
Detection of a DNA Methylation Signature for the Intellectual Developmental Disorder, X-Linked, Syndromic, Armfield Type

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Cohorts
2.2. Methylation Data Analysis
2.3. Probe Selection, Dimension Reduction, and Constructing a Supervised Classifier
3. Results
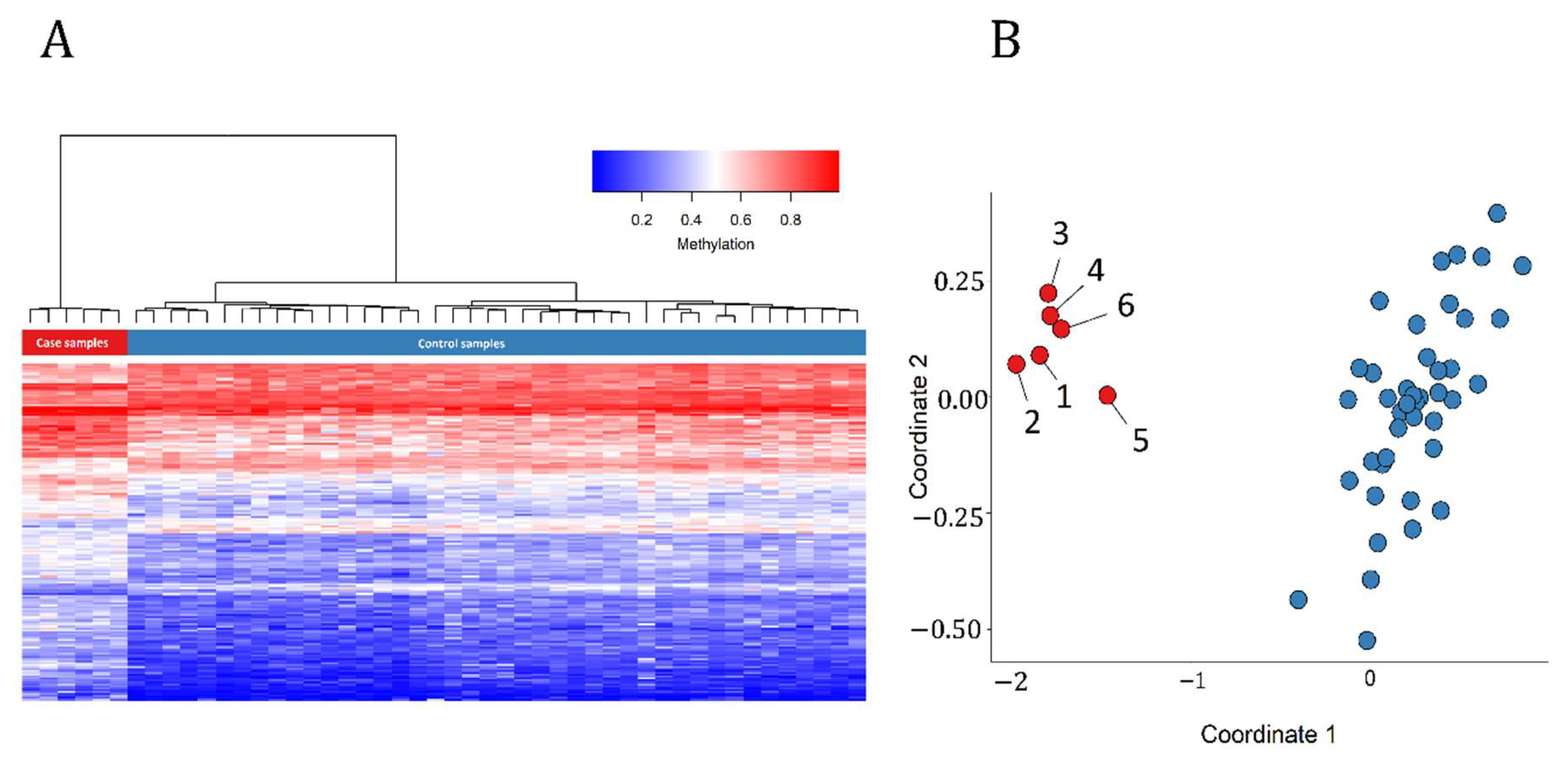
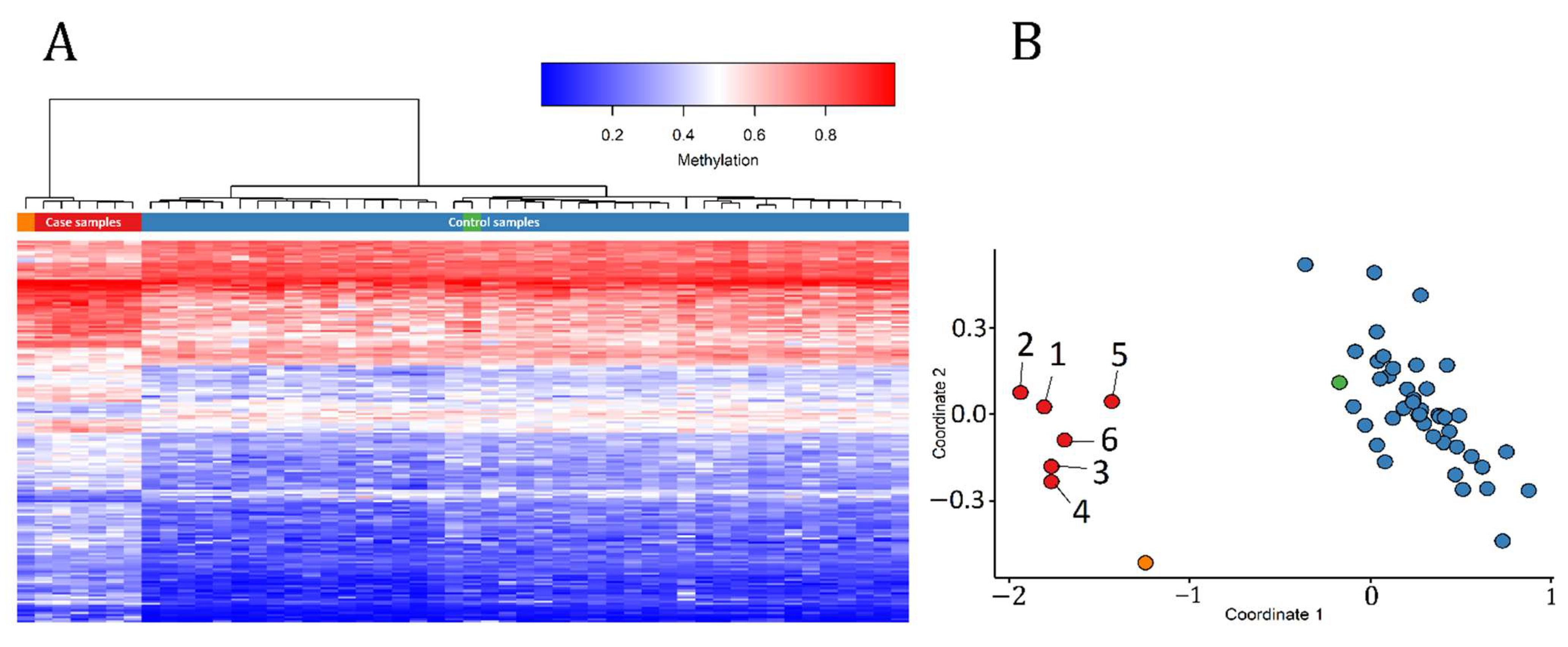
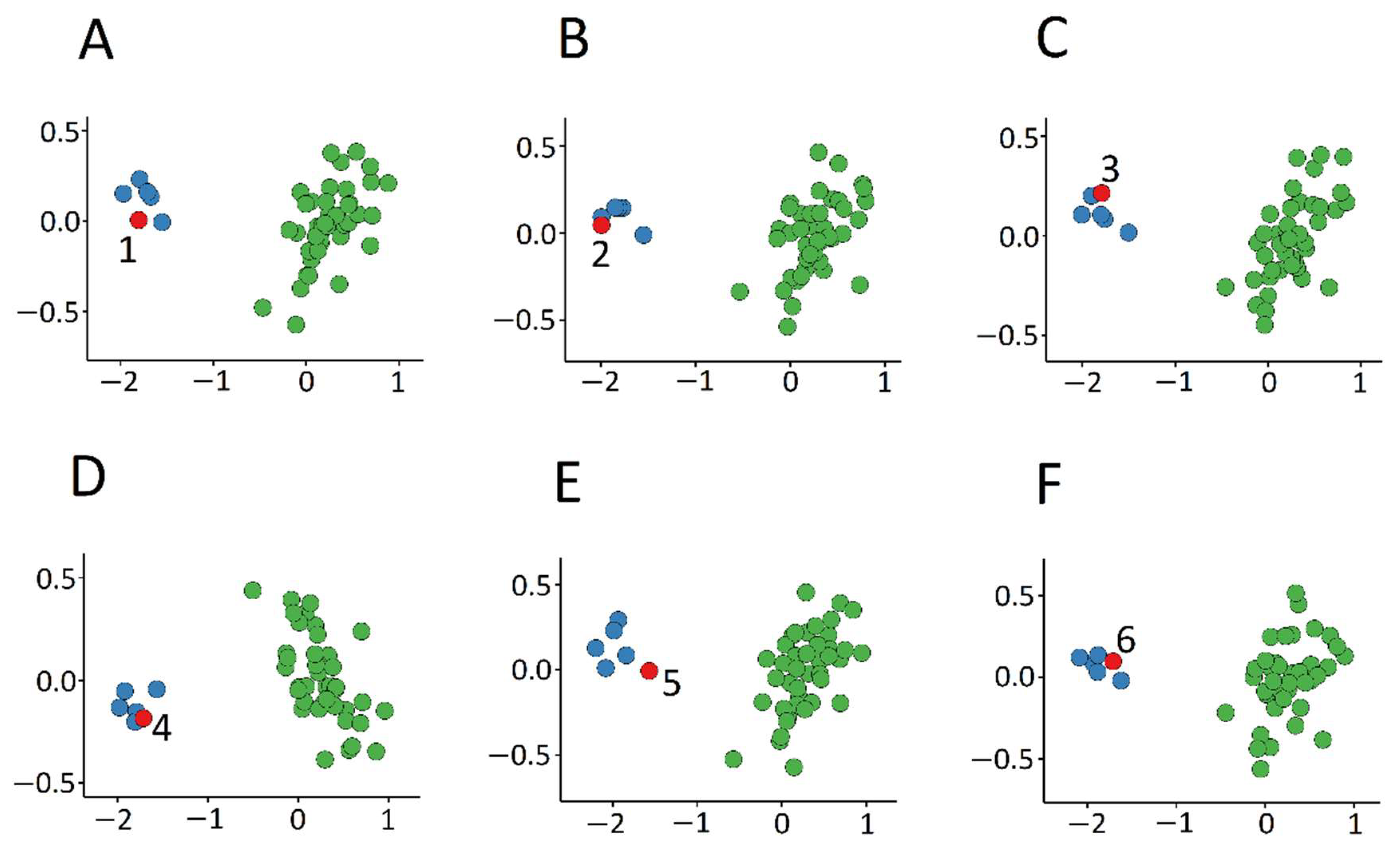
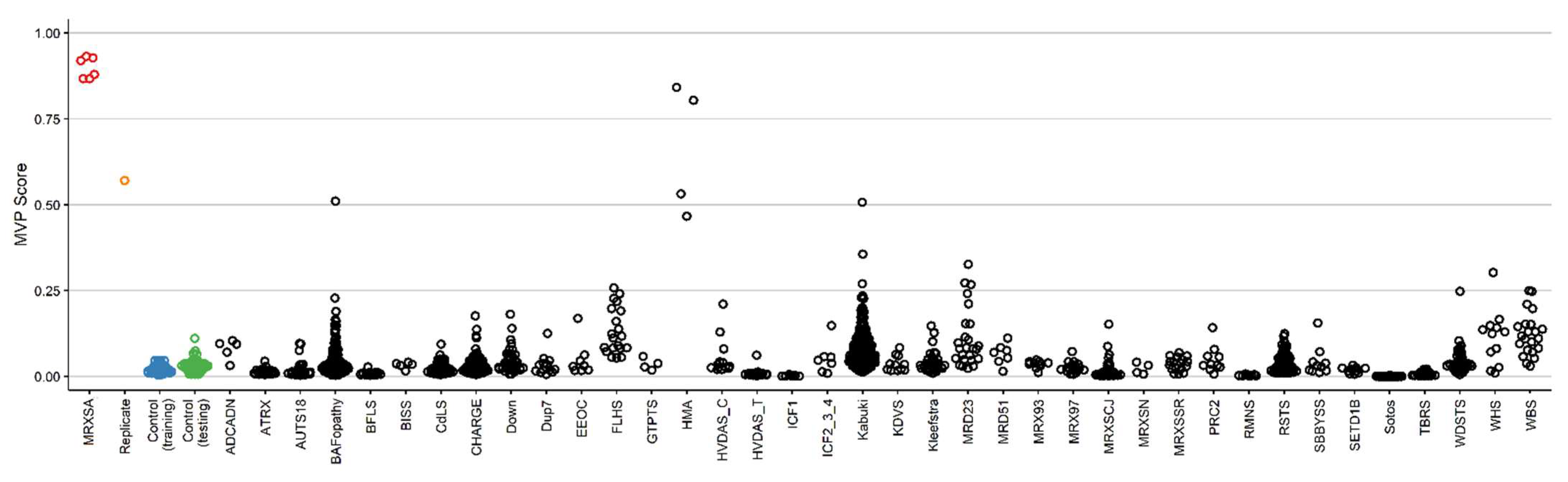
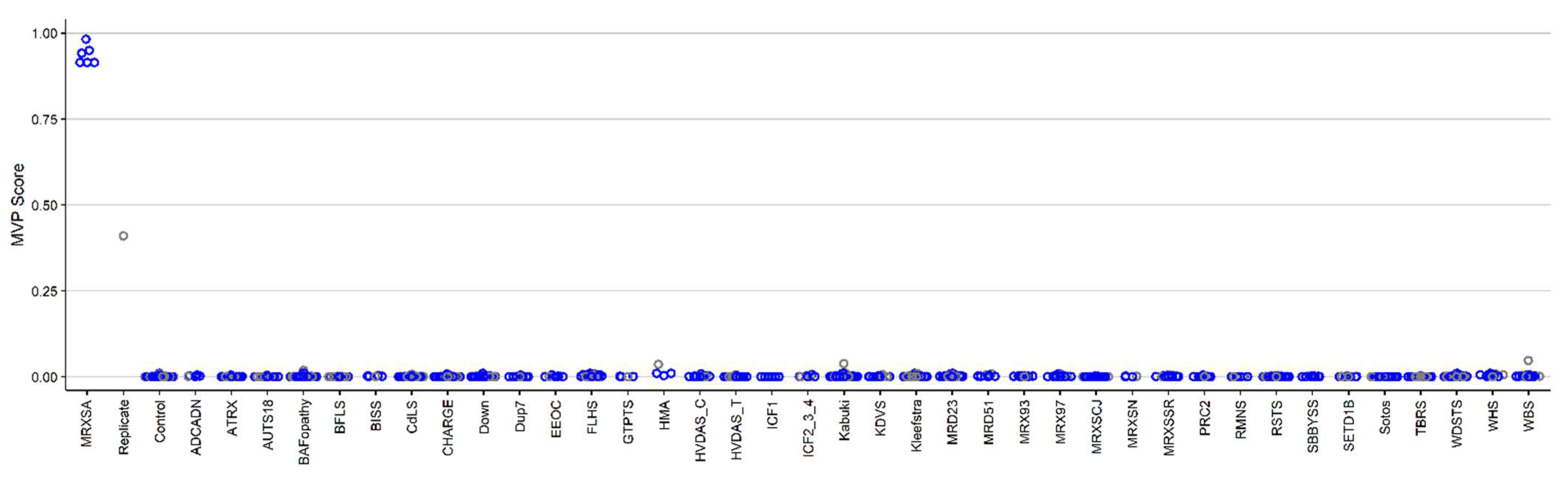
3.1. Detection and Verification of an Episignature for MRXSA
3.3. Identification of the Regions of Differential Methylation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethics
References
- Bland, A.; Harrington, E.A.; Dunn, K.; Pariani, M.; Platt, J.C.K.; Grove, M.E.; Caleshu, C. Clinically impactful differences in variant interpretation between clinicians and testing laboratories: A single-center experience. Genet. Med. 2018, 20, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Christianson, A.; Howson, C.P.; Modell, B. March of Dimes. Global Report on Birth Defect. The Hidden Toll of Dying and Disabled Children; March of Dimes Birth Defects Foundation: Arlington, VI, USA, 2006. [Google Scholar]
- Baird, P.A.; Anderson, T.W.; Newcombe, H.B.; Lowry, R.B. Genetic disorders in children and young adults: A population study. Am. J. Hum. Genet. 1988, 42, 677–693. [Google Scholar] [PubMed]
- Aref-Eshghi, E.; Bend, E.G.; Colaiacovo, S.; Caudle, M.; Chakrabarti, R.; Napier, M.; Brick, L.; Brady, L.; Carere, D.A.; Levy, M.A.; et al. Diagnostic Utility of Genome-wide DNA Methylation Testing in Genetically Unsolved Individuals with Suspected Hereditary Conditions. Am. J. Hum. Genet. 2019, 104, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjornsson, H.T. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015, 25, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadikovic, B.; Levy, M.A.; Aref-Eshghi, E. Functional annotation of genomic variation: DNA methylation episignatures in neurodevelopmental Mendelian disorders. Hum. Mol. Genet. 2020, 29, R27–R32. