
Preventive Effect of Canstatin against Ventricular Arrhythmia Induced by Ischemia/Reperfusion Injury: A Pilot Study


Abstract
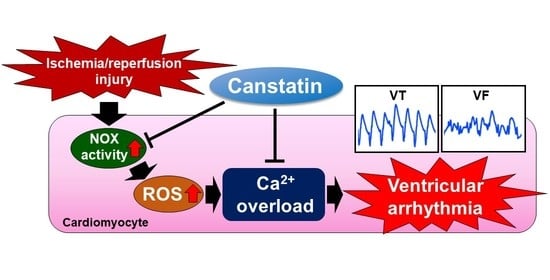
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
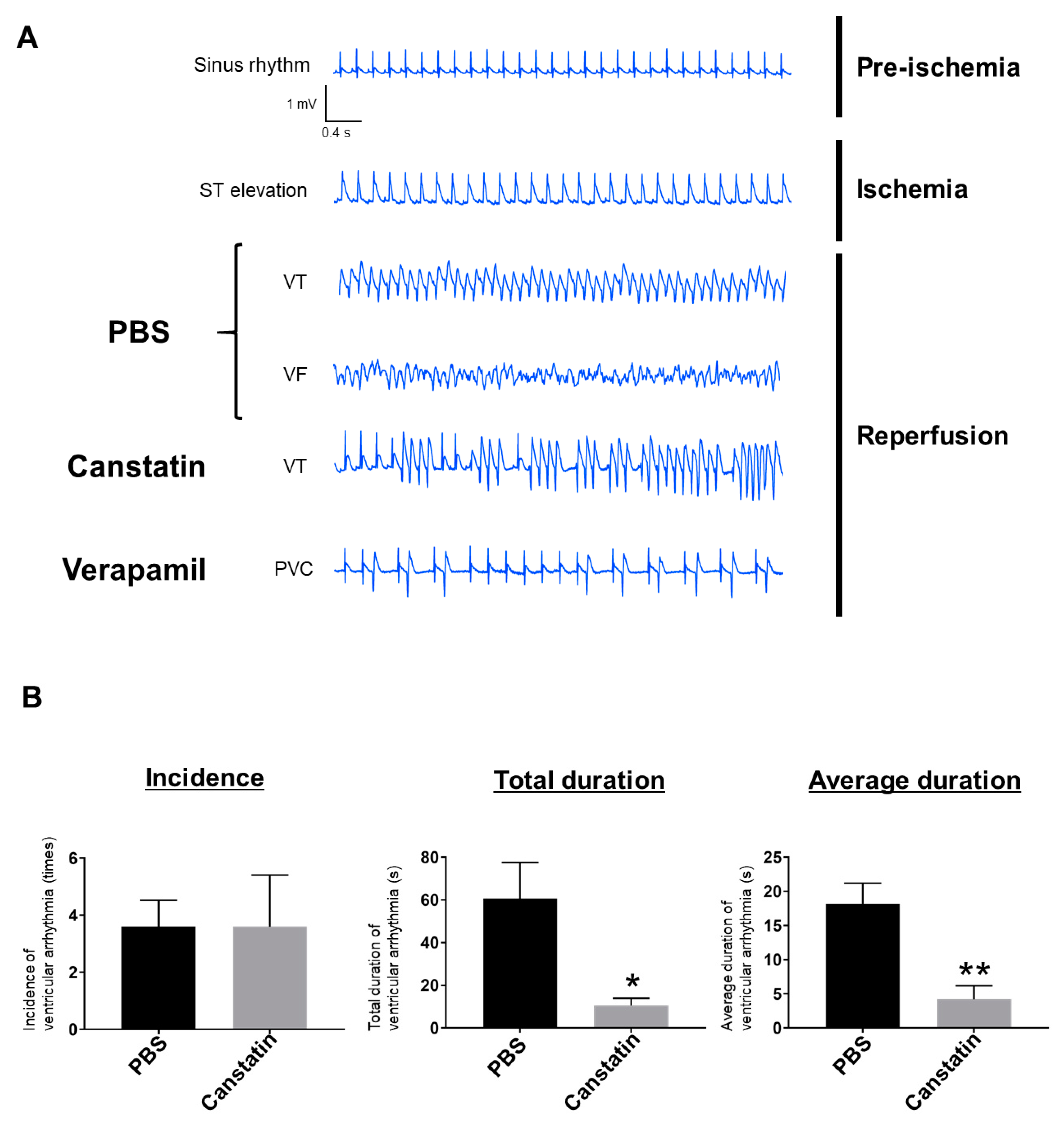
2.1. Canstatin Inhibited I/R-Induced Ventricular Arrhythmia
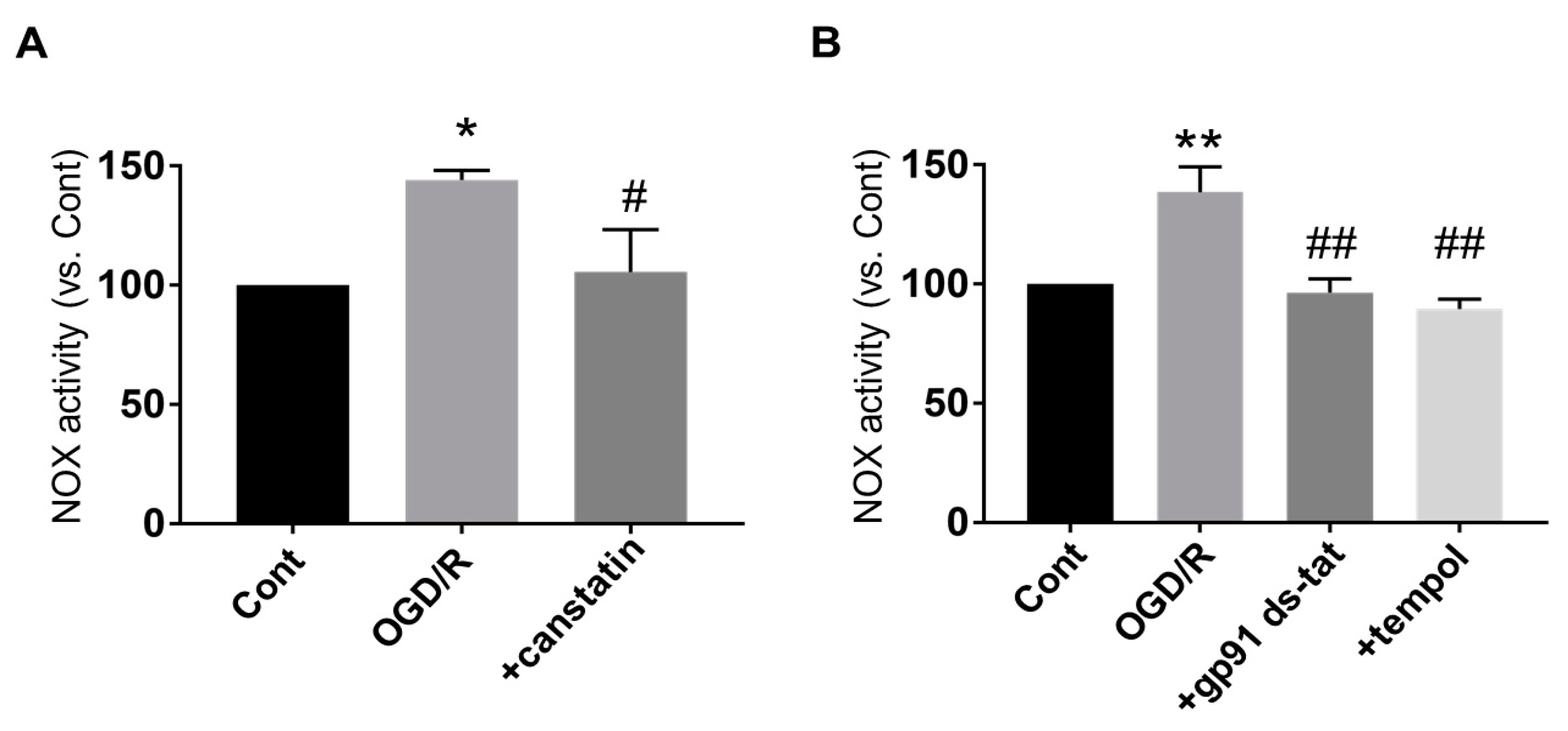
2.2. Canstatin Inhibited Oxygen and Glucose Deprivation Followed by Reoxygenation (OGD/R)-Induced Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase (NOX) Activation in NRCMs
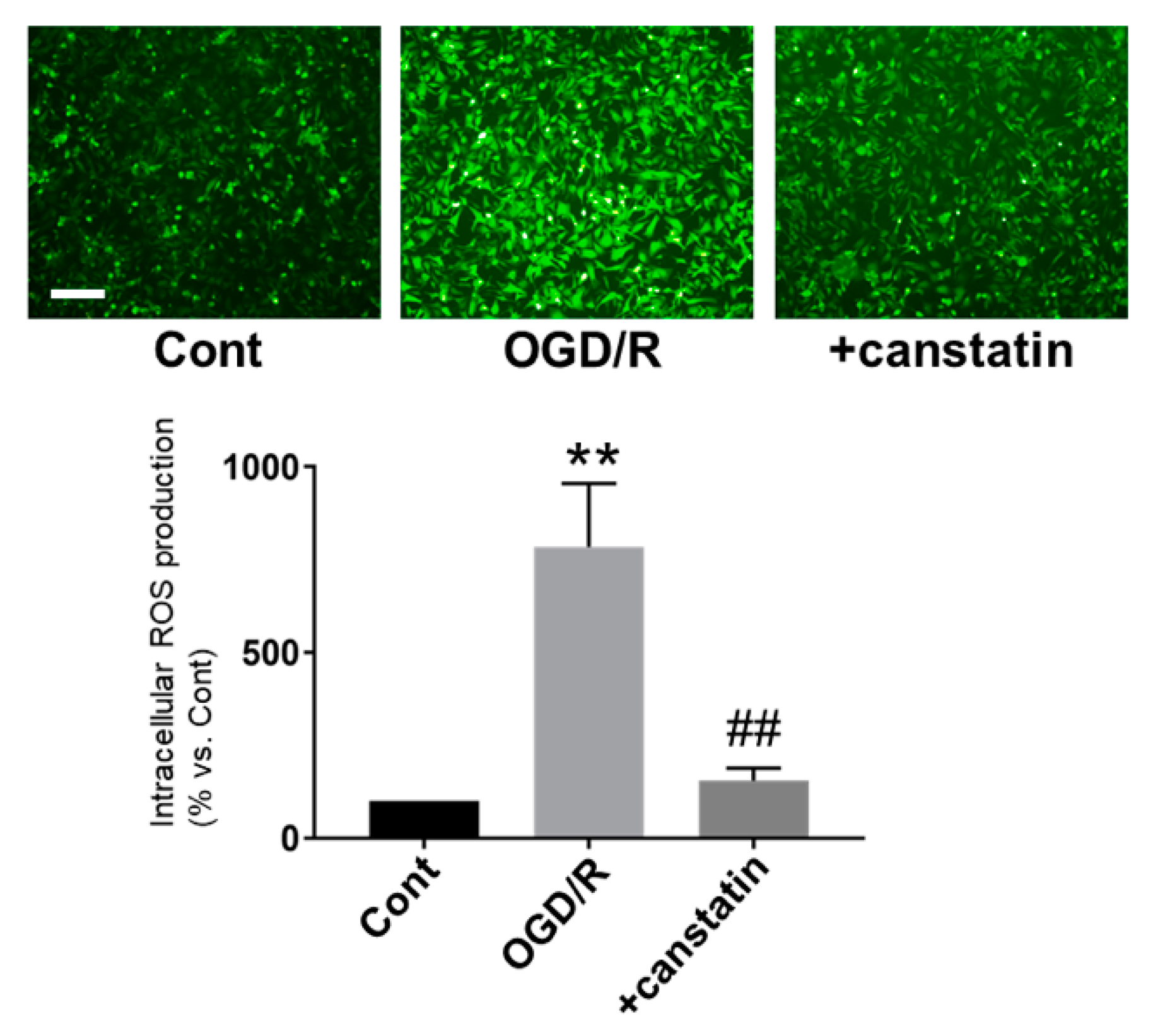
2.3. Canstatin Inhibited OGD/R-Induced ROS Production in NRCMs
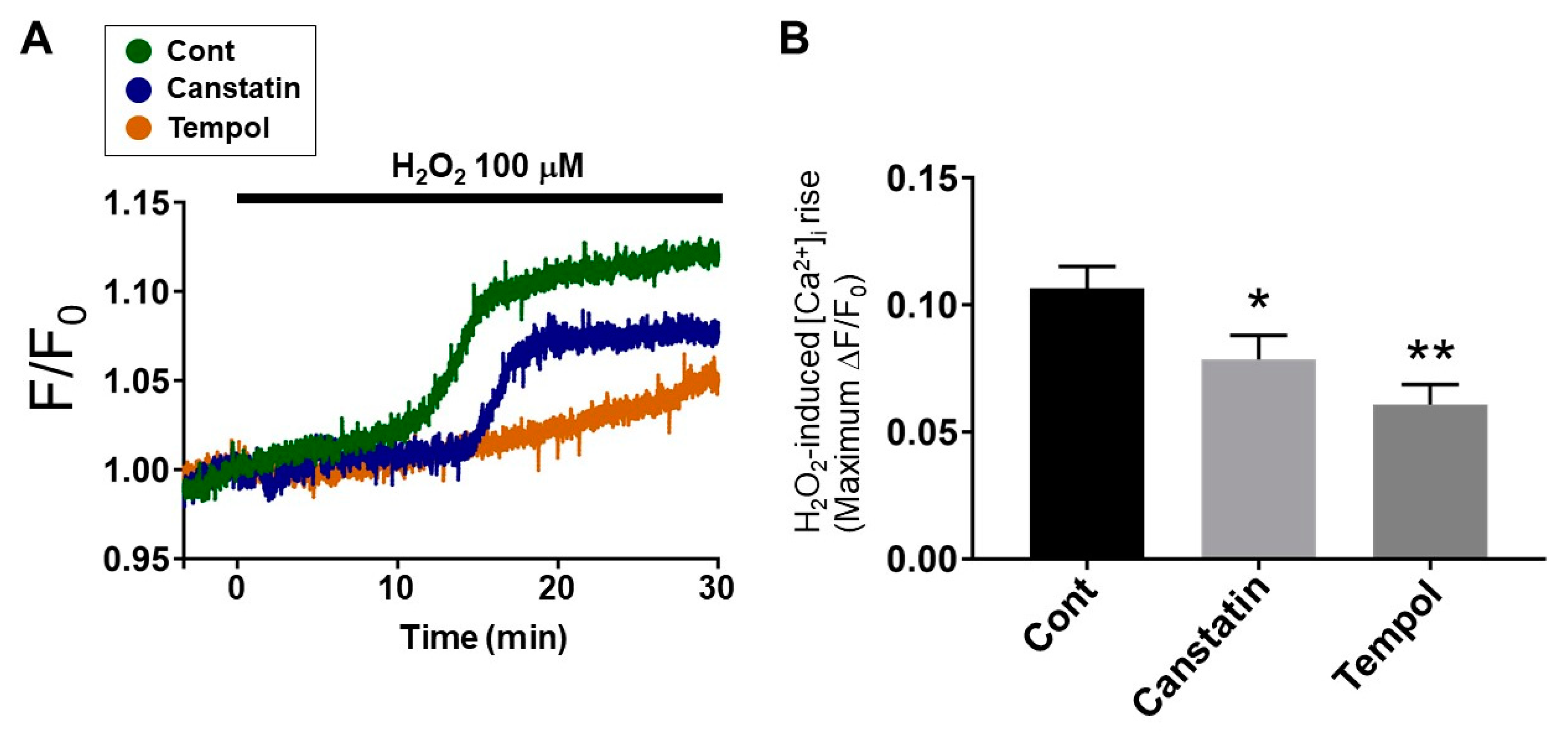
2.4. Canstatin Inhibited H2O2-Induced [Ca2+]i Rise in NRCMs
3. Discussion
4. Materials and Methods
4.1. Regents
4.2. Animals
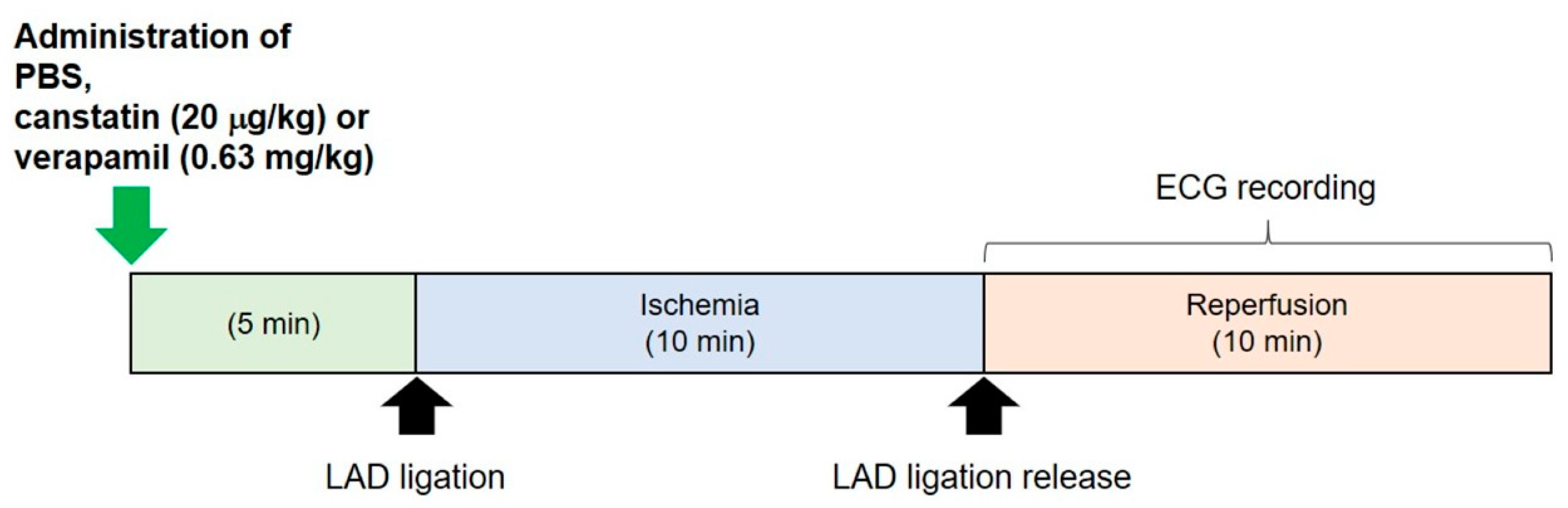
4.3. In Vivo Model of I/R-Induced Ventricular Arrhythmia
4.4. Isolation of NRCMs
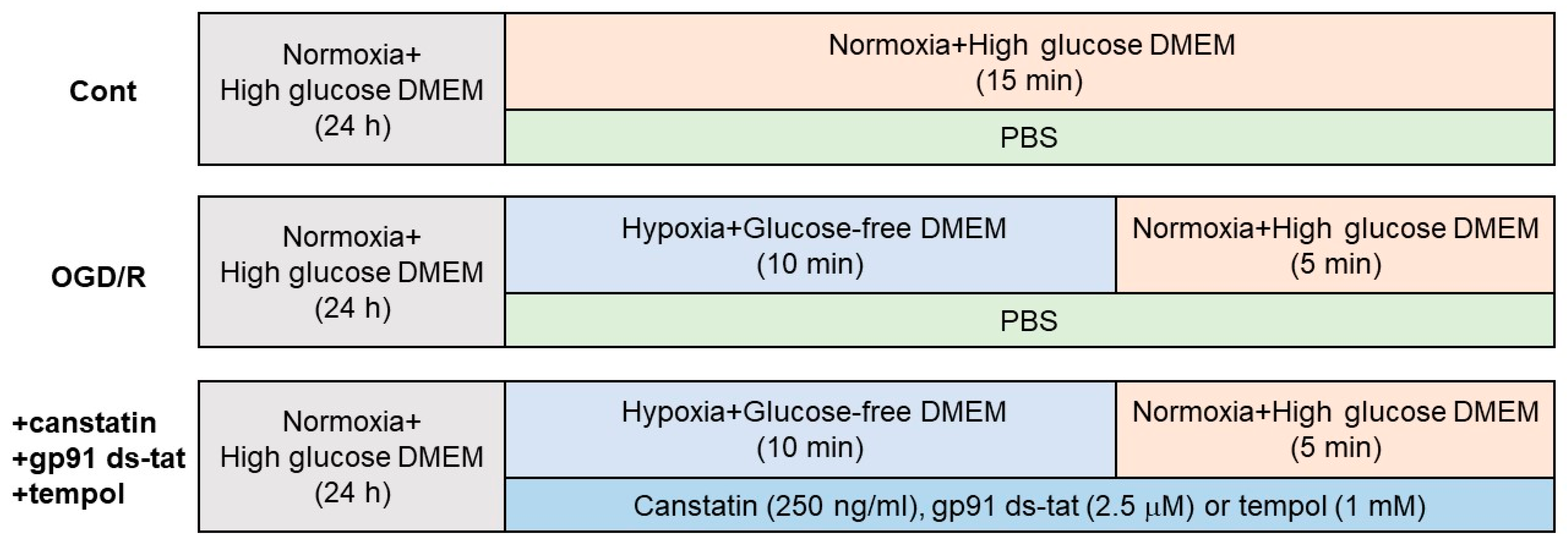
4.5. OGD/R
4.6. Lucigenin Assay
4.7. DCF-DA Staining
4.8. Measurement of [Ca2+]i in NRCMs
