Analysis of m6A RNA Methylation-Related Genes in Liver Hepatocellular Carcinoma and Their Correlation with Survival


Abstract
:1. Introduction
2. Results
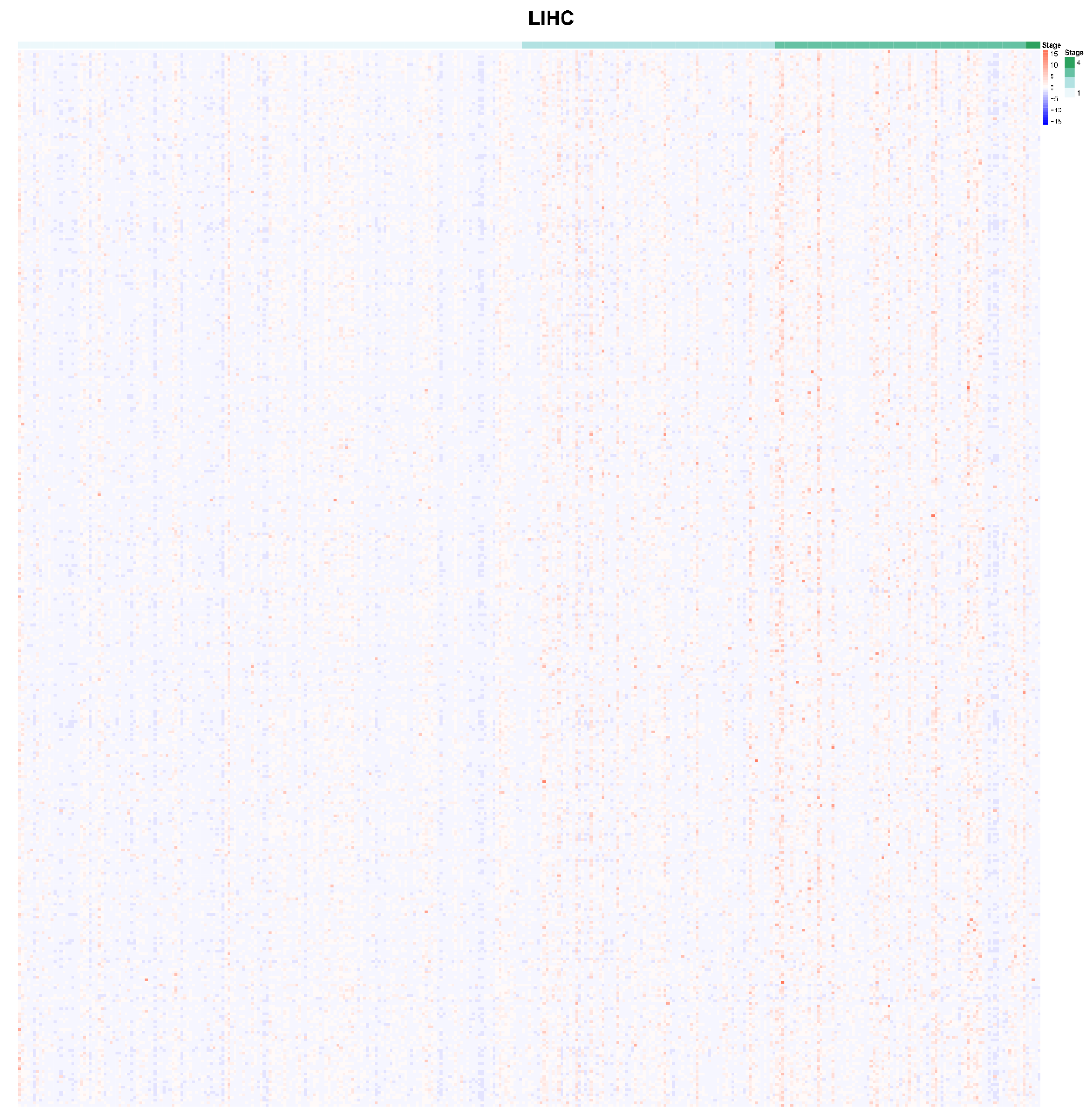
2.1. Identification of m6A-Related Genes which were Correlated with AJCC Stage
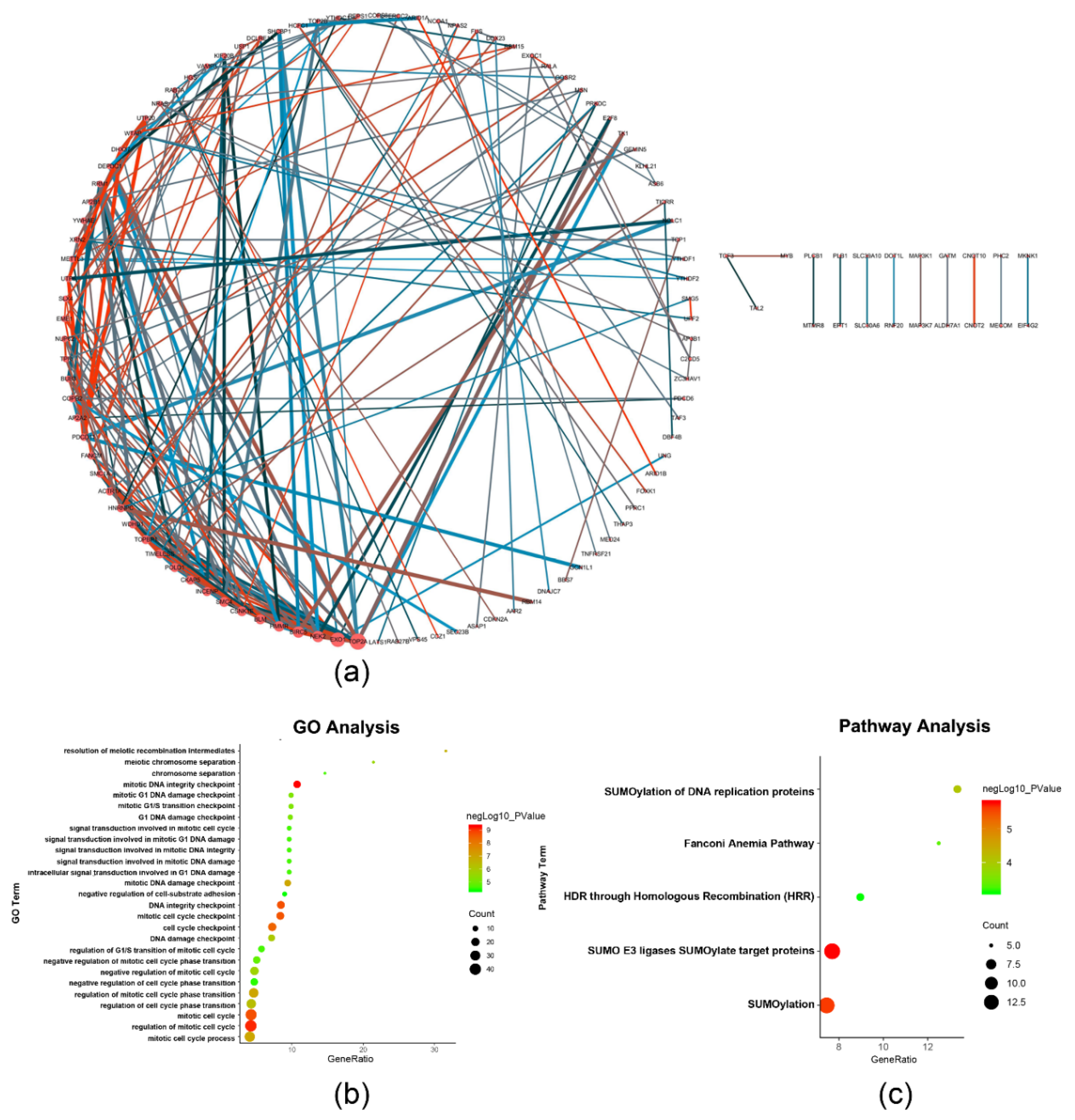