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Kernohan, K.D.; McBride, A.; Reina, D.; Hodge, A.; Ainsworth, P.J.; Rodenhiser, D.I.; Pare, G.; Bérubé, N.G.; Skinner, C.; et al. Identification of epigenetic signature associated with alpha thalassemia/mental retardation X-linked syndrome. Epigenet. Chromatin 2017, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Aref-Eshghi, E.; Rodenhiser, D.I.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Hood, R.L.; Bulman, D.E.; Kernohan, K.D.; et al. Genomic DNA Methylation Signatures Enable Concurrent Diagnosis and Clinical Genetic Variant Classification in Neurodevelopmental Syndromes. Am. J. Hum. Genet. 2018, 102, 156–174. [Google Scholar] [CrossRef] [Green Version]
- Butcher, D.T.; Cytrynbaum, C.; Turinsky, A.L.; Siu, M.T.; Inbar-Feigenberg, M.; Mendoza-Londono, R.; Chitayat, D.; Walker, S.; Machado, J.; Caluseriu, O.; et al. CHARGE and Kabuki Syndromes: Gene-Specific DNA Methylation Signatures Identify Epigenetic Mechanisms Linking These Clinically Overlapping Conditions. Am. J. Hum. Genet. 2017, 100, 773–788. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Schwartz, C.; Skinner, C.; Rodenhiser, D.I.; Ainsworth, P.J.; Pare, G.; Sadikovic, B. Clinical Validation of Fragile X Syndrome Screening by DNA Methylation Array. J. Mol. Diagn. 2016, 18, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Schenkel, L.C.; Aref-Eshghi, E.; Skinner, C.; Ainsworth, P.; Lin, H.; Paré, G.; Rodenhiser, D.I.; Schwartz, C.; Sadikovic, B. Peripheral blood epi-signature of Claes-Jensen syndrome enables sensitive and specific identification of patients and healthy carriers with pathogenic mutations in KDM5C. Clin. Epigenet. 2018, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, J.A.; Sears, R.L.; Gao, F.; Klein, E.D.; Karydas, A.; Geschwind, M.D.; Rosen, H.J.; Boxer, A.L.; Guo, W.; et al. An Epigenetic Signature in Peripheral Blood Associated with the Haplotype on 17q21.31, a Risk Factor for Neurodegenerative Tauopathy. PLoS Genet. 2014, 10, e1004211. [Google Scholar] [CrossRef] [PubMed]
