
Cardiomyocytes Derived from Induced Pluripotent Stem Cells as a Disease Model for Propionic Acidemia

 , and
, and
Abstract
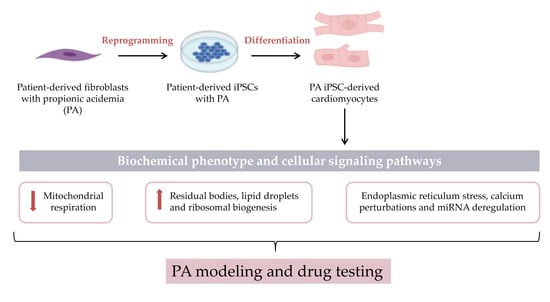
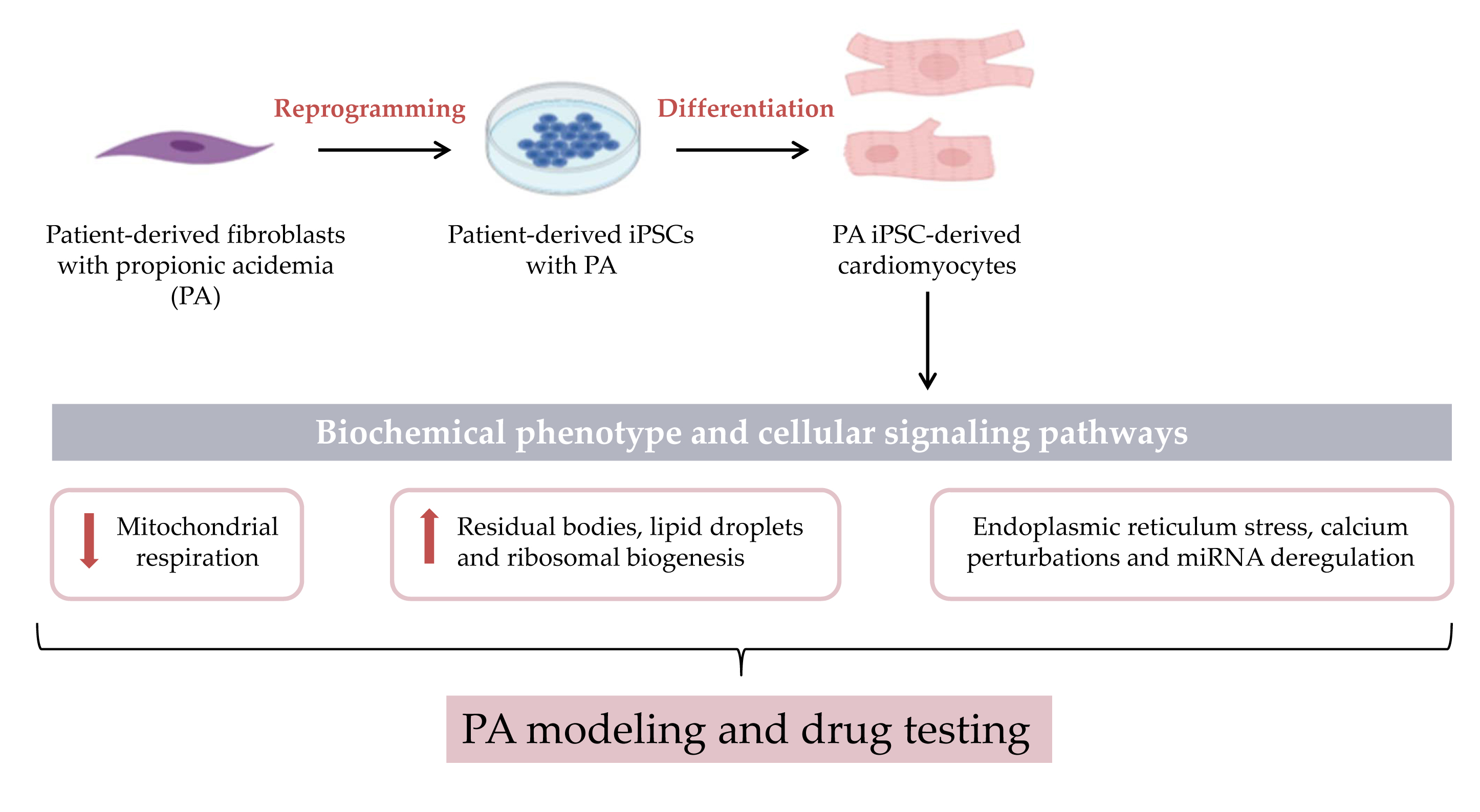
:
{kind=link}
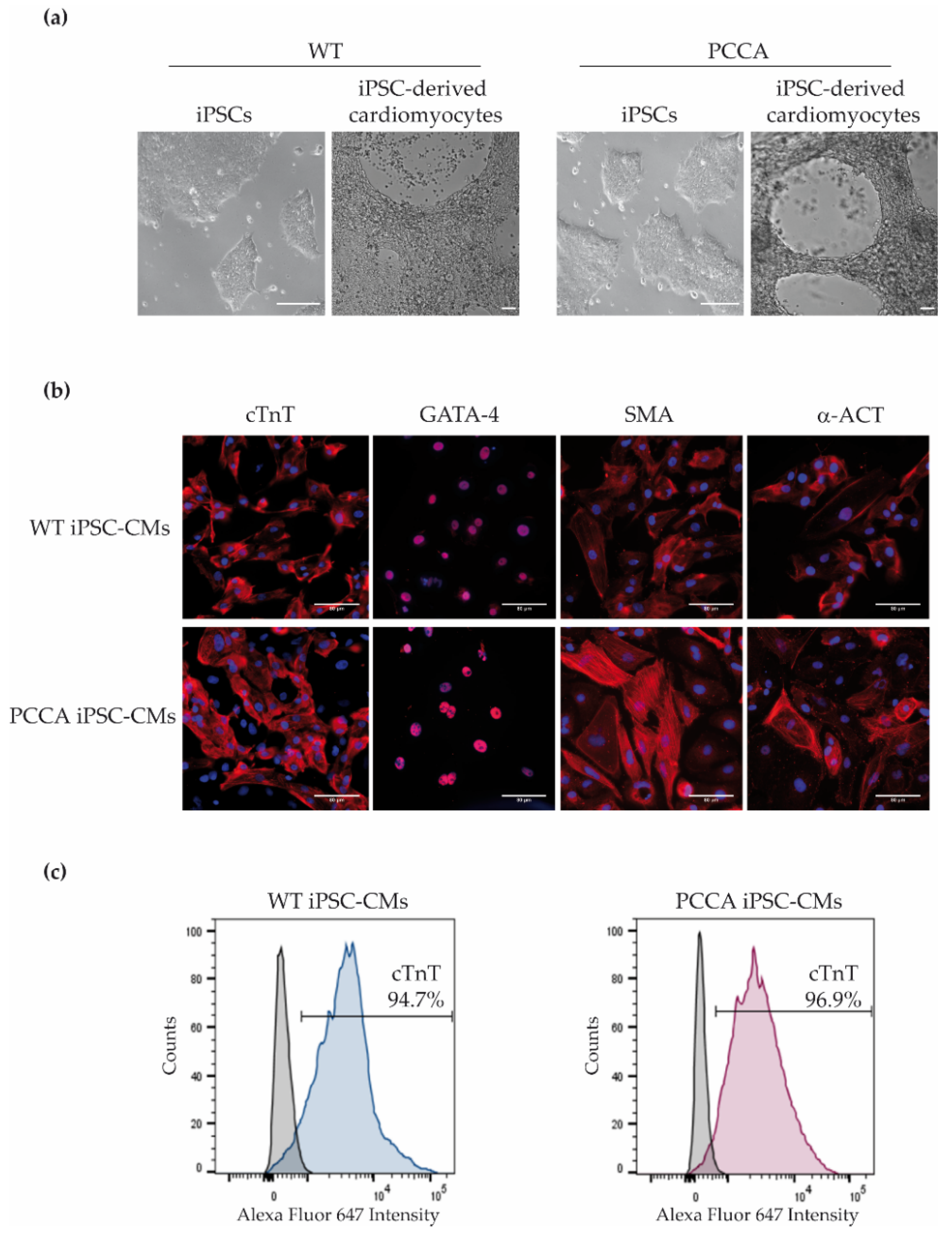{kind=link}
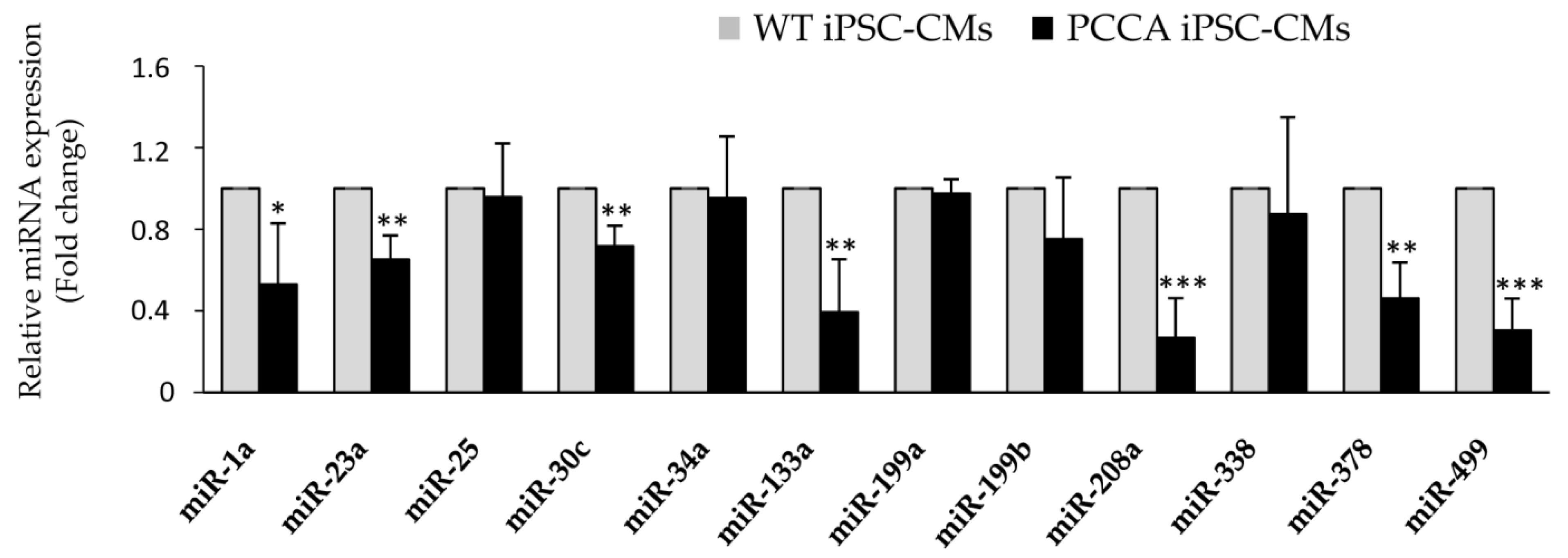{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
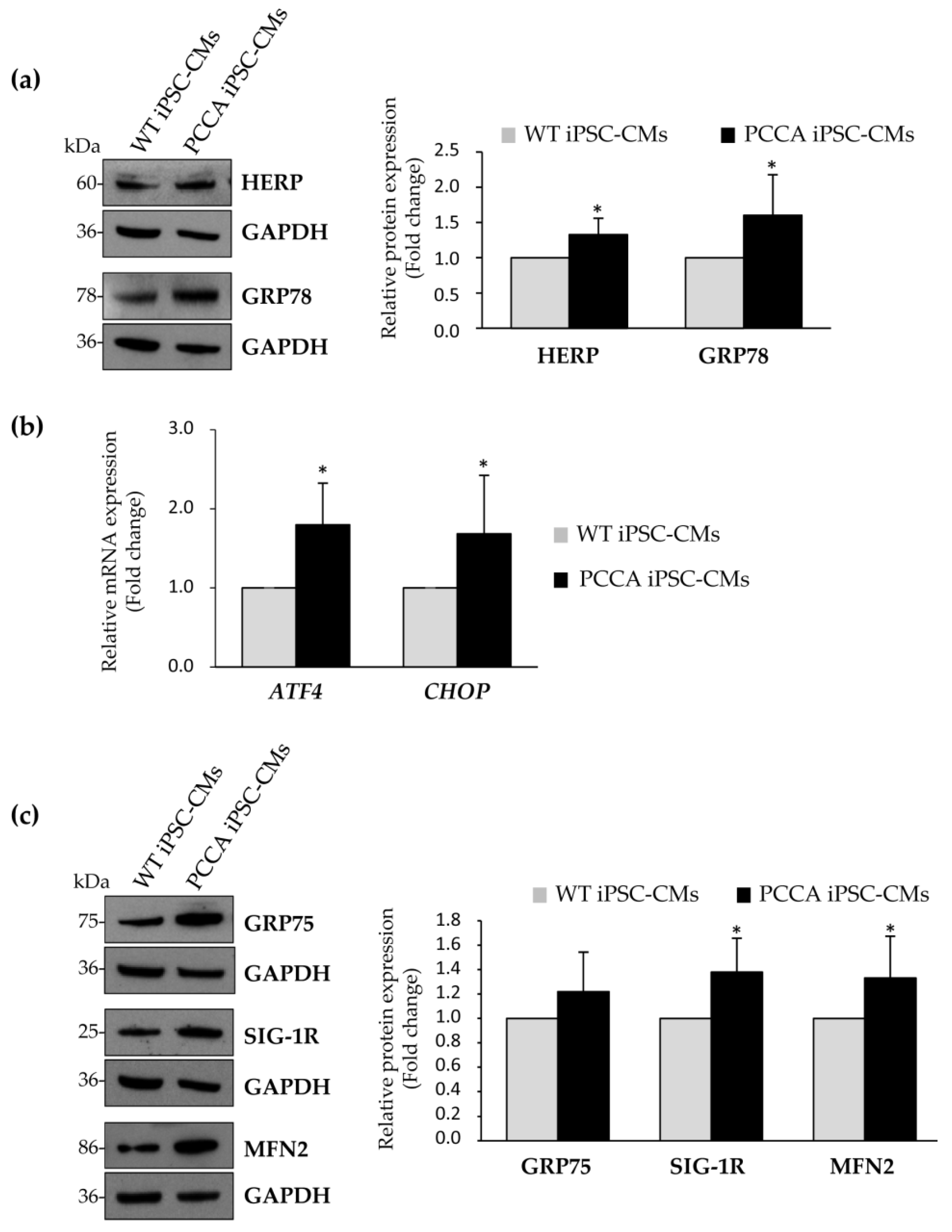
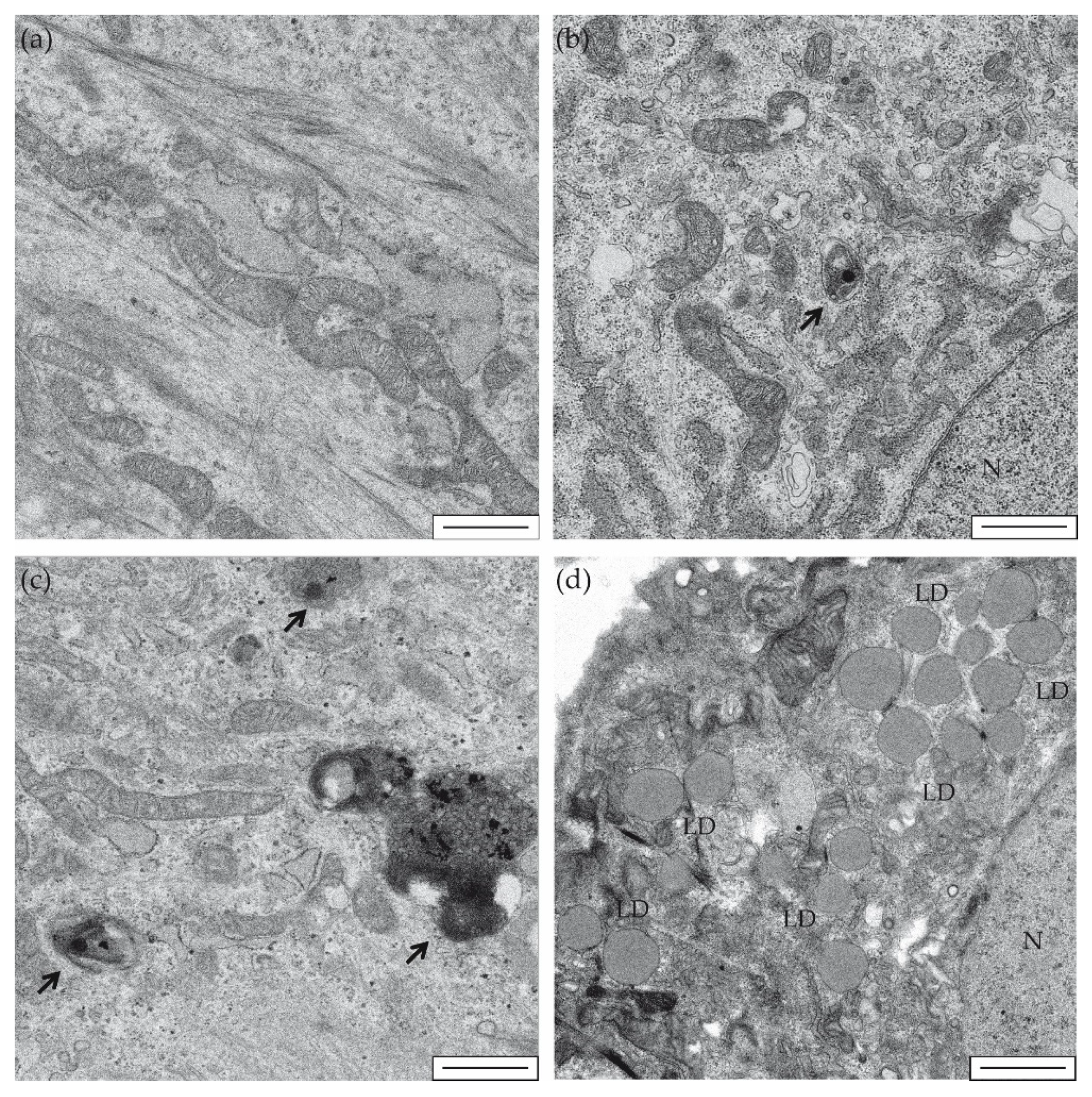
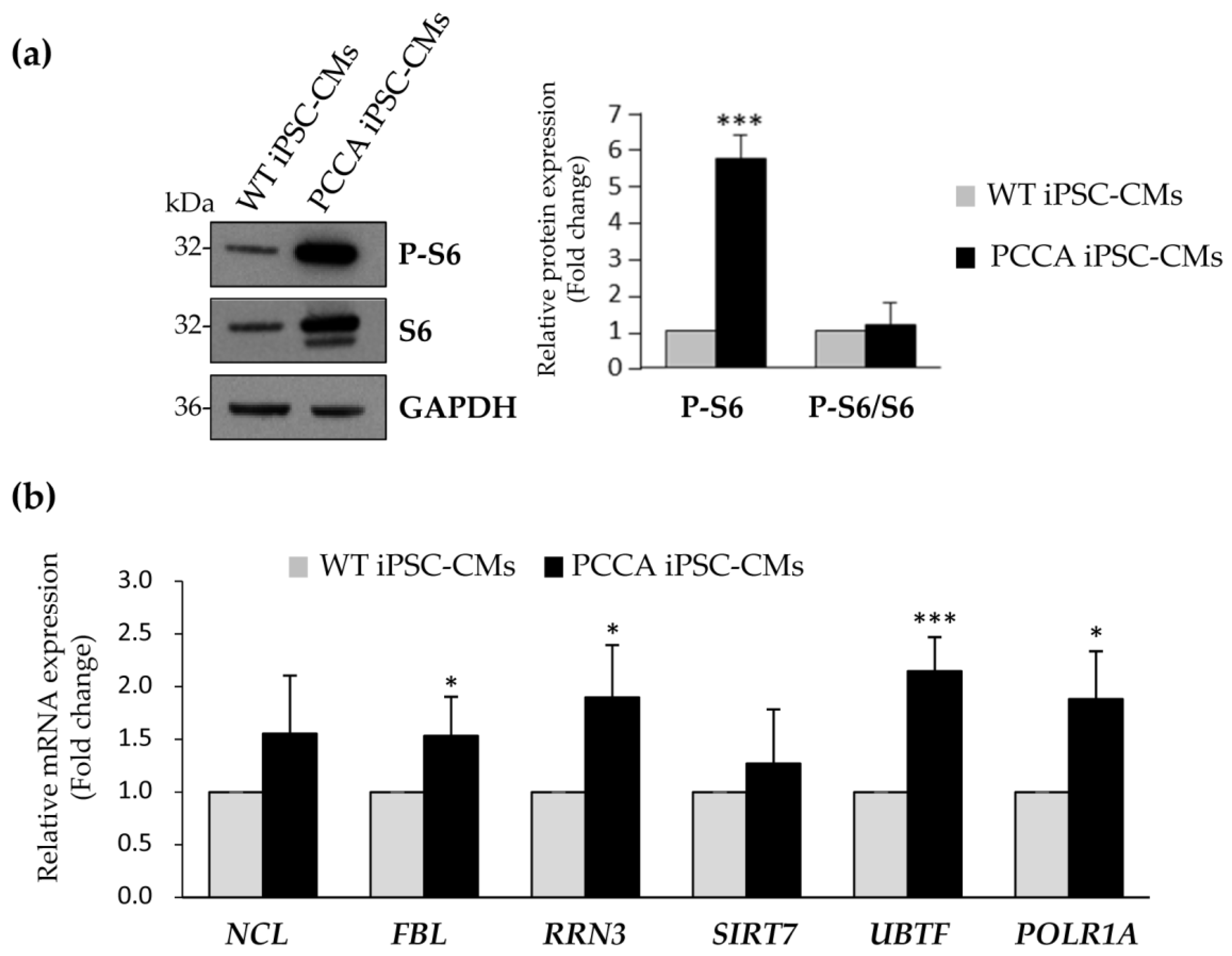
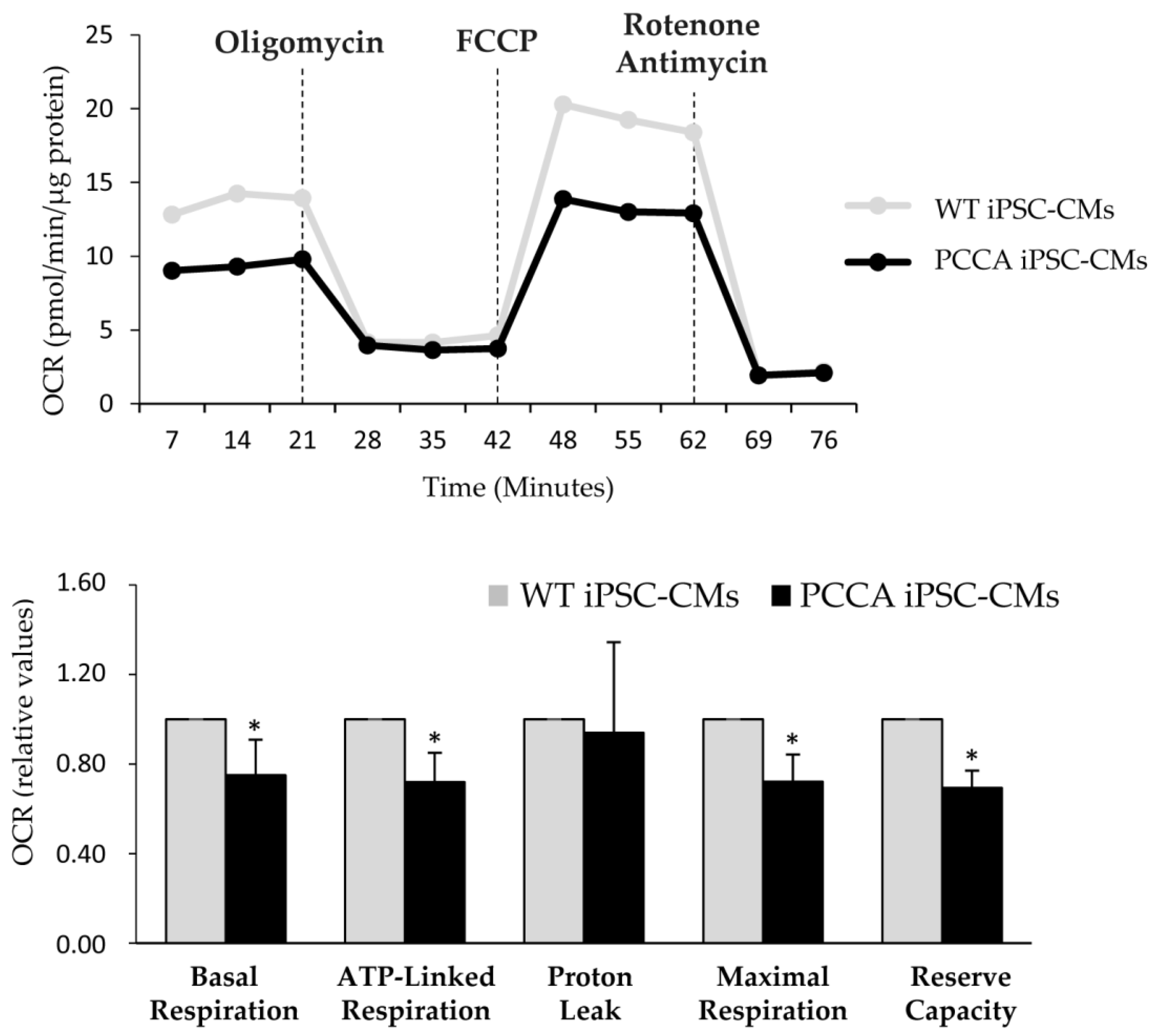
2. Results and Discussion
3. Materials and Methods
3.1. Cell Lines
3.2. Cardiomyocyte Differentiation
3.3. Immunostaining
3.4. Flow Cytometry
3.5. mRNA and miRNA Analysis
3.6. Immunoblotting
3.7. Electron Microscopy
3.8. Extracellular Flux Assay
3.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-ACT | α-Actinin |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| cTnT | Cardiac troponin T |
| ER | Endoplasmic reticulum |
| FBL | rRNA methyltransferase fibrillarin |
| GRP75 | 75-kDa Glucose regulated protein |
| GRP78 | 78-kDa Glucose-regulated protein |
| HERP | Homocysteine-inducible ER stress protein |
| iPSCs | Induced pluripotent stem cells |
| iPSC-CMs | iPSC-derived cardiomyocytes |
| MAMs | Mitochondria-associated membranes |
| MFN2 | Mitofusin-2 |
| NCL | Nucleolin |
| OA | Organic acidemia/aciduria |
| OCR | Oxygen consumption rate |
| PA | Propionic acidemia |
| PBS | Phosphate buffered saline |
| PCC | Propionyl-CoA carboxylase |
| POLR1A | Large subunit of Pol I |
| PPAR | Peroxisome proliferator activated receptors |