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palasubramaniam, J.; Wang, X.; Peter, K. Myocardial infarction-From atherosclerosis to thrombosis: Uncovering new diagnostic and therapeutic approaches. Arterioscler. Thromb. Vasc. Biol. 2019, 39, E176–E185. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/reperfusion injury following acute myocardial infarction: A critical issue for clinicians and forensic pathologists. Mediat. Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F. Clinical manifestations and basic mechanisms of myocardial ischemia/reperfusion injury. Tzu Chi Med. J. 2018, 30, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sovari, A.A. Cellular and molecular mechanisms of arrhythmia by oxidative stress. Cardiol. Res. Pract. 2016, 2016, 9656078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kistamás, K.; Veress, R.; Horváth, B.; Bányász, T.; Nánási, P.P.; Eisner, D.A. Calcium handling defects and cardiac arrhythmia syndromes. Front. Pharmacol. 2020, 11, 72. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium signaling and cardiac arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Bloma, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the Europe. Europace 2015, 17, 1601–1687. [Google Scholar]
- Brogden, R.N.; Benfield, P. Verapamil: A review of its pharmacological properties and therapeutic use in coronary artery disease. Drugs 1996, 51, 792–819. [Google Scholar] [CrossRef]
- Kamphaus, G.D.; Colorado, P.C.; Panka, D.J.; Hopfer, H.; Ramchandran, R.; Torre, A.; Maeshima, Y.; Mier, J.W.; Sukhatme, V.P.; Kalluri, R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J. Biol. Chem. 2000, 275, 1209–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, A.; Mitsui, A.; Okada, M.; Yamawaki, H. Cathepsin S degrades arresten and canstatin in infarcted area after myocardial infarction in rats. J. Vet. Med. Sci. 2019, 81, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, A.; Okada, M.; Yamawaki, H. Pathophysiological roles of canstatin on myofibroblasts after myocardial infarction in rats. Eur. J. Pharmacol. 2017, 807, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Imoto, K.; Hirakawa, M.; Okada, M.; Yamawaki, H. Canstatin modulates L-type calcium channel activity in rat ventricular cardiomyocytes. Biochem. Biophys. Res. Commun. 