- Choufani, S.; Cytrynbaum, C.; Chung, B.H.Y.; Turinsky, A.L.; Grafodatskaya, D.; Chen, Y.A.; Cohen, A.S.A.; Dupuis, L.; Butcher, D.T.; Siu, M.T.; et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aref-Eshghi, E.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Siu, V.; Rodenhiser, D.; Schwartz, C.; Sadikovic, B. Clinical Validation of a Genome-Wide DNA Methylation Assay for Molecular Diagnosis of Imprinting Disorders. J. Mol. Diagn. 2017, 19, 848–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, R.L.; Schenkel, L.C.; Nikkel, S.M.; Ainsworth, P.J.; Pare, G.; Boycott, K.M.; Bulman, D.E.; Sadikovic, B. The defining DNA methylation signature of Floating-Harbor Syndrome. Sci. Rep. 2016, 6, 38803. [Google Scholar] [CrossRef]
- Guastafierro, T.; Bacalini, M.G.; Marcoccia, A.; Gentilini, D.; Pisoni, S.; Di Blasio, A.M.; Corsi, A.; Franceschi, C.; Raimondo, D.; Spanò, A.; et al. Genome-wide DNA methylation analysis in blood cells from patients with Werner syndrome. Clin. Epigenet. 2017, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Aref-Eshghi, E.; Bend, E.G.; Hood, R.L.; Schenkel, L.C.; Carere, D.A.; Chakrabarti, R.; Nagamani, S.C.S.; Cheung, S.W.; Campeau, P.M.; Prasad, C.; et al. BAFopathies’ DNA methylation epi-signatures demonstrate diagnostic utility and functional continuum of Coffin–Siris and Nicolaides–Baraitser syndromes. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Bend, E.G.; Aref-Eshghi, E.; Everman, D.B.; Rogers, R.C.; Cathey, S.S.; Prijoles, E.J.; Lyons, M.J.; Davis, H.; Clarkson, K.; Gripp, K.W.; et al. Gene domain-specific DNA methylation episignatures highlight distinct molecular entities of ADNP syndrome. Clin. Epigenet. 2019, 11, 64. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; Dubourg, C.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef]
- Armfield, K.; Nelson, R.; Lubs, H.A.; Häne, B.; Schroer, R.J.; Arena, F.; Schwartz, C.E.; Stevenson, R.E. X-linked mental retardation syndrome with short stature, small hands and feet, seizures, cleft palate, and glaucoma is linked to Xq28. Am. J. Med. Genet. 1999, 85, 236–242. [Google Scholar] [CrossRef]