| qRT-PCR | Real-time quantitative reverse transcription- polymerase chain reaction |
| RRN3 | pol I-pecific transcription initiation factor |
| SIG-1R | Sigma-1 receptor |
| SIRT7 | Pol I-activating NAD-dependent histone deacetylase sirtuin 7 |
| SMA | Alpha-smooth muscle actin |
| UBTF | Ribosomal RNA upstream binding transcription factor |
| UPR | Unfolded protein response |
| WT | Wild-type |
References
- Fenton, W.A.; Gravel, R.A.; Rosenberg, L.E. Disorders of propionate and methylmalonate metabolism. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2165–2190. [Google Scholar]
- Baumgartner, D.; Scholl-Burgi, S.; Sass, J.O.; Sperl, W.; Schweigmann, U.; Stein, J.I.; Karall, D. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J. Pediatr. 2007, 150, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Pena, L.; Franks, J.; Chapman, K.A.; Gropman, A.; Mew, N.A.; Chakrapani, A.; Island, E.; MacLeod, E.; Matern, D.; Smith, B.; et al. Natural history of propionic acidemia. Mol. Genet. Metab. 2012, 105, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Valayannopoulos, V.; Touati, G.; Jais, J.P.; Rabier, D.; de Keyzer, Y.; Bonnet, D.; de Lonlay, P. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J. Pediatr. 2010, 156, 128–134. [Google Scholar] [CrossRef]
- Guenzel, A.J.; Hofherr, S.E.; Hillestad, M.; Barry, M.; Weaver, E.; Venezia, S.; Kraus, J.P.; Matern, D.; Barry, M.A. Generation of a hypomorphic model of propionic acidemia amenable to gene therapy testing. Mol. Ther. 2013, 21, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego-Villar, L.; Rivera-Barahona, A.; Cuevas-Martin, C.; Guenzel, A.; Perez, B.; Barry, M.A.; Murphy, M.P.; Logan, A.; Gonzalez-Quintana, A.; Martin, M.A.; et al. In vivo evidence of mitochondrial dysfunction and altered redox homeostasis in a genetic mouse model of propionic acidemia: Implications for the pathophysiology of this disorder. Free Radic. Biol. Med. 2016, 96, 1–12. [Google Scholar] [CrossRef]
- Tamayo, M.; Fulgencio-Covian, A.; Navarro-Garcia, J.A.; Val-Blasco, A.; Ruiz-Hurtado, G.; Gil-Fernandez, M.; Martin-Nunes, L.; Lopez, J.A.; Desviat, L.R.; Delgado, C.; et al. Intracellular calcium mishandling leads to cardiac dysfunction and ventricular arrhythmias in a mouse model of propionic acidemia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165586. [Google Scholar] [CrossRef]
- Fulgencio-Covian, A.; Alonso-Barroso, E.; Guenzel, A.J.; Rivera-Barahona, A.; Ugarte, M.; Perez, B.; Barry, M.A.; Perez-Cerda, C.; Richard, E.; Desviat, L.R. Pathogenic implications of dysregulated miRNAs in propionic acidemia related cardiomyopathy. Transl. Res. 2020, 218, 43–56. [Google Scholar] [CrossRef]
- Ulmer, B.M.; Eschenhagen, T. Human pluripotent stem cell-derived cardiomyocytes for studying energy metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118471. [Google Scholar] [CrossRef]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef]
