Human Plasma and Recombinant Hemopexins: Heme Binding Revisited

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
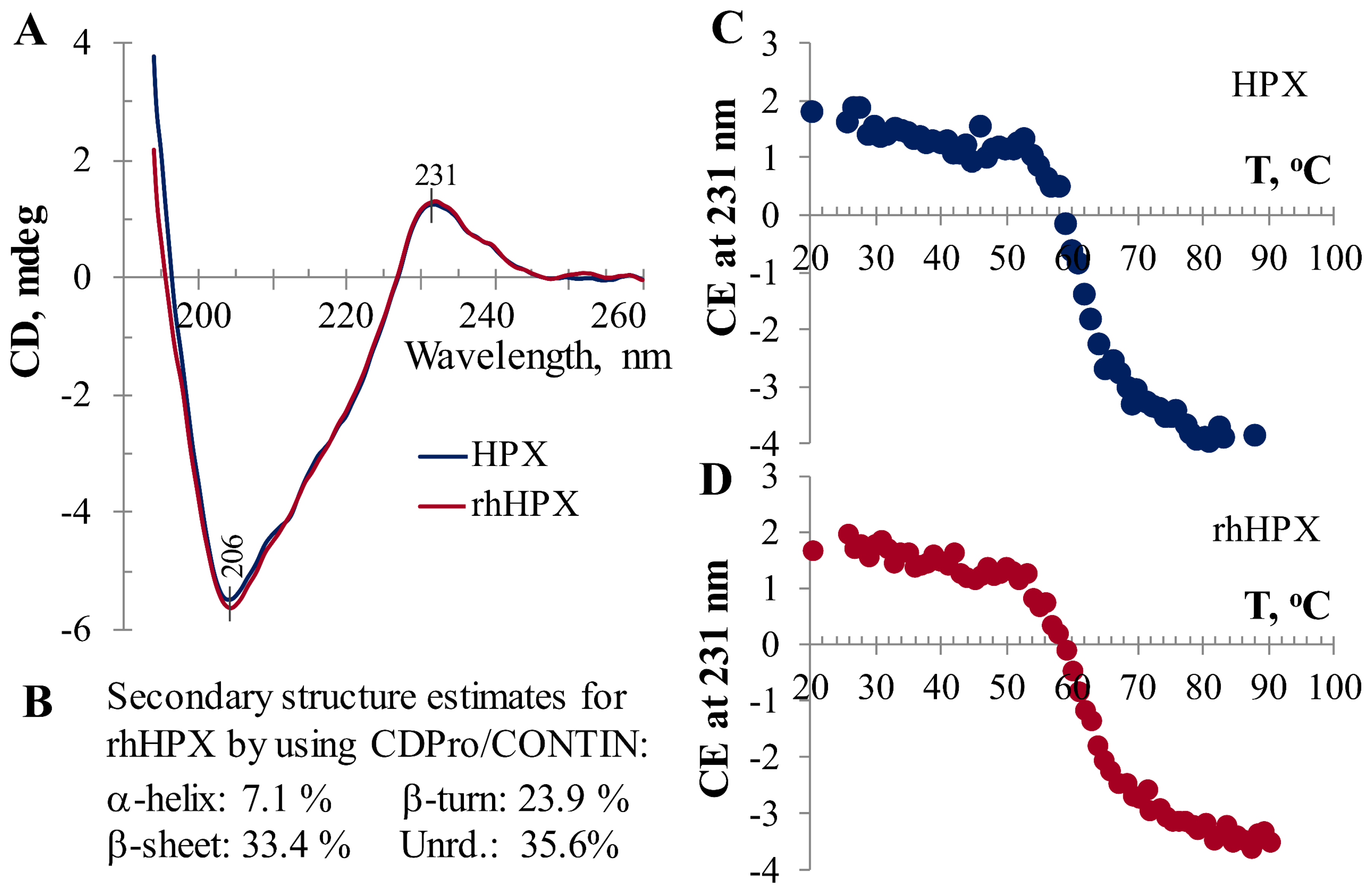
2.1. Characterization of rhHPX
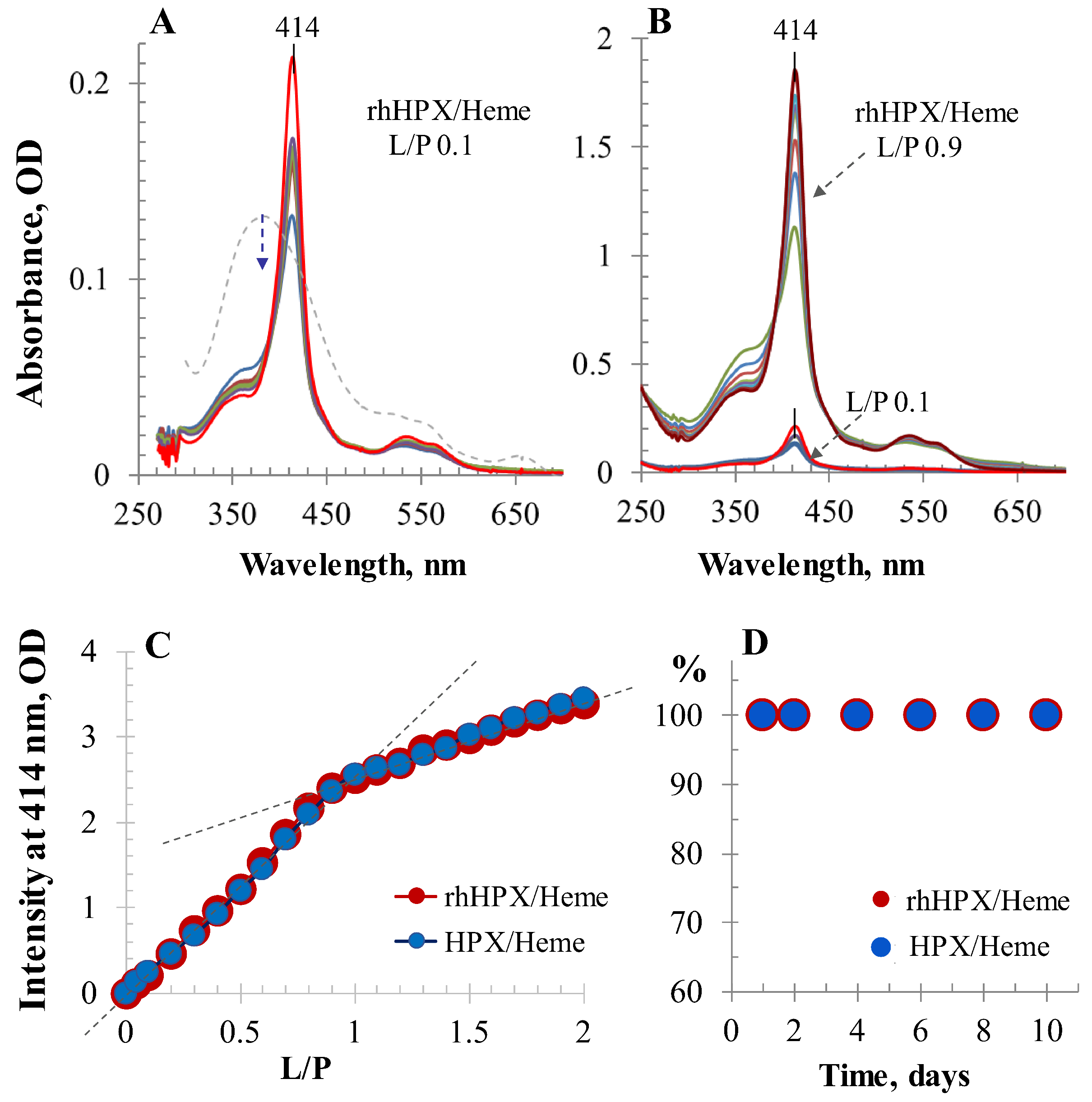
2.2. UV/Vis Titration
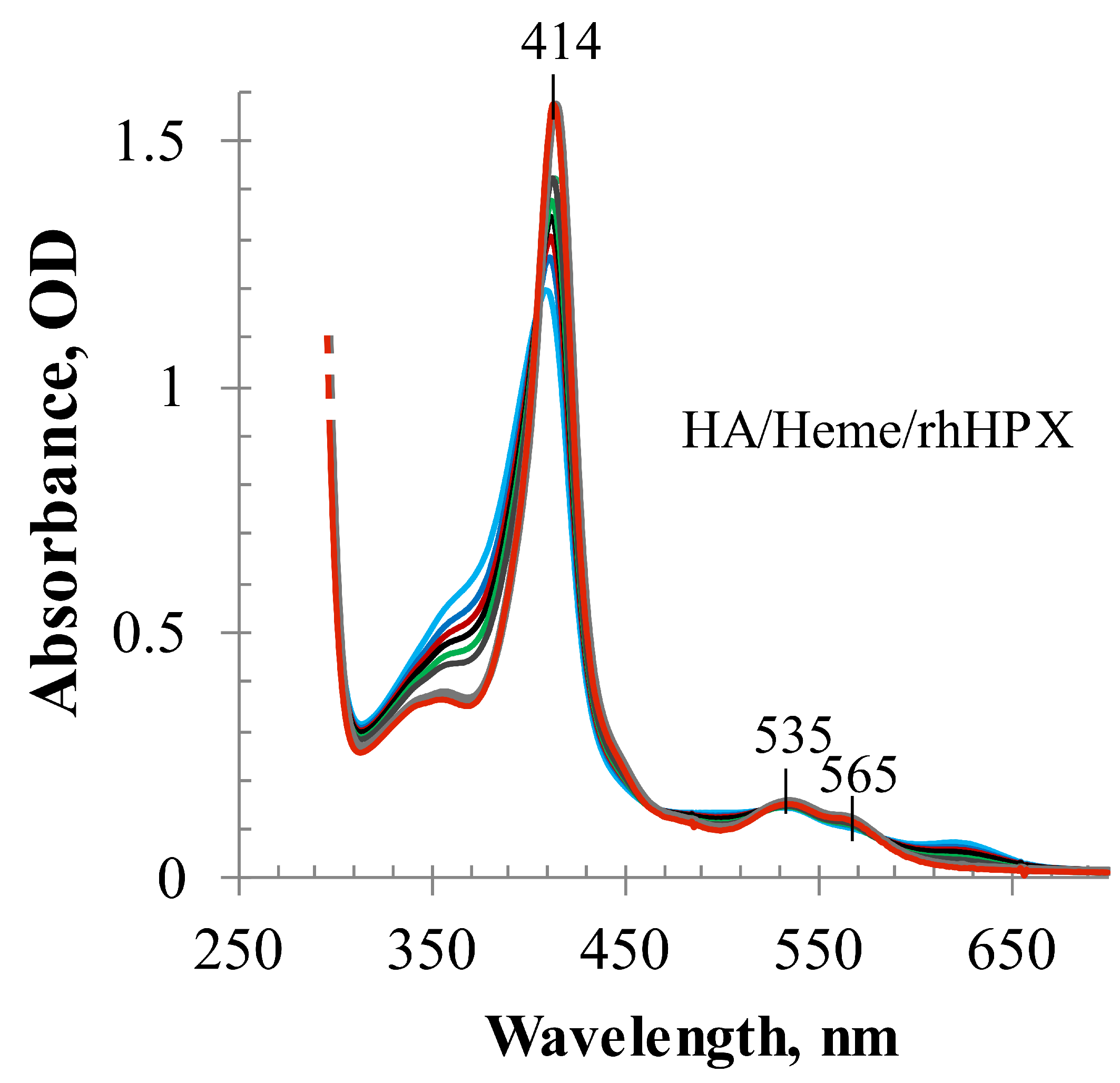
2.3. Heme Transfer from Methemalbumin to HPX
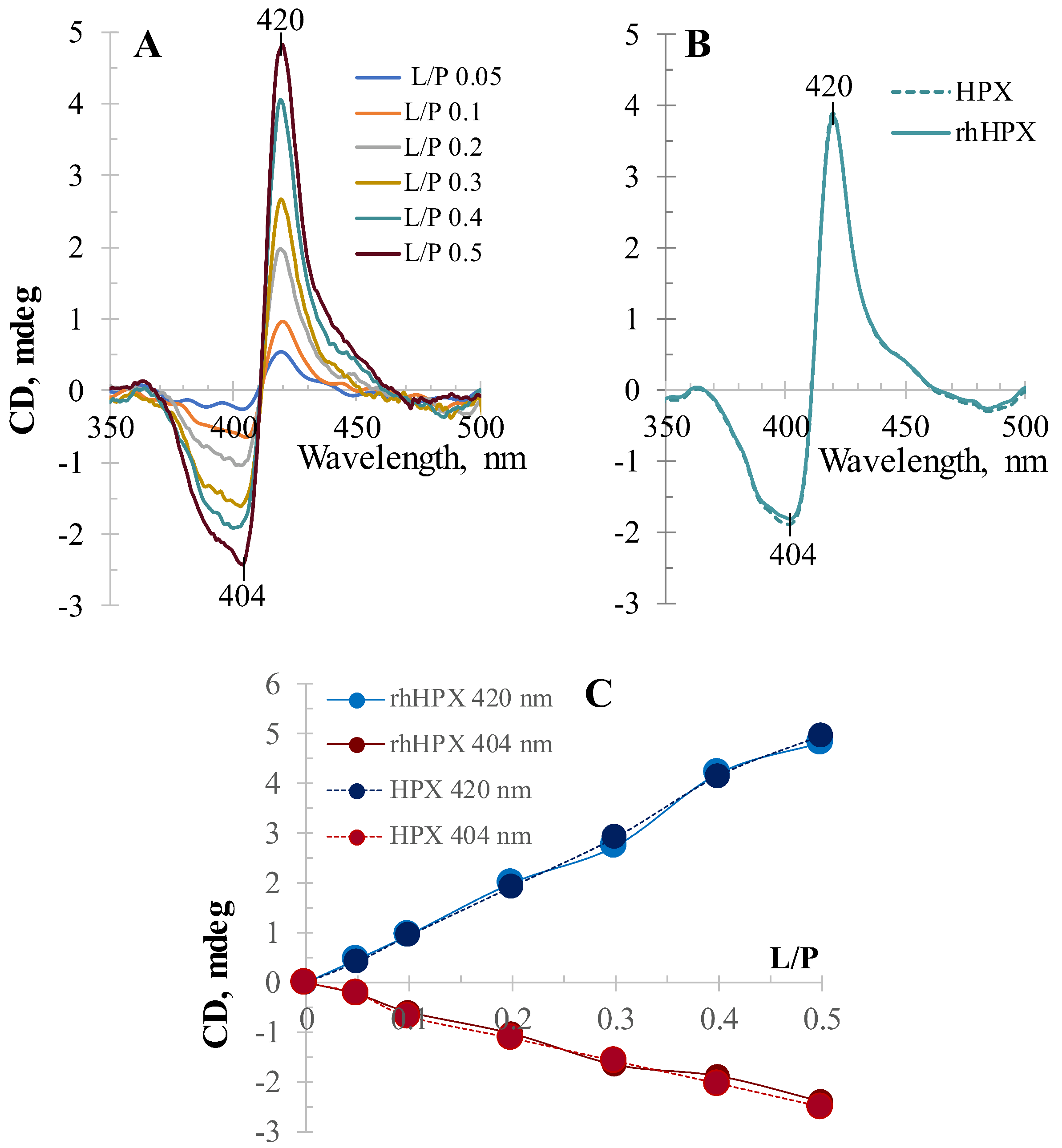
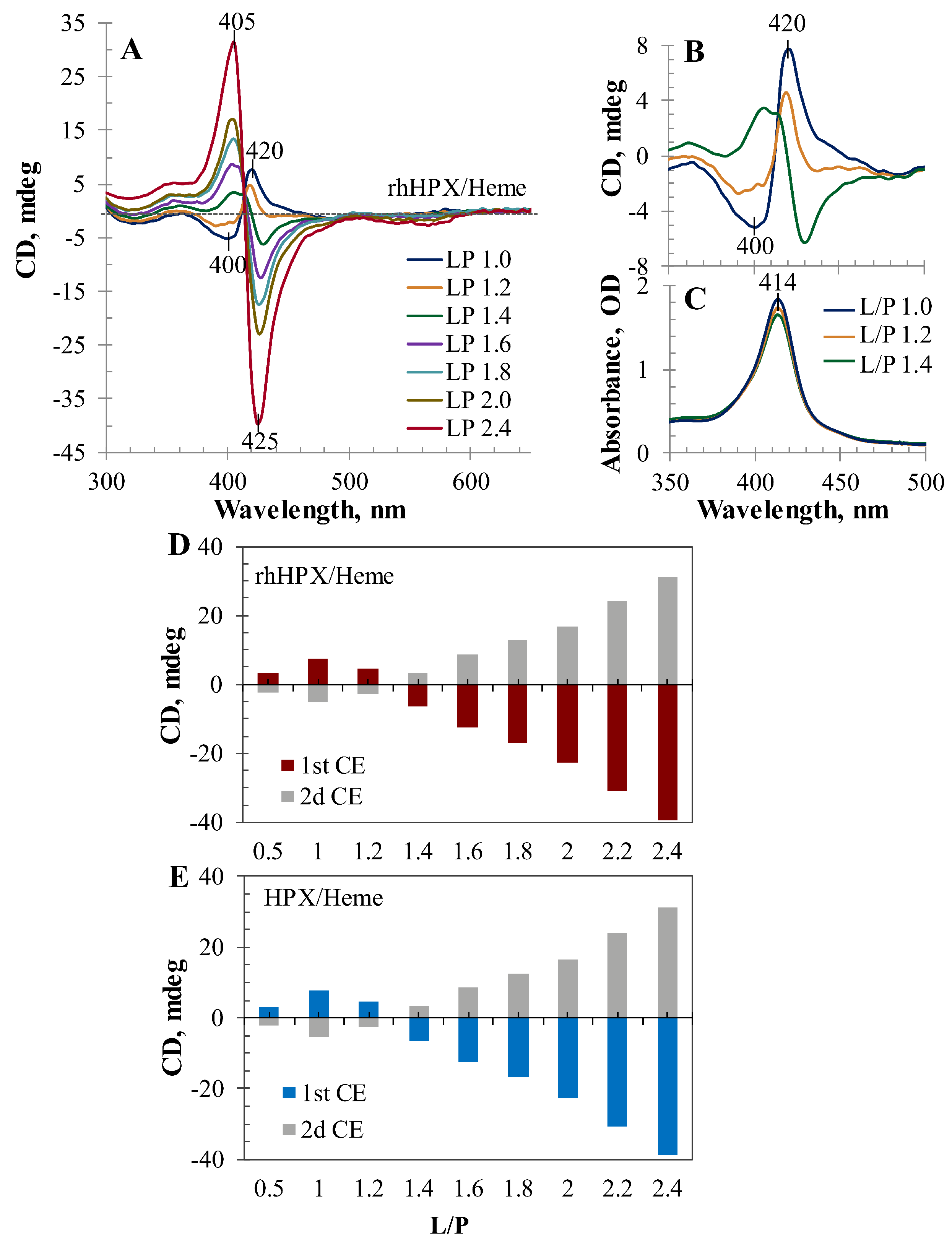
2.4. CD Titration
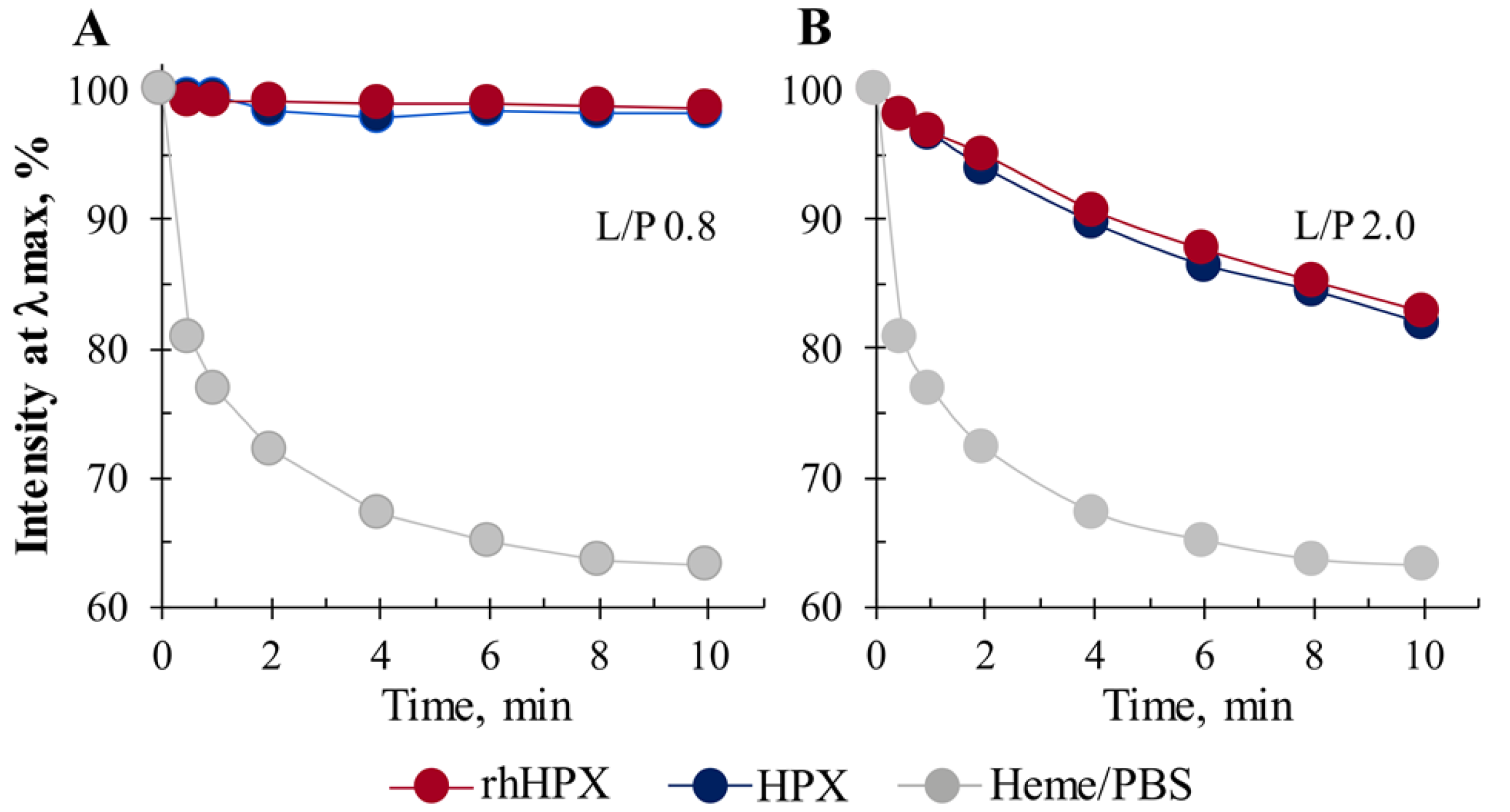
2.5. Heme Accessibility to Hydrogen Peroxide
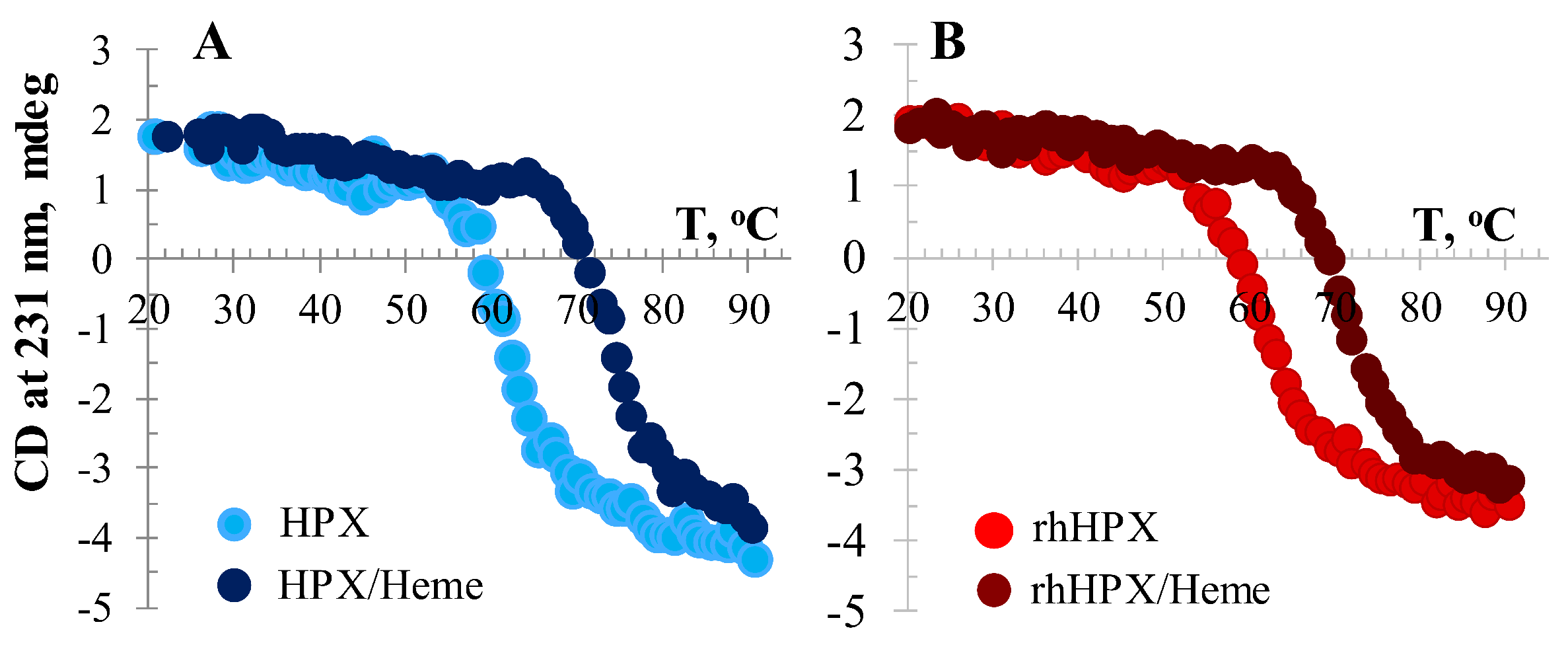
2.6. Temperature-Induced CD Assessment
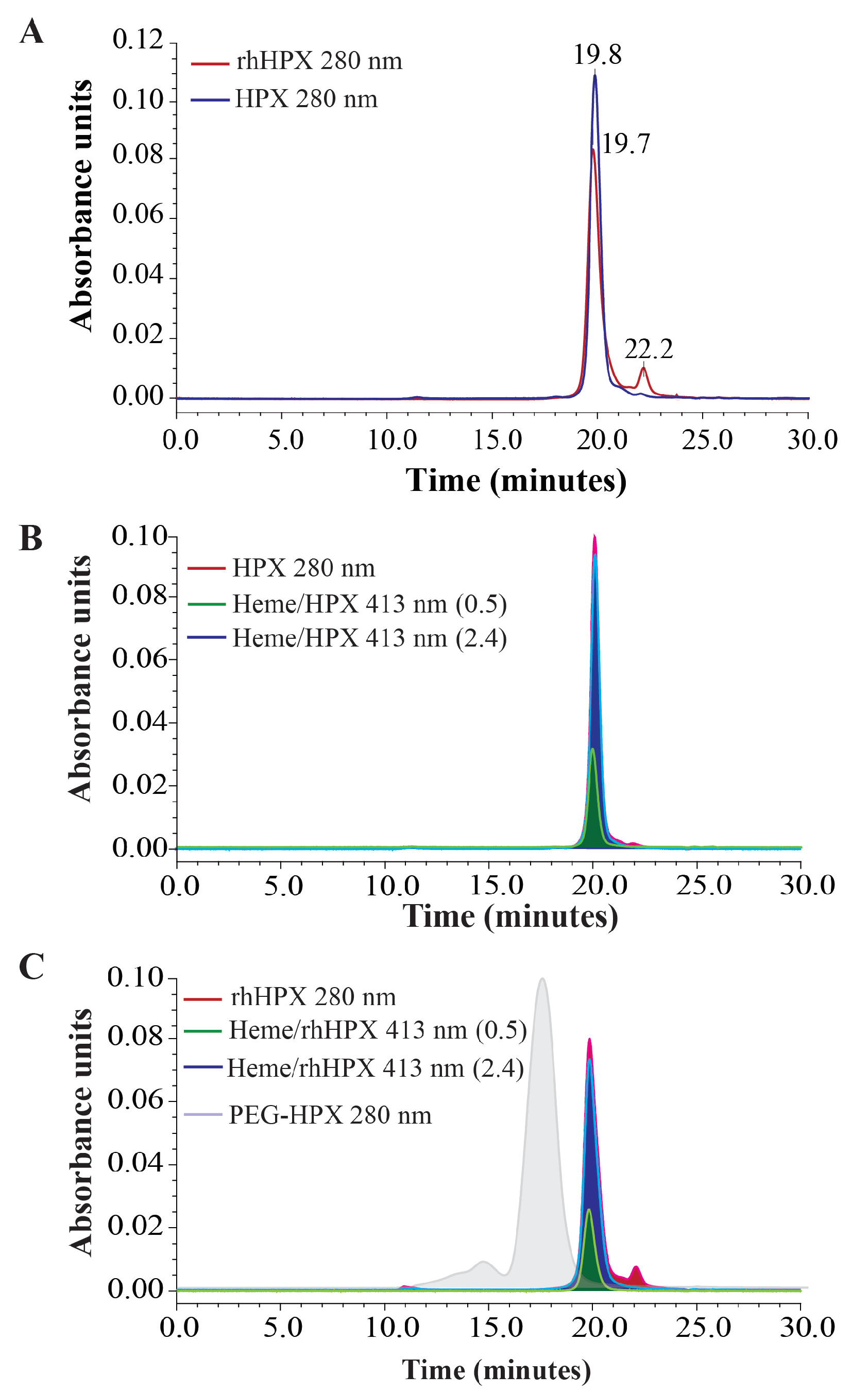
2.7. SEC-HPLC Assessment
3. Material and Methods
3.1. Materials
3.2. Generation of rhHPX Expression Plasmid and Transient Transfections
3.3. Purification of rhHPX
3.4. PEG-HPX Preparation
3.5. Stock Solutions of Heme, rhHPX, and Plasma HPX
3.6. Titration of Plasma HPX and rhHPX with Heme
3.7. UV/Vis Measurements
3.8. CD Measurements
3.9. Stability to Hydrogen Peroxide
3.10. SEC-HPLC
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CD | Circular dichroism |
| CE | Cotton effect |
| DMSO | Dimethylsulfoxide |
| EC | Exciton coupling |
| HA | Human albumin |
| HA/Heme | Methemalbumin, HA/Heme complex |
| Heme | Iron-protoporphyrin IX |
| HPX | Hemopexin |
| HPX/Heme | Hemopexin/Heme complex |
| L/P | Ligand-to-protein (molar) ratio |
| λmax | Wavelength of absorption maximum |
| PBS | Phosphate buffer saline |
| SEC-HPLC | Size-exclusion high-performance liquid chromatography |
References
- Muller-Eberhard, U.; Javid, H.; Liem, H.H.; Hanstein, A.; Hanns, M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood 1968, 32, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Vinchi, F.; Ingoglia, G.; Tolosano, E.; Buehler, P.W. Haptoglobin, hemopexin, and related defense pathways-basic science, clinical perspectives, and drug development. Front. Physiol. 