- Lee, Y.R.; Khan, K.; Armfield-Uhas, K.; Srikanth, S.; Thompson, N.A.; Pardo, M.; Yu, L.; Norris, J.W.; Peng, Y.; Gripp, K.W.; et al. Mutations in FAM50A suggest that Armfield XLID syndrome is a spliceosomopathy. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, D.E.; Imai, K.; King, G.; Stuart, E.A. Matching as nonparametric preprocessing for reducing model dependence in parametric causal inference. Polit. Anal. 2007, 15, 199–236. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, T.J.; Buckley, M.J.; Statham, A.L. De novo identification of differentially methylated regions in the human genome. Epigenet. Chromatin 2015, 8. [Google Scholar] [CrossRef] [Green Version]
- Kernohan, K.D.; Schenkel, L.C.; Huang, L.; Smith, A.; Pare, G.; Ainsworth, P.; Care4Rare Canada Consortium; Boycott, K.M.; Warman-Chardon, J.; Sadikovic, B. Identification of a methylation profile for DNMT1-associated autosomal dominant cerebellar ataxia, deafness, and narcolepsy. Clin. Epigenet. 2016, 8, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Siu, M.T.; Butcher, D.T.; Turinsky, A.L.; Cytrynbaum, C.; Stavropoulos, D.J.; Walker, S.; Caluseriu, O.; Carter, M.; Lou, Y.; Nicolson, R.; et al. Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants. Clin. Epigenet. 2019, 11, 103. [Google Scholar] [CrossRef]
- Cappuccio, G.; Sayou, C.; Le Tanno, P.; Tisserant, E.; Bruel, A.L.; El Kennani, S.; Sá, J.; Low, K.J.; Dias, C.; Havlovicová, M.; et al. De novo SMARCA2 variants clustered outside the helicase domain cause a new recognizable syndrome with intellectual disability and blepharophimosis distinct from Nicolaides–Baraitser syndrome. Genet. Med. 2020, 22, 1838–1850. [Google Scholar] [CrossRef]
- Bacalini, M.G.; Gentilini, D.; Boattini, A.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Scurti, M.; Remondini, D.; Capri, M.; et al. Identification of a DNA methylation signature in blood cells from persons with Down syndrome. Aging 2015, 7, 82–96. [Google Scholar] [CrossRef] [Green Version]
- Strong, E.; Butcher, D.T.; Singhania, R.; Mervis, C.B.; Morris, C.A.; De Carvalho, D.; Weksberg, R.; Osborne, L.R. Symmetrical Dose-Dependent DNA-Methylation Profiles in Children with Deletion or Duplication of 7q11.23. Am. J. Hum. Genet. 2015, 97, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Aref-Eshghi, E.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Rodenhiser, D.; Schwartz, C.; Sadikovic, B. The defining DNA methylation signature of Kabuki syndrome enables functional assessment of genetic variants of unknown clinical significance. Epigenetics 2017, 12, 923–933. [Google Scholar] [CrossRef]