- Alonso-Barroso, E.; Brasil, S.; Briso-Montiano, A.; Navarrete, R.; Perez-Cerda, C.; Ugarte, M.; Perez, B.; Desviat, L.R.; Richard, E. Generation and characterization of a human iPSC line from a patient with propionic acidemia due to defects in the PCCA gene. Stem Cell Res. 2017, 23, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Barahona, A.; Fulgencio-Covian, A.; Perez-Cerda, C.; Ramos, R.; Barry, M.A.; Ugarte, M.; Perez, B.; Richard, E.; Desviat, L.R. Dysregulated miRNAs and their pathogenic implications for the neurometabolic disease propionic acidemia. Sci. Rep. 2017, 7, 5727. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; Sedmera, D. Developmental anatomy of the heart: A tale of mice and man. Physiol. Genom. 2003, 15, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, R. Heart failure and mouse models. Dis. Model. Mech. 2010, 3, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, S.; Takahashi, K. [Induction of pluripotent stem cells from mouse fibroblast cultures]. Tanpakushitsu Kakusan Koso 2006, 51, 2346–2351. [Google Scholar]
- Jimenez-Tellez, N.; Greenway, S.C. Cellular models for human cardiomyopathy: What is the best option? World J. Cardiol. 2019, 11, 221–235. [Google Scholar] [CrossRef]
- Mardach, R.; Verity, M.A.; Cederbaum, S.D. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol. Genet. Metab. 2005, 85, 286–290. [Google Scholar] [CrossRef]
- Lee, T.M.; Addonizio, L.J.; Barshop, B.A.; Chung, W.K. Unusual presentation of propionic acidaemia as isolated cardiomyopathy. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), S97–S101. [Google Scholar] [CrossRef] [Green Version]