2014, 5, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buehler, P.W.; Karnaukhova, E. When might transferrin, hemopexin or haptoglobin administration be of benefit following the transfusion of red blood cells? Curr. Opin. Hematol. 2018, 25, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, S.; Clark, J.; Adamides, A.A.; Ziogas, J. The role of haptoglobin and hemopexin in the prevention of delayed cerebral ischaemia after aneurysmal subarachnoid haemorrhage: A review of current literature. Neurosurg. Rev. 2019, 43, 1273–1288. [Google Scholar] [CrossRef]
- Van Avondt, K.; Nur, E.; Zeerleder, S. Mechanisms of haemolysis-induced kidney injury. Nat. Rev. Nephrol. 2019, 15, 671–692. [Google Scholar] [CrossRef]
- Morgan, W.T.; Liem, H.H.; Sutor, R.P.; Muller-Ebergard, U. Transfer of heme from heme-albumin to hemopexin. Biochim. Biophys. Acta 1976, 444, 435–445. [Google Scholar] [CrossRef]
- Tolosano, E.; Altruda, F. Hemopexin: Structure, function, and regulation. DNA Cell. Biol. 2002, 21, 297–306. [Google Scholar] [CrossRef]
- Tolosano, E.; Fagoonee, S.; Morello, N.; Vinchi, F.; Fiorito, V. Heme scavenging and the other facets of hemopexin. Antiox. Redox Signal. 2010, 12, 305–320. [Google Scholar] [CrossRef]
- Buehler, P.W.; Karnaukhova, E.; Gelderman, M.P.; Alayash, A.I. Blood aging, safety, and transfusion: Capturing the “radical” menace. Antiox. Redox Signal. 2011, 14, 1713–1728. [Google Scholar] [CrossRef]
- Hahl, P.; Davis, T.; Washburn, C.; Rogers, J.T.; Smith, A. Mechanisms of neuroprotection by hemopexin: Modeling the control of heme and iron homeostasis in brain neurons in inflammatory states. J. Neurochem. 