- Ciolfi, A.; Aref-Eshghi, E.; Pizzi, S.; Pedace, L.; Miele, E.; Kerkhof, J.; Flex, E.; Martinelli, S.; Radio, F.C.; Ruivenkamp, C.A.L.; et al. Frameshift mutations at the C-terminus of HIST1H1E result in a specific DNA hypomethylation signature. Clin. Epigenet. 2020, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Krzyzewska, I.M.; Maas, S.M.; Henneman, P.; Lip, K.V.D.; Venema, A.; Baranano, K.; Chassevent, A.; Aref-Eshghi, E.; Van Essen, A.J.; Fukuda, T.; et al. A genome-wide DNA methylation signature for SETD1B-related syndrome. Clin. Epigenet. 2019, 11, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Kim, T.; Long, Q.; Liu, J.; Wang, P.; Zhou, Y.; Ding, Y.; Prasain, J.; Wood, P.A.; Yang, Q. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 2012, 126, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Nishino, I. Megaconial congenital muscular dystrophy due to loss-of-function mutations in choline kinase β. Curr. Opin. Neurol. 2013, 26, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.; Kerkhof, J.; Sadikovic, B. DNA Methylation Signatures in Mendelian Neurodevelopmental Disorders as a Diagnostic Link Between a Genotype and Phenotype. Adv. in Mol. Pathol. 2020, 3, 29–39. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Rodenhiser, D.I.; Ainsworth, P.J.; Paré, G.; Sadikovic, B. DNA methylation analysis in constitutional disorders: Clinical implications of the epigenome. Crit. Rev. Clin. Lab. Sci. 2016, 53, 147–165. [Google Scholar] [CrossRef]
- Martin-Herranz, D.E.; Aref-Eshghi, E.; Bonder, M.J.; Stubbs, T.M.; Stegle, O.; Sadikovic, B.; Reik, W.; Thornton, J.M. Screening for genes that accelerate the epigenetic ageing clock in humans reveals a role for the H3K36 methyltransferase NSD1. Genome Biol. 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Choufani, S.; Gibson, W.T.; Turinsky, A.L.; Chung, B.H.Y.; Wang, T.; Garg, K.; Vitriolo, A.; Cohen, A.S.A.; Cyrus, S.; Goodman, S.; et al. DNA Methylation Signature for EZH2 Functionally Classifies Sequence Variants in Three PRC2 Complex Genes. Am. J. Hum. Genet. 2020, 106, 596–610. [Google Scholar] [CrossRef]