- Park, K.C.; Krywawych, S.; Richard, E.; Desviat, L.R.; Swietach, P. Cardiac complications of propionic and other inherited organic acidemias. Front. Cardiovasc. Med. 2020, 7, 1–20. [Google Scholar] [CrossRef]
- Raut, S.K.; Singh, G.B.; Rastogi, B.; Saikia, U.N.; Mittal, A.; Dogra, N.; Singh, S.; Prasad, R.; Khullar, M. miR-30c and miR-181a synergistically modulate p53–p21 pathway in diabetes induced cardiac hypertrophy. Mol. Cell. Biochem. 2016, 417, 191–203. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kaminska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 2017, 26, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Chen, Y.; He, R.; Shi, Y.; Su, L. Rescuing infusion of miRNA-1 prevents cardiac remodeling in a heart-selective miRNA deficient mouse. Biochem. Biophys. Res. Commun. 2018, 495, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Kambis, T.N.; Shahshahan, H.R.; Kar, S.; Yadav, S.K.; Mishra, P.K. Transgenic Expression of miR-133a in the Diabetic Akita Heart Prevents Cardiac Remodeling and Cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Breschi, A.; Gingeras, T.R.; Guigo, R. Comparative transcriptomics in human and mouse. Nat. Rev. Genet. 2017, 18, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Q.; Wei, C.; Zhao, L.; Guo, X.; Cui, X.; Shao, L.; Long, J.; Gu, J.; Zhao, M. MiR-378 modulates energy imbalance and apoptosis of mitochondria induced by doxorubicin. Am. J. Transl. Res. 2018, 10, 3600–3609. [Google Scholar] [PubMed]
- Wu, S.; Zou, M.H. Mitochondria-associated endoplasmic reticulum membranes in the heart. Arch. Biochem. Biophys. 2019, 662, 201–212. [Google Scholar] [CrossRef]