2013, 125, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Deuel, J.W.; Vallelian, F.; Schaer, C.A.; Puglia, M.; Buehler, P.W.; Schaer, D.J. Different target specificities of haptoglobin and hemopexin define a sequential protection system against vascular hemoglobin toxicity. Free Rad. Biol. Med. 2015, 89, 931–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Hanstein, A.; Muller-Eberhard, U. Concentrations of serum hemopexin in healthy children and adults and those with a variety of hematological disorders. J. Lab. Clin. Med. 1968, 71, 232–239. [Google Scholar] [CrossRef]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef] [Green Version]
- Wagener, F.A.; Volk, H.D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003, 55, 551–571. [Google Scholar] [CrossRef] [Green Version]
- Yalamanoglu, A.; Deuel, J.W.; Hunt, R.C.; Baek, J.H.; Hassell, K.; Redinius, K.; Irwin, D.C.; Schaer, D.J.; Buehler, P.W. Depletion of haptoglobin and hemopexin promote hemoglobin-mediated lipoprotein oxidation in sickle cell disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L765–L774. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Kwak, Y.H.; Sammy, F.; He, P.; Thundivalappil, S.; Sun, G.; Chao, W.; Warren, H.S. Synergistic inflammation is induced by blood degradation products with microbial Toll-like receptor agonists and is blocked by hemopexin. J. Infect. Dis. 2010, 202, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Sammy, F.; Yang, H.; Thundivalappil, S.; Hellman, J.; Tracey, K.J.; Warren, H.S. Identification of hemopexin as an anti-inflammatory factor that inhibits synergy of hemoglobin with HMGB1 in sterile and infectious inflammation. J. Immunol. 2012, 189, 2017–2022. [Google Scholar] [CrossRef] [Green Version]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassu, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef]
- Immenschuh, S.; Vijayan, V.; Janciauskiene, S.; Gueler, F. Heme as a Target for Therapeutic Interventions. Front. Pharmacol. 2017, 8, 146. [Google Scholar] [CrossRef] [Green Version]