- Jaiswal, S.K.; Kumar, A.; Ali, A.; Rai, A.K. Co-occurrence of mosaic supernumerary isochromosome 18p and intermittent 2q13 deletions in a child with multiple congenital anomalies. Gene 2015, 559, 94–98. [Google Scholar] [CrossRef]
- Powers, N.R.; Parvanov, E.D.; Baker, C.L.; Walker, M.; Petkov, P.M.; Paigen, K. The Meiotic Recombination Activator PRDM9 Trimethylates Both H3K36 and H3K4 at Recombination Hotspots In Vivo. PLoS Genet. 2016, 12, e1006146. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Xue, X.; Yan, Y.; Li, G. Highlight article: Dysfunctional Cav1.2 channel in Timothy syndrome, from cell to bedside. Exp. Biol. Med. 2019, 244, 960–971. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Kindred | Age | FAM50A Variant | Clinical Features |
|---|---|---|---|---|
| 1 | 8100 | 62 | c.764A>G; p.Asp255Gly inherited | Global developmental delay (GDD), glaucoma, cataracts, short stature, speech problems, and craniofacial anomalies |
| 2 | 8100 | 50 | c.764A>G; p.Asp255Gly inherited | Short stature, dysmorphic facial features, and a left inguinal hernia |
| 3 | 8100 | 45 | c.764A>G; p.Asp255Gly inherited | GDD, speech problems, seizures, short stature, craniofacial anomalies, glaucoma, and small hands and feet |
| 4 | 8100 | 28 | c.764A>G; p.Asp255Gly inherited | GDD, dysmorphic facial features, strabismus, and small feet |
| 5 | 9656 | 10 | c.761A>G; p.Glu254Gly de novo | GDD, strabismus, short stature, and dysmorphic facial features |
| 6 | 9677 | 26 | c.763G>A; p.Asp255Asn de novo | GDD, dysmorphic facial features, and exotropia |
| Syndrome | Syndrome Abbreviation | Underlying Gene/Location | Phenotype MIM Number | Signature Published |
|---|---|---|---|---|
| Cerebellar ataxia, deafness, and narcolepsy, autosomal dominant | ADCADN | DNMT1 | 604121 | Yes [4,8,19,27] |
| Alpha-thalassemia mental retardation syndrome | ATRX | ATRX | 301040 | Yes [4,7,8,19] |
| Autism, susceptibility to, 18 | AUTS18 | CHD8 | 615032 | Yes [19,28] |
