSH directly or use extracellular neuronal aminopeptidase N to form glycine and cysteine (black arrow). In glutamate uptake and recycling, glutamate from the synaptic space enters astrocytes through EAAT and is converted by glutamine synthetase (GS) to inactive glutamine. After its release and import into neurons, glutamine can be re-converted to glutamate (red arrow). Recycled ascorbate can directly scavenge ROS and act as a cofactor for the recycling of oxidized vE and GSH. Astrocytes take up dehydroascorbic acid (DHA), an oxidation product of ascorbate, from the extracellular space and recycle it back to ascorbic acid. Astrocytes capture and transport excess extracellular K+ to the astrocytic syncytium through Na+/K+ ATPase. Nrf2 induction of glutamate cysteine ligase (GCL) increases GSH synthesis in astrocytes, and GSH is subsequently exported to the extracellular medium. Astrocytes also participate in metal sequestration in the brain to prevent the generation of free radicals by redox active metals. Microglia-neuron: Microglia contain a high cellular GSH concentration and express and upregulate diverse antioxidant enzymes. The expression of classical antioxidant proteins are controlled by Nrf2 in microglia. Heme oxygenase-1 (HO-1), an antioxidant enzyme upregulated by Nrf2, inhibits NOX2 activation. Fractalkine (FKN) is predominantly expressed on neuronal cells, and microglia and neurons exclusively express the fractalkine receptor (CX3CR1); this is an interesting signaling axis for communication. Abbreviations: ARE, antioxidant response element; ASC, ascorbate; ApoE, apolipoprotein E; xCT, cysteine-glutamate exchanger; Cys, cysteine; DHA, dehydroascorbic acid; DMT1, divalent metal transporter; EAAT, excitatory amino acid transporter; mFKN, membrane-anchored fractalkine; sFKN, soluble fractalkine; CX3CR1, fractalkine receptor; Glc, glucose; GLUT, glucose transporter; Glu, glutamate; Gln, glutamine; GSH, glutathione; GCL, glutamate-cysteine ligase; GS, glutamine synthetase; GLAST, glutamate aspartate transporter; GLT1, glutamate transporter 1; Gly, glycine; HO-1, heme oxygenase-1; JNK, c-Jun amino terminal kinase; LRP, lipoprotein receptor-related protein; MCT, monocarboxylate transporter; Nrf2, nuclear erythroid-related factor 2; Pyr, pyruvate; SVTC-2, sodium-dependent transporter; TRPC, transient receptor potential canonical.
Figure 3.
This diagram represents neuron–glia crosstalk involved in neuroprotection and the antioxidant defense mechanism. Astrocyte-neuron: Astrocytes contain a variety of antioxidant molecules, including glutathione (GSH), ascorbate, and vitamin E (vE), and ROS-detoxifying enzymes, such as GSH S-transferase, GSH peroxidase, thioredoxin reductase, and catalase. Astrocytes project the end-feet processes onto brain capillary surface so that astrocytes control the movement of molecules and cells between the vascular compartments and the brain. In the lactate shuttle, astrocytes support neurons by regulating glucose transformation into lactate, which ensures the energetic support of neurons. Neuronal activity triggers glucose metabolism in astrocytes. Glucose is converted to pyruvate by glycolysis and to lactate, which is released from astrocytes and taken up by neurons (blue arrow). Astrocytes can synthesize GSH via activation of Nrf2 and can shuttle GSH precursors to neurons for GSH synthesis. Astrocytes release GSH into the extracellular space and neurons take up the GSH directly or use extracellular neuronal aminopeptidase N to form glycine and cysteine (black arrow). In glutamate uptake and recycling, glutamate from the synaptic space enters astrocytes through EAAT and is converted by glutamine synthetase (GS) to inactive glutamine. After its release and import into neurons, glutamine can be re-converted to glutamate (red arrow). Recycled ascorbate can directly scavenge ROS and act as a cofactor for the recycling of oxidized vE and GSH. Astrocytes take up dehydroascorbic acid (DHA), an oxidation product of ascorbate, from the extracellular space and recycle it back to ascorbic acid. Astrocytes capture and transport excess extracellular K+ to the astrocytic syncytium through Na+/K+ ATPase. Nrf2 induction of glutamate cysteine ligase (GCL) increases GSH synthesis in astrocytes, and GSH is subsequently exported to the extracellular medium. Astrocytes also participate in metal sequestration in the brain to prevent the generation of free radicals by redox active metals. Microglia-neuron: Microglia contain a high cellular GSH concentration and express and upregulate diverse antioxidant enzymes. The expression of classical antioxidant proteins are controlled by Nrf2 in microglia. Heme oxygenase-1 (HO-1), an antioxidant enzyme upregulated by Nrf2, inhibits NOX2 activation. Fractalkine (FKN) is predominantly expressed on neuronal cells, and microglia and neurons exclusively express the fractalkine receptor (CX3CR1); this is an interesting signaling axis for communication. Abbreviations: ARE, antioxidant response element; ASC, ascorbate; ApoE, apolipoprotein E; xCT, cysteine-glutamate exchanger; Cys, cysteine; DHA, dehydroascorbic acid; DMT1, divalent metal transporter; EAAT, excitatory amino acid transporter; mFKN, membrane-anchored fractalkine; sFKN, soluble fractalkine; CX3CR1, fractalkine receptor; Glc, glucose; GLUT, glucose transporter; Glu, glutamate; Gln, glutamine; GSH, glutathione; GCL, glutamate-cysteine ligase; GS, glutamine synthetase; GLAST, glutamate aspartate transporter; GLT1, glutamate transporter 1; Gly, glycine; HO-1, heme oxygenase-1; JNK, c-Jun amino terminal kinase; LRP, lipoprotein receptor-related protein; MCT, monocarboxylate transporter; Nrf2, nuclear erythroid-related factor 2; Pyr, pyruvate; SVTC-2, sodium-dependent transporter; TRPC, transient receptor potential canonical.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413315
AMA Style
Lee KH, Cha M, Lee BH. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. International Journal of Molecular Sciences. 2021; 22(24):13315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413315
Chicago/Turabian StyleLee, Kyung Hee, Myeounghoon Cha, and Bae Hwan Lee. 2021. "Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection" International Journal of Molecular Sciences 22, no. 24: 13315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413315
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.