2018, 499, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Okada, M.; Yamawaki, H. Canstatin suppresses isoproterenol-induced cardiac hypertrophy through inhibition of calcineurin/nuclear factor of activated T-cells pathway in rats. Eur. J. Pharmacol. 2020, 871, 172849. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.M.; Liu, M.; Sturdy, M.; Gao, G.; Varghese, S.T.; Sovari, A.A.; Dudley, S.C., Jr. Metabolic stress, reactive oxygen species, and arrhythmia. J. Mol. Cell. Cardiol. 2012, 52, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedighi, M.; Nazari, A.; Faghihi, M.; Rafieian-Kopaei, M.; Karimi, A.; Moghimian, M.; Mozaffarpur, S.A.; Rashidipour, M.; Namdari, M.; Cheraghi, M.; et al. Protective effects of cinnamon bark extract against ischemia–reperfusion injury and arrhythmias in rat. Phyther. Res. 2018, 32, 1983–1991. [Google Scholar] [CrossRef]
- Dodo, K.; Shimizu, T.; Sasamori, J.; Aihara, K.; Terayama, N.; Nakao, S.; Iuchi, K.; Takahashi, M.; Sodeoka, M. Indolylmaleimide derivative IM-17 shows cardioprotective effects in ischemia-reperfusion injury. ACS Med. Chem. Lett. 2018, 9, 182–187. [Google Scholar] [CrossRef]