| BAFopathies: Coffin–Siris 1–4 (CSS1–4) and Nicolaides-Baraitser (NCBRS) syndromes | BAFopathy | ARID1A, ARID1B, SMARCB1, SMARCA4, SMARCA2 | 614607, 135900, 614609, 614608, 601358 | Yes [4,17,19] |
| Börjeson-Forssman-Lehmann syndrome | BFLS | PHF6 | 301900 | Yes [19] |
| Blepharophimosis intellectual disability syndrome | BIS | SMARCA2 | NA | Yes [29] |
| Cornelia de Lange syndrome 1–4 | CdLS | NIPBL, RAD21, SMC3, SMC1A | 122470, 614701, 610759, 300590 | Yes [4,19] |
| CHARGE syndrome | CHARGE | CHD7 | 214800 | Yes [4,8,9,19] |
| Down syndrome | Down | Chr21 trisomy | 190685 | Yes [4,19,30] |
| Chr7q11.23 duplication syndrome | Dup7 | Chr7q11.23 Duplication | 609757 | Yes [4,19,31] |
| Epileptic encephalopathy, childhood-onset | EEOC | CHD2 | 615369 | Yes [19] |
| Floating-Harbor syndrome | FLHS | SRCAP | 136140 | Yes [4,8,15,19] |
| Genitopatellar syndrome | GTPTS | KAT6B | 606170 | Yes [4,8,19] |
| Hunter McAlpine craniosynostosis syndrome | HMA | Chr5q35-qter duplication involving NSD1 | 601379 | Yes [19] |
| Helsmoortel-van der Aa syndrome (ADNP syndrome [Central]) | HVDAS_C | ADNP (c.2000-2340) | 615873 | Yes [4,19] |
| Helsmoortel-van der Aa syndrome (ADNP syndrome [Terminal]) | HVDAS_T | ADNP (outside c.2000-2340) | 615873 | Yes [4,19] |
| Immunodeficiency-centromeric instability-facial anomalies syndrome 1 | ICF1 | DNMT3B | 242860 | Yes [19] |
| Immunodeficiency-centromeric instability-facial anomalies syndrome 2–4 | ICF2-4 | CDCA7, ZBTB24, HELLS | 614069, 616910, 616911 | Yes [19] |
| Kabuki syndrome 1 and 2 | Kabuki | KMT2D, KDM6A | 147920, 300867 | Yes [4,8,9,19,32] |
| Koolen de Vries syndrome | KDVS | KANSL1 | 610443 | Yes [19] |
| Kleefstra syndrome 1 | Kleefstra1 | EHMT1 | 610253 | Yes [19] |
| Mental retardation, autosomal dominant 23 | MRD23 | SETD5 | 615761 | No |
| Mental retardation, autosomal dominant 51 | MRD51 | KMT5B | 617788 | Yes [19] |
| Mental retardation, X-linked 93 | MRX93 | BRWD3 | 300659 | Yes [19] |
| Mental retardation, X-linked 97 | MRX97 | ZNF711 | 300803 | Yes [19] |
| Mental retardation, X-linked, syndromic, Claes-Jensen type | MRXSCJ | KDM5C | 300534 | Yes [4,8,11,19] |