- McMahon, M.; Samali, A.; Chevet, E. Regulation of the unfolded protein response by noncoding RNA. Am. J. Physiol. Cell Physiol. 2017, 313, C243–C254. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Yan, Z.; Zhu, Z. Mitochondria-Associated Endoplasmic Reticulum Membranes in Cardiovascular Diseases. Front. Cell Dev. Biol. 2020, 8, 604240. [Google Scholar] [CrossRef]
- West, G.; Gullmets, J.; Virtanen, L.; Li, S.P.; Keinanen, A.; Shimi, T.; Mauermann, M.; Helio, T.; Kaartinen, M.; Ollila, L.; et al. Deleterious assembly of the lamin A/C mutant p.S143P causes ER stress in familial dilated cardiomyopathy. J. Cell Sci. 2016, 129, 2732–2743. [Google Scholar] [CrossRef] [Green Version]
- Schwab, M.A.; Sauer, S.W.; Okun, J.G.; Nijtmans, L.G.; Rodenburg, R.J.; van den Heuvel, L.P.; Drose, S.; Brandt, U.; Hoffmann, G.F.; Ter Laak, H.; et al. Secondary mitochondrial dysfunction in propionic aciduria: A pathogenic role for endogenous mitochondrial toxins. Biochem. J. 2006, 398, 107–112. [Google Scholar] [CrossRef]
- Miyazaki, T.; Ohura, T.; Kobayashi, M.; Shigematsu, Y.; Yamaguchi, S.; Suzuki, Y.; Hata, I.; Aoki, Y.; Yang, X.; Minjares, C.; et al. Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene. J. Biol. Chem. 2001, 276, 35995–35999. [Google Scholar] [CrossRef] [Green Version]
- Simpson, L.J.; Reader, J.S.; Tzima, E. Mechanical Regulation of Protein Translation in the Cardiovascular System. Front. Cell Dev. Biol. 2020, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.A.; Shanware, N.P.; Chang, H.; Tibbetts, R.S. Regulation