- Lu, H.R.; Yang, P.; Remeysen, P.; Saels, A.; Dai, D.Z.; De Clerck, F. Ischemia/reperfusion-induced arrhythmias in anaesthetized rats: A role of Na+ and Ca2+ influx. Eur. J. Pharmacol. 1999, 365, 233–239. [Google Scholar] [CrossRef]
- Gorenek, B.; Blomström Lundqvist, C.; Brugada Terradellas, J.; Camm, A.J.; Hindricks, G.; Huber, K.; Kirchhof, P.; Kuck, K.H.; Kudaiberdieva, G.; Lin, T.; et al. Cardiac arrhythmias in acute coronary syndromes: Position paper from the joint EHRA, ACCA, and EAPCI task force. EuroIntervention 2015, 10, 1095–1108. [Google Scholar] [CrossRef]
- Magnon, C.; Galaup, A.; Mullan, B.; Rouffiac, V.; Bidart, J.M.; Griscelli, F.; Opolon, P.; Perricaudet, M. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with αvβ3 and αvβ5 integrins. Cancer Res. 2005, 65, 4353–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honoré, S.; Kovacic, H.; Pichard, V.; Briand, C.; Rognoni, J.B. α2β1-Integrin signaling by itself controls G1/S transition in a human adenocarcinoma cell line (Caco-2): Implication of NADPH oxidase-dependent production of ROS. Exp. Cell Res. 2003, 285, 59–71. [Google Scholar] [CrossRef]