| Mental retardation, X-linked syndromic, Nascimento-type | MRXSN | UBE2A | 300860 | Yes [19] |
| Mental retardation, X-linked, Snyder- Robinson type | MRXSSR | SMS | 309583 | Yes [19] |
| PRC2: Cohen-Gibson syndrome (COGIS) and Weaver syndrome (WVS) | PRC2 | EED, EZH2 | 617561, 277590 | No |
| Rahman syndrome | RMNS | HIST1H1E | 617537 | Yes [19,33] |
| Rubinstein-Taybi syndrome 1 and 2 | RSTS | CREBBP, EP300 | 180849, 613684 | Yes [19] |
| Ohdo syndrome, SBBYS variant | SBBYSS | KAT6B | 603736 | Yes [8,19] |
| SETD1B-related syndrome | SETD1B | SETD1B | N/A | Yes [34] |
| Sotos syndrome | Sotos | NSD1 | 117550 | Yes [4,8,13,19] |
| Tatton-Brown-Rahman syndrome | TBRS | DNMT3A | 615879 | Yes [19] |
| Wiedemann-Steiner syndrome | WDSTS | KMT2A | 605130 | Yes [19] |
| Williams-Beuren syndrome | WBS | Chr7q11.23 deletion | 194050 | Yes [4,19,31] |
| Wolf-Hirschhorn syndrome | WHS | Chr4p16.3 deletion | 194190 | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haghshenas, S.; Levy, M.A.; Kerkhof, J.; Aref-Eshghi, E.; McConkey, H.; Balci, T.; Siu, V.M.; Skinner, C.D.; Stevenson, R.E.; Sadikovic, B.; et al. Detection of a DNA Methylation Signature for the Intellectual Developmental Disorder, X-Linked, Syndromic, Armfield Type. Int. J. Mol. Sci. 2021, 22, 1111. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031111
Haghshenas S, Levy MA, Kerkhof J, Aref-Eshghi E, McConkey H, Balci T, Siu VM, Skinner CD, Stevenson RE, Sadikovic B, et al. Detection of a DNA Methylation Signature for the Intellectual Developmental Disorder, X-Linked, Syndromic, Armfield Type. International Journal of Molecular Sciences. 2021; 22(3):1111. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031111
Chicago/Turabian StyleHaghshenas, Sadegheh, Michael A. Levy, Jennifer Kerkhof, Erfan Aref-Eshghi, Haley McConkey, Tugce Balci, Victoria Mok Siu, Cindy D. Skinner, Roger E. Stevenson, Bekim Sadikovic, and et al. 2021. "Detection of a DNA Methylation Signature for the Intellectual Developmental Disorder, X-Linked, Syndromic, Armfield Type" International Journal of Molecular Sciences 22, no. 3: 1111. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031111