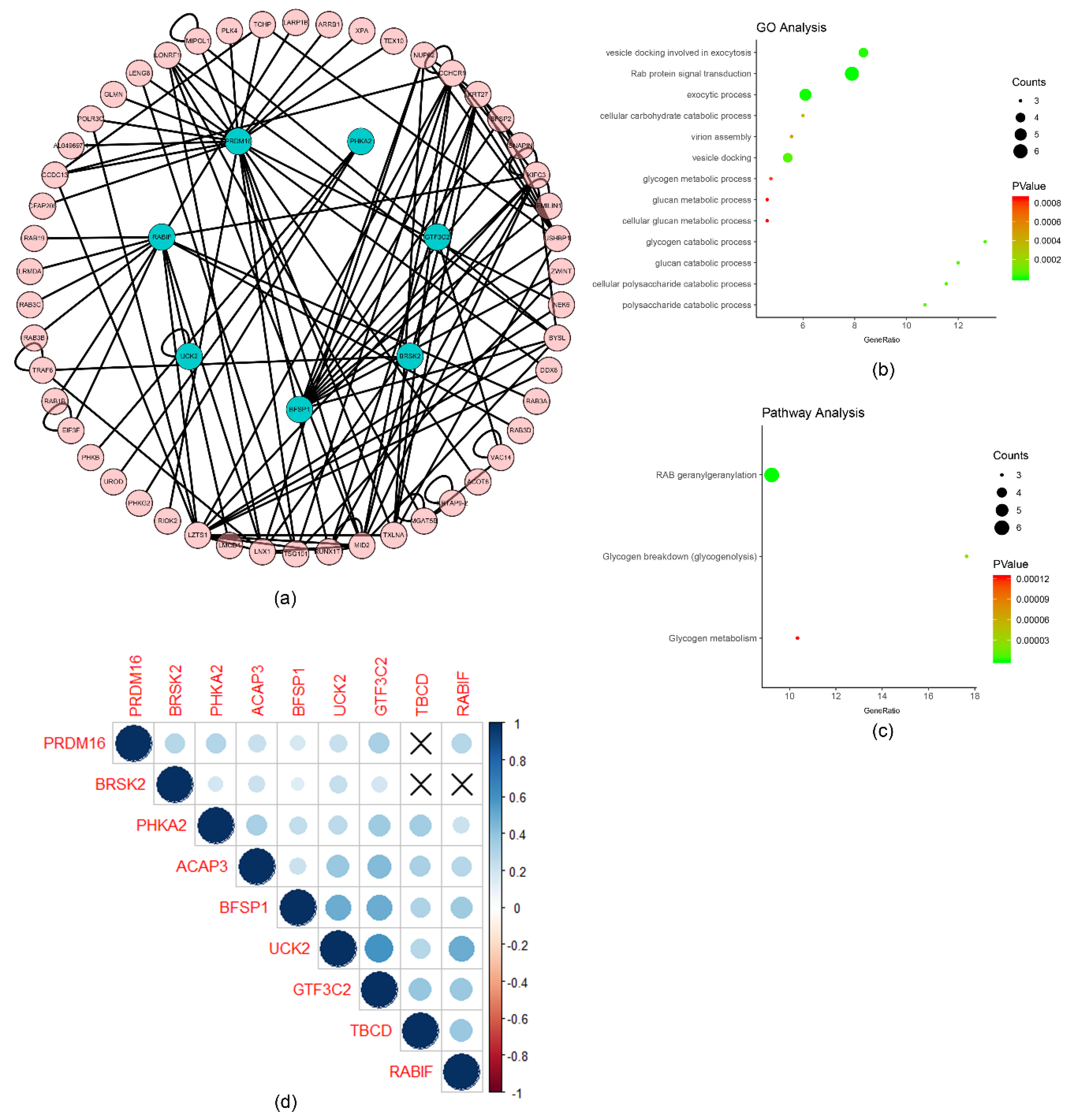
2.2. Functional Annotation of the Survival-Related m6A RNA Methylation-Related Genes
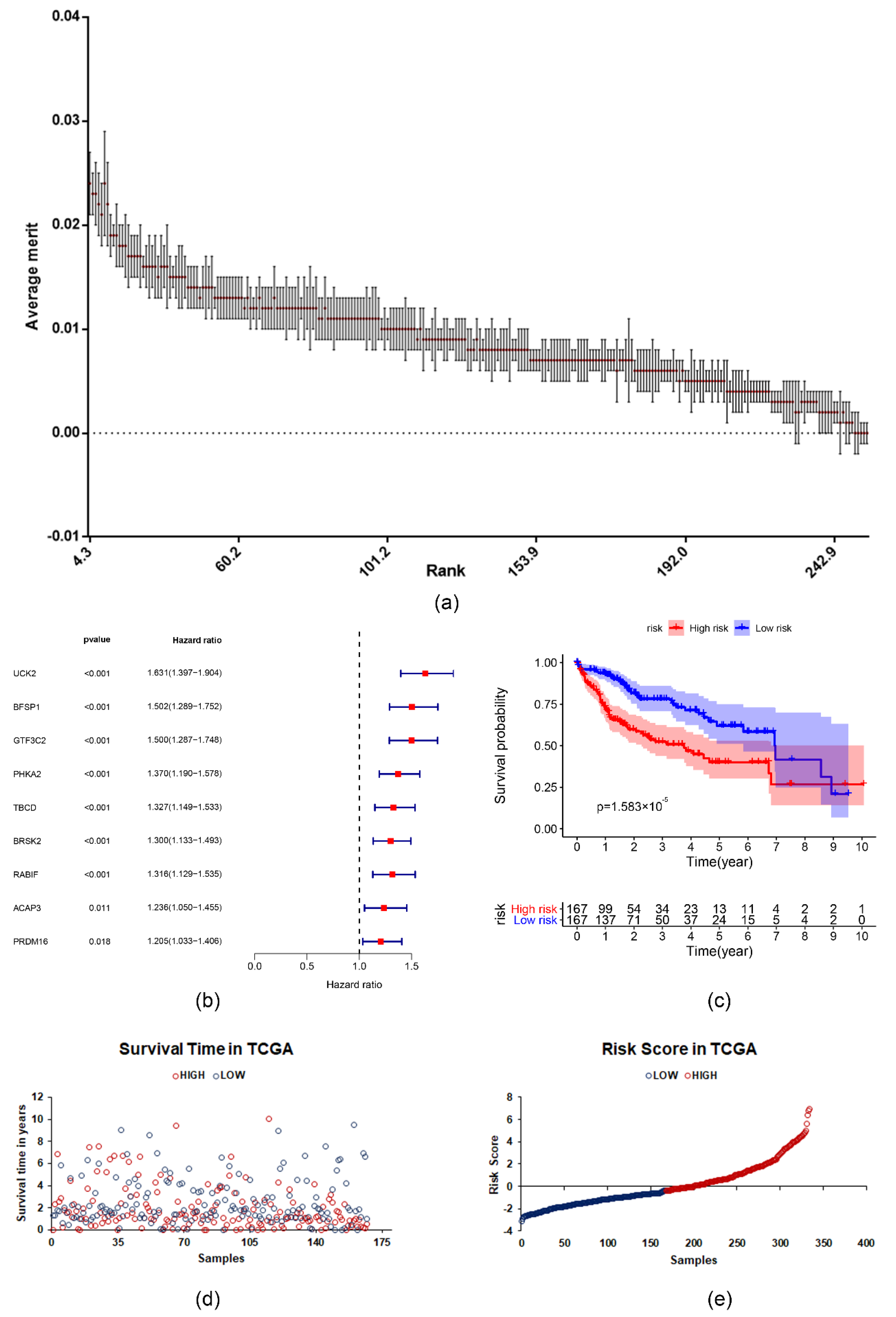
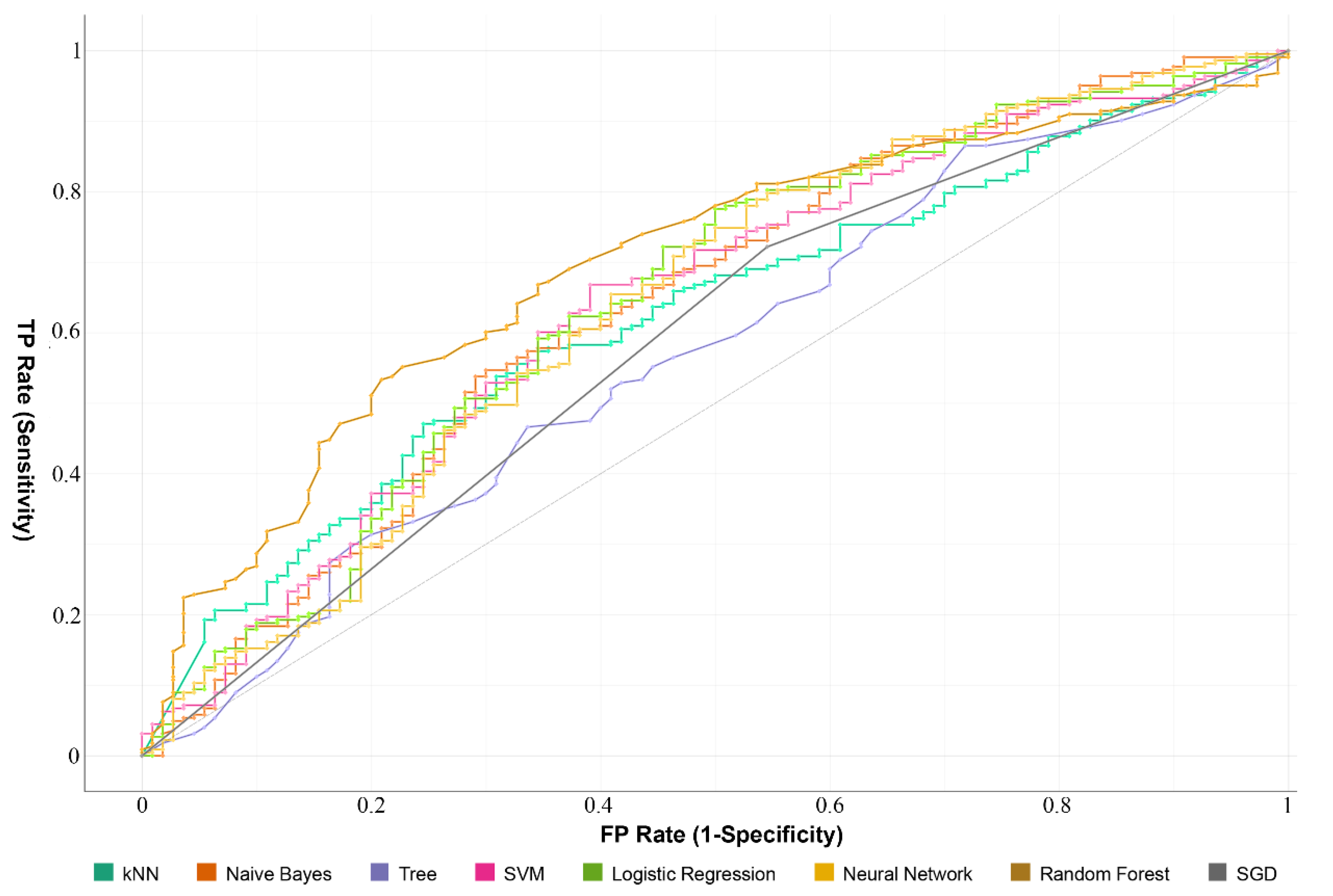
2.3. The Prognostic Value of the Survival-Related m6A RNA Methylation-Related Genes and a Risk