- Heide, K.; Haupt, H.; Störiko, K.; Schultze, H.E. On the heme-binding capacity of hemopexin. Clin. Chim. Acta 1964, 10, 460–469. [Google Scholar] [CrossRef]
- Paoli, M.; Anderson, B.F.; Baker, H.M.; Morgan, W.T.; Smith, A.; Baker, E.N. Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two beta-propeller domains. Nat. Struct. Biol. 1999, 6, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Morgan, W.T. Haem transport to the liver by haemopexin. Receptor-mediated uptake with recycling of the protein. Biochem. J. 1979, 182, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.; Morgan, W.T. Hemopexin-mediated transport of heme into isolated rat hepatocytes. J. Biol. Chem. 1981, 256, 10902–10909. [Google Scholar] [CrossRef]
- Smith, A.; Alam, J.; Escriba, P.V.; Morgan, W.T. Regulation of heme oxygenase and metallothionein gene expression by the heme analogs, cobalt-, and tin-protoporphyrin. J. Biol. Chem. 1993, 268, 7365–7371. [Google Scholar] [CrossRef]
- Morgan, W.T.; Smith, A. Domain structure of rabbit hemopexin. Isolation and characterization of a heme-binding glycopeptide. J. Biol. Chem. 1984, 259, 12001–12006. [Google Scholar] [CrossRef]
- Morgan, W.T.; Muster, P.; Tatum, F.; Kao, S.; Alam, J.; Smith, A. Identification of the histidine residues of hemopexin that coordinate with heme-iron and of a receptor-binding region. J. Biol. Chem. 1993, 268, 6256–6262. [Google Scholar] [CrossRef]
- Eskew, J.D.; Vanacore, R.M.; Sung, L.; Morales, P.J.; Smith, A. Cellular protection mechanisms against extracellular heme. heme-hemopexin, but not free heme, activates the N-terminal c-jun kinase. J. Biol. Chem. 1999, 274, 638–648. [Google Scholar] [CrossRef] [Green Version]