of ribosomal protein S6 phosphorylation by casein kinase 1 and protein phosphatase 1. J. Biol. Chem. 2011, 286, 8688–8696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Xia, Y.; Zhang, F.; Xiong, Z.; Li, Y.; Yan, W.; Chen, X.; Wang, W.; Wang, H.; Gao, E.; et al. Nucleostemin dysregulation contributes to ischemic vulnerability of diabetic hearts: Role of ribosomal biogenesis. J. Mol. Cell. Cardiol. 2017, 108, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Chaillou, T.; Kirby, T.J.; McCarthy, J.J. Ribosome biogenesis: Emerging evidence for a central role in the regulation of skeletal muscle mass. J. Cell. Physiol. 2014, 229, 1584–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razeghi, P.; Buksinska-Lisik, M.; Palanichamy, N.; Stepkowski, S.; Frazier, O.H.; Taegtmeyer, H. Transcriptional regulators of ribosomal biogenesis are increased in the unloaded heart. FASEB J. 2006, 20, 1090–1096. [Google Scholar] [CrossRef]
- de Keyzer, Y.; Valayannopoulos, V.; Benoist, J.F.; Batteux, F.; Lacaille, F.; Hubert, L.; Chretien, D.; Chadefeaux-Vekemans, B.; Niaudet, P.; Touati, G.; et al. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr. Res. 2009, 66, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Fragaki, K.; Cano, A.; Benoist, J.F.; Rigal, O.; Chaussenot, A.; Rouzier, C.; Bannwarth, S.; Caruba, C.; Chabrol, B.; Paquis-Flucklinger, V. Fatal heart failure associated with CoQ10 and multiple OXPHOS deficiency in a child with propionic acidemia. Mitochondrion 2011, 11, 533–536. [Google Scholar] [CrossRef]