Signature Built Using Nine Selected Survival-Related m6A RNA Methylation-Related Genes
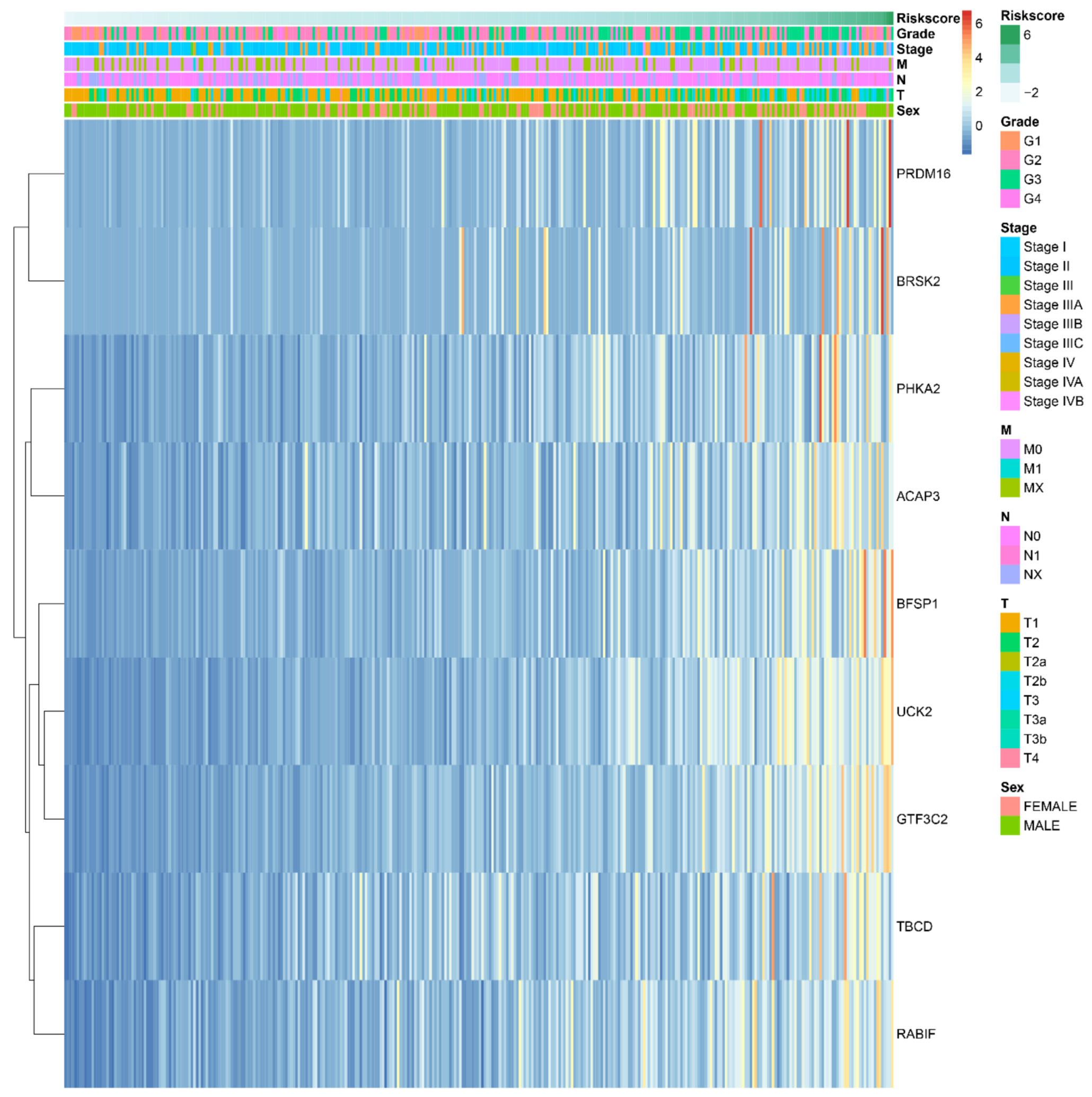
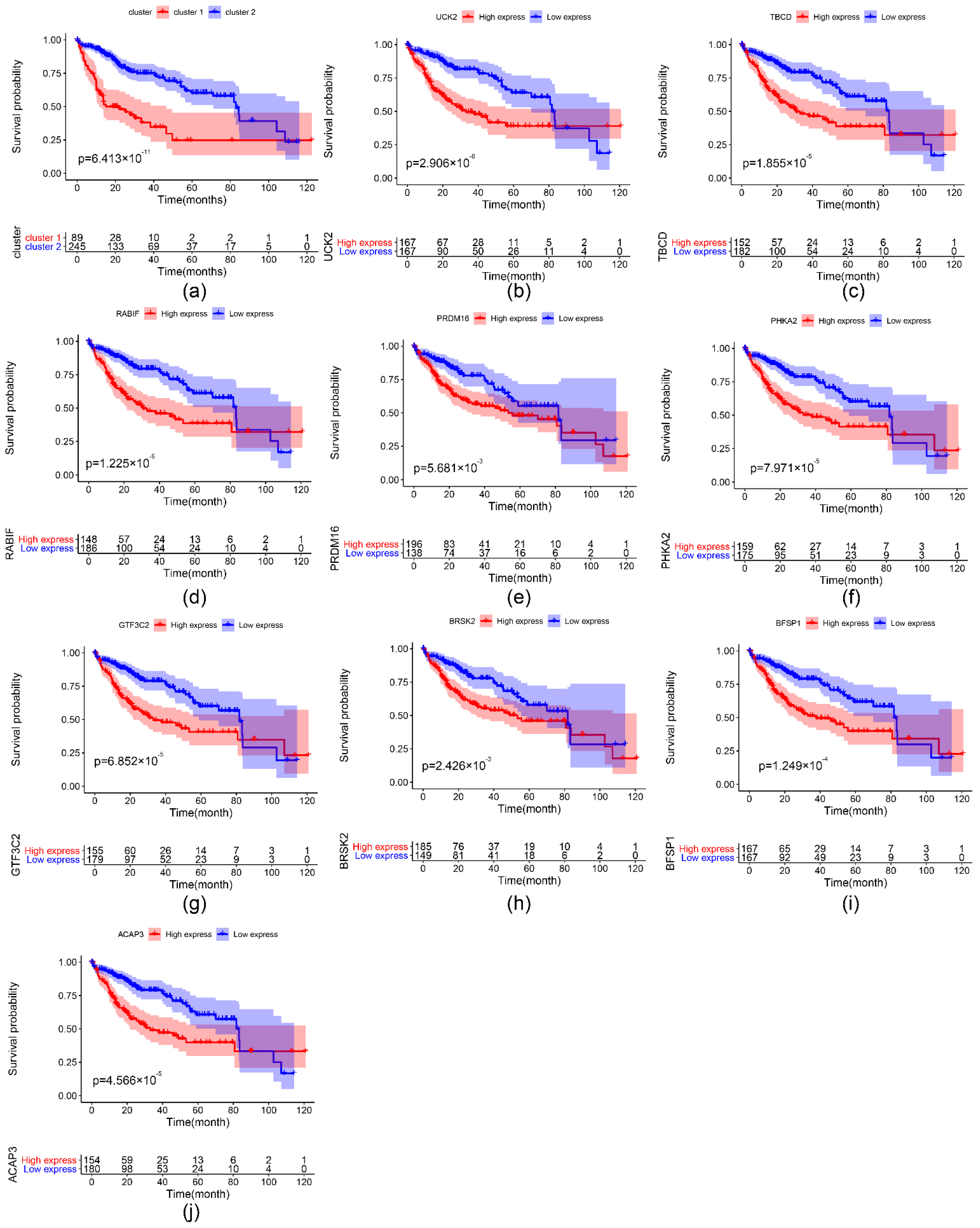
2.4. The Prognostic Value of the Important m6A-Related Genes for Liver Hepatocellular Carcinoma with Distinct Clinical Outcomes
3. Discussion