- Shipulina, N.; Hunt, R.C.; Shaklai, N.; Smith, A. Coordination of nitric oxide by heme-hemopexin. J. Protein Chem. 1998, 17, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Shipulina, N.; Smith, A.; Morgan, W.T. Heme binding by hemopexin: Evidence for multiple modes of binding and functional implications. J. Protein Chem. 2000, 19, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Morgan, W.T.; Vickery, L.E. Magnetic and natural circular dichroism of metalloporphyrin complexes of human and rabbit hemopexin. J. Biol. Chem. 1978, 253, 2940–2945. [Google Scholar] [CrossRef]
- Mauk, M.R.; Rosell, F.I.; Lelj-Garolla, B.; Moore, G.R.; Mauk, A.G. Metal ion binding to human hemopexin. Biochemistry 2005, 44, 1864–1871. [Google Scholar] [CrossRef]
- Mauk, M.R.; Rosell, F.I.; Mauk, A.G. Chromatographically distinguishable heme insertion isoforms of human hemopexin. Biochemistry 2007, 46, 15033–15041. [Google Scholar] [CrossRef]
- Morgan, W.T.; Müller-Eberhard, U. Interactions of porphyrins with rabbit hemopexin. J. Biol. Chem. 1972, 247, 7181–7187. [Google Scholar] [CrossRef]
- Bernard, N.; Lombart, C.; Waks, M. Modification of rat hemopexin properties upon heme binding. Eur. J. Biochem. 1980, 103, 271–276. [Google Scholar] [CrossRef]
- Wu, M.L.; Morgan, W.T. Characterization of hemopexin and its interaction with heme by differential scanning calorimetry and circular dichroism. Biochemistry 1993, 32, 7216–7222. [Google Scholar] [CrossRef]
- Wu, M.L.; Morgan, W.T. Conformational analysis of hemopexin by Fourier-transform infrared and circular dichroism spectroscopy. Proteins 1994, 20, 185–190. [Google Scholar] [CrossRef]
- Woody, R.W. Contributions of tryptophan side chains to the far-ultraviolet circular dichroism of proteins. Eur. Biophys. J. 1994, 23, 253–262. [Google Scholar] [CrossRef]
- Hider, R.C.; Kupryszewski, G.; Rekowski, P.; Lammek, B. Origin of the positive 225-230 nm circular dichroism band in proteins. Its application to conformational analysis. Biophys. Chem. 1988, 31, 45–51. [Google Scholar] [CrossRef]