- Mc Guire, P.J.; Parikh, A.; Diaz, G.A. Profiling of oxidative stress in patients with inborn errors of metabolism. Mol. Genet. Metab. 2009, 98, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Villar, L.; Perez, B.; Ugarte, M.; Desviat, L.R.; Richard, E. Antioxidants successfully reduce ROS production in propionic acidemia fibroblasts. Biochem. Biophys. Res. Commun. 2014, 452, 457–461. [Google Scholar] [CrossRef]
- Rivera-Barahona, A.; Alonso-Barroso, E.; Perez, B.; Murphy, M.P.; Richard, E.; Desviat, L.R. Treatment with antioxidants ameliorates oxidative damage in a mouse model of propionic acidemia. Mol. Genet. Metab. 2017, 122, 43–50. [Google Scholar] [CrossRef]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Barroso, E.; Pérez, B.; Desviat, L.R.; Richard, E. Cardiomyocytes Derived from Induced Pluripotent Stem Cells as a Disease Model for Propionic Acidemia. Int. J. Mol. Sci. 2021, 22, 1161. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031161
Alonso-Barroso E, Pérez B, Desviat LR, Richard E. Cardiomyocytes Derived from Induced Pluripotent Stem Cells as a Disease Model for Propionic Acidemia. International Journal of Molecular Sciences. 2021; 22(3):1161. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031161
Chicago/Turabian StyleAlonso-Barroso, Esmeralda, Belén Pérez, Lourdes Ruiz Desviat, and Eva Richard. 2021. "Cardiomyocytes Derived from Induced Pluripotent Stem Cells as a Disease Model for Propionic Acidemia" International Journal of Molecular Sciences 22, no. 3: 1161. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031161