- Kim, E.Y.; Roshanravan, H.; Dryer, S.E. Syndecan-4 ectodomain evokes mobilization of podocyte TRPC6 channels and their associated pathways: An essential role for integrin signaling. Biochim. Biophys. Acta. 2015, 1853, 2610–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowden Dahl, K.D.; Robertson, S.E.; Weaver, V.M.; Simon, M.C. Hypoxia-inducible factor regulates αvβ3 integrin cell surface expression. Mol. Biol. Cell 2005, 16, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Monferran, S.; Delmas, C.; Favre, G.; Bonnet, J.; Toulas, C.; Moyal, E.C.J. αvβ3/αvβ5 Integrins-FAK-RhoB: A novel pathway for hypoxia regulation in glioblastoma. Cancer Res. 2009, 69, 3308–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanazawa, H.; Imoto, K.; Okada, M.; Yamawaki, H. Canstatin inhibits hypoxia-induced apoptosis through activation of integrin/focal adhesion kinase/Akt signaling pathway in H9c2 cardiomyoblasts. PLoS ONE 2017, 12, e0173051. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Mogford, J.E.; Platts, S.H.; Davis, G.E.; Meininger, G.A.; Davis, M.J. Modulation of calcium current in arteriolar smooth muscle by αvβ3 and α5β1 integrin ligands. J. Cell Biol. 1998, 143, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Lai, N.C.; Kawaraguchi, Y.; Liao, P.; Copps, J.; Sugano, Y.; Okada-Maeda, S.; Banerjee, I.; Schilling, J.M.; Gingras, A.R.; et al. Integrins protect cardiomyocytes from ischemia/reperfusion injury. J. Clin. Investig. 2013, 123, 4294–4308. [Google Scholar] [CrossRef] [Green Version]
- Page, R.L.; Joglar, J.A.; Caldwell, M.A.; Calkins, H.; Conti, J.B.; Deal, B.J.; Estes, N.A.M.; Field, M.E.; Goldberger, Z.D.; Hammill, S.C.; et al. 2015 ACC/AHA/HRS guideline for the management of adult patients with supraventricular tachycardia: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2016, 133, e506–e574. [Google Scholar] [CrossRef]
- Sugiyama, A.; Ito, R.; Okada, M.; Yamawaki, H. Long-term administration of recombinant canstatin prevents adverse cardiac remodeling after myocardial infarction. Sci. Rep. 2020, 10, 12881. [Google Scholar] [CrossRef]
- Yasuda, J.; Okada, M.; Yamawaki, H. Protective effect of T3 peptide, an active fragment of tumstatin, against ischemia/reperfusion injury in rat heart. J. Pharmacol. Sci. 2019, 139, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Mukohda, M.; Morita, T.; Okada, M.; Hara, Y.; Yamawaki, H. Long-term methylglyoxal treatment impairs smooth muscle contractility in organ-cultured rat mesenteric artery. Pharmacol. Res. 2012, 65, 91–99. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugiyama, A.; Shimizu, Y.; Okada, M.; Otani, K.; Yamawaki, H. Preventive Effect of Canstatin against Ventricular Arrhythmia Induced by Ischemia/Reperfusion Injury: A Pilot Study. Int. J. Mol. Sci. 2021, 22, 1004. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031004
Sugiyama A, Shimizu Y, Okada M, Otani K, Yamawaki H. Preventive Effect of Canstatin against Ventricular Arrhythmia Induced by Ischemia/Reperfusion Injury: A Pilot Study. International Journal of Molecular Sciences. 2021; 22(3):1004. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031004
Chicago/Turabian StyleSugiyama, Akira, Yurie Shimizu, Muneyoshi Okada, Kosuke Otani, and Hideyuki Yamawaki. 2021. "Preventive Effect of Canstatin against Ventricular Arrhythmia Induced by Ischemia/Reperfusion Injury: A Pilot Study" International Journal of Molecular Sciences 22, no. 3: 1004. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031004