- Kelly, S.M.; Price, N.C. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 2000, 1, 349–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Hrkal, Z.; Muller-Eberhard, U. Partial characterization of the heme-binding serum glycoproteins rabbit and human hemopexin. Biochemistry 1971, 10, 1746–1750. [Google Scholar] [CrossRef] [PubMed]
- Berova, N.; Borhan, B.; Dong, J.G.; Huang, X.; Karnaukhova, E.; Kawamura, A.; Lou, J.; Matile, S.; Nakanishi, K.; Rickman, B.; et al. Solving challenging bioorganic problems by exciton coupled CD. Pure Appl. Chem. 1998, 70, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Berova, N.; Nakanishi, K. Exciton Chirality Method: Principles and Applications. In Circular Dichroism: Principles and Applications, 2nd ed.; Berova, N., Nakanishi, K., Woody, R.W., Eds.; Wiley-VCH: New York, NY, USA, 2000; pp. 337–382. ISBN 9780471330035. [Google Scholar]
- Huang, X.; Nakanishi, K.; Berova, N. Porphyrins and metalloporphyrins: Versatile circular dichroic reporter groups for structural studies. Chirality 2000, 12, 237–255. [Google Scholar] [CrossRef]
- Harada, N.; Nakanishi, K.; Berova, N. Electronic CD Exciton Chirality Method: Principles and Applications. In Comprehensive Chiroptical Spectroscopy; Berova, N., Polavarapu, P.L., Nakanishi, K., Woody, R.W., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 115–166. ISBN 9781118012932. [Google Scholar]
- Hsu, M.C.; Woody, R.W. The origin of the heme Cotton effects in myoglobin and hemoglobin. J. Am. Chem. Soc. 1971, 93, 3515–3525. [Google Scholar] [CrossRef] [PubMed]
- Woody, R.W. Circular dichroism probes of hemoglobin structure. In Biochemical and Clinical Aspects of Hemoglobin Abnormalities; Caughey, W.S., Ed.; Academic Press: New York, NY, USA, 1978; pp. 279–298. ISBN 9780323142748. [Google Scholar]
- Myer, Y.P.; Pande, A. Circular dichroism studies of hemoproteins and heme models. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; Volume 3, pp. 271–345. ISBN 9780323143905. [Google Scholar]
- Blauer, G.; Sreerama, N.; Woody, R.W. Optical activity of hemoproteins in the Soret region. Circular dichroism of the heme undecapeptide of cytochrome c in aqueous solution. Biochemistry 1993, 32, 6674–6679. [Google Scholar] [CrossRef]
- Woody, R.W. Circular dichroism of peptides and proteins. In Circular Dichroism. Principles and Applications; Nakanishi, K., Berova, N., Woody, R.W., Eds.; VCH: New York, NY, USA, 1994; pp. 473–496. ISBN 156081618X. [Google Scholar]
- Sreerama, N.; Woody, R.W. Computation and analysis of protein circular dichroism spectra. Methods Enzymol. 2004, 383, 318–351. [Google Scholar] [CrossRef]
- Nagai, M.; Nagai, Y.; Imai, K.; Neya, S. Circular dichroism of hemoglobin and myoglobin. Chirality 2014, 26, 438–442. [Google Scholar] [CrossRef]
- Karnaukhova, E.; Rutardottir, S.; Rajabi, M.; Wester Rosenlöf, L.; Alayash, A.I.; Åkerström, B. Characterization of heme binding to recombinant α1-microglobulin. Front. Physiol. 2014, 5, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, S.H.; Grady, R.W.; Snider, J.M.; Muller-Eberhard, U. The influence of heme-binding proteins in heme-catalyzed oxidations. Arch. Biochem. Biophys. 1988, 265, 539–550. [Google Scholar] [CrossRef]
- Rosell, F.I.; Mauk, M.R.; Grant Mauk, A.G. Effects of metal ion binding on structural dynamics of human hemopexin. Biochemistry 2007, 46, 9301–9309. [Google Scholar] [CrossRef]
- Majuri, R. Behaviour in a silica-based high-performance liquid gel permeation chromatographic column of the apo- and holo-forms of the haem-binding proteins haemopexin, histidine-rich glycoprotein, globin and albumin. J. Chromatogr. 1987, 387, 281–290. [Google Scholar] [CrossRef]
- Cox, M.C.; Le Brun, N.; Thomson, A.J.; Smith, A.; Morgan, W.T.; Moore, G.R. MCD, EPR and NMR spectroscopic studies of rabbit hemopexin and its heme binding domain. Biophys. Biochim. Acta 1995, 1253, 215–223. [Google Scholar] [CrossRef]
- Kozak, M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987, 15, 8125–8148. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, P.M.; Abdo, M.; Butcher, R.E.; Yap, M.Y.; Scotney, P.D.; Ramunno, M.L.; Martin-Roussety, G.; Owczarek, C.; Hardy, M.P.; Chen, C.G.; et al. A robust robotic high-throughput antibody purification platform. J. Chromatogr. A 2016, 1455, 9–19. [Google Scholar] [CrossRef]
- Collier, G.S.; Pratt, J.M.; de Wet, C.R.; Tshabalala, C.F. Studies on haemin in dimethyl sulphoxide/water mixtures. Biochem. J. 1979, 179, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Seery, V.L.; Hathaway, G.; Muller-Eberhard, U. Hemopexin of human and rabbit: Molecular weight and extinction coefficient. Arch. Biochem. Biophys. 1972, 150, 269–272. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Woollard, A.C.S.; Wolff, S.P. Hydrogen peroxide production during experimental protein glycation. FEBS Lett. 1990, 268, 69–71. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karnaukhova, E.; Owczarek, C.; Schmidt, P.; Schaer, D.J.; Buehler, P.W. Human Plasma and Recombinant Hemopexins: Heme Binding Revisited. Int. J. Mol. Sci. 2021, 22, 1199. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031199
Karnaukhova E, Owczarek C, Schmidt P, Schaer DJ, Buehler PW. Human Plasma and Recombinant Hemopexins: Heme Binding Revisited. International Journal of Molecular Sciences. 2021; 22(3):1199. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031199
Chicago/Turabian StyleKarnaukhova, Elena, Catherine Owczarek, Peter Schmidt, Dominik J. Schaer, and Paul W. Buehler. 2021. "Human Plasma and Recombinant Hemopexins: Heme Binding Revisited" International Journal of Molecular Sciences 22, no. 3: 1199. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031199