4. Materials and Methods
4.1. Data Preparation
4.2. The Differentially Expressed Genes (DEGs) Screening and Enrichment Analysis
4.3. The Filter of Survival-Related m6A RNA Methylation-Related Genes and the Evaluation of Risk Score
4.4. Construction and Evaluation of Prognostic Models Based on the Important Survival-Related Genes
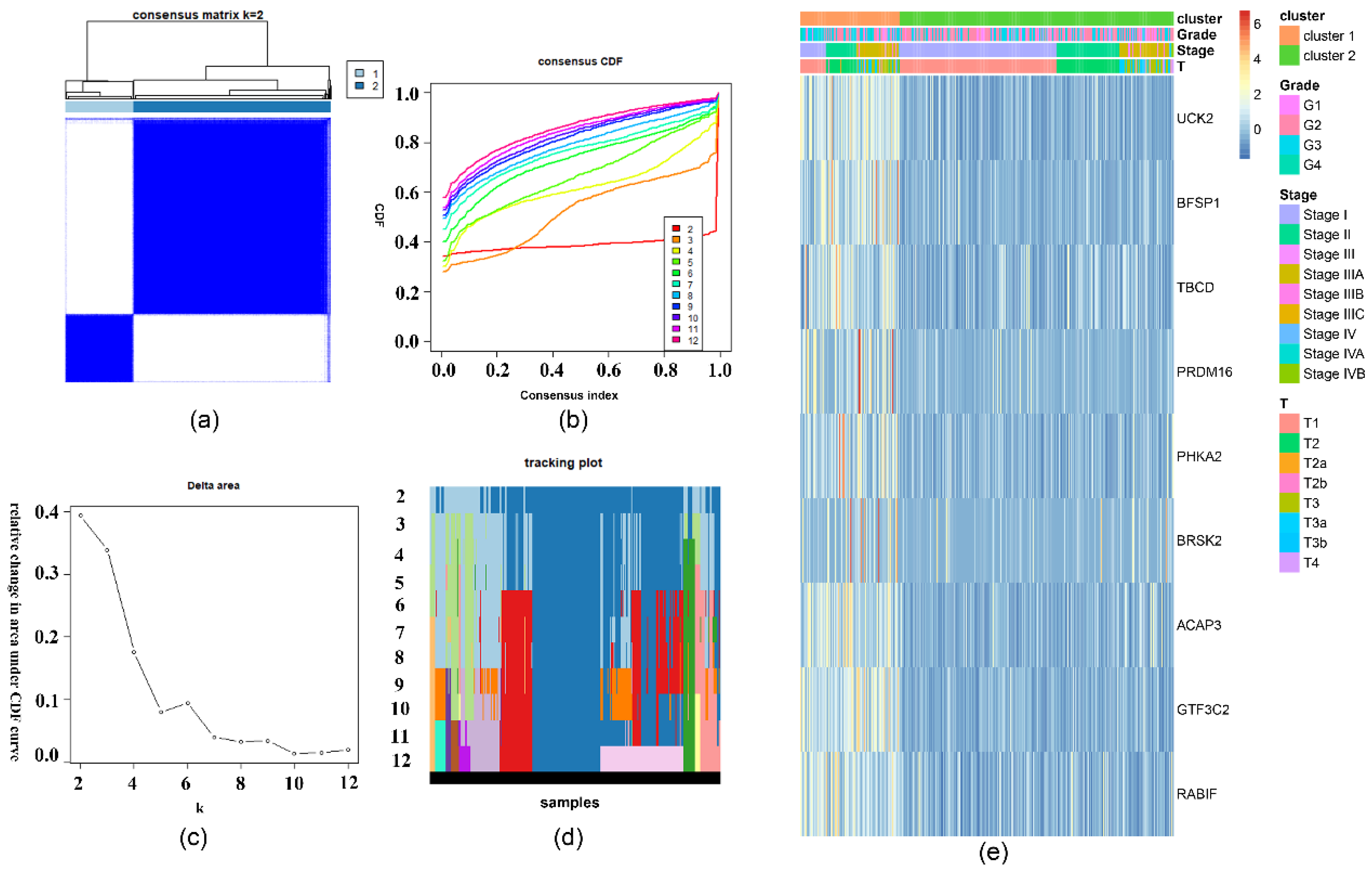
4.5. Consensus Clustering for the Important Survival-Related m6A RNA Methylation-Related Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACAP3 | Arf-GAP with coiled-coil |
| AJCC | American Joint Committee on Cancer |
| AUC | Area Under Curve |
| AURKB | Aurora kinase B |
| BFSP1 | Filensin |
| BIRC5 | Baculoviral IAP repeat-containing protein 5 |
| BLM | Bloom syndrome protein |
| BRSK2 | Serine/threonine-protein kinase BRSK2 |
| CDK1 | Cyclin-dependent kinase 1 |
| CKAP5 | Cytoskeleton-associated protein 5 |
| CSNK1E | Casein kinase I isoform epsilon |
| DEGs | Differentially expressed genes |
| DT | Decision Tree |
| EGFR | Epidermal growth factor receptor |
| EXO1 | Exodeoxyribonuclease 1 |
| FTO | Fat mass and obesity-associated protein |
| GO | Gene Ontology |
| GTF3C2 | General transcription factor 3C polypeptide 2 |
| HCC | Hepatocellular Carcinoma |
| HMMR | Hyaluronan mediated motility receptor |
| HNRNPC | Heterogeneous nuclear ribonucleoprotein C |
| HSP90 | Heat shock protein HSP 90 |
| INCENP | Inner centromere protein |
| KNN | k-Nearest Neighbor |
| LG | Logistic Regression |
| LIHC | Liver hepatocellular carcinoma |
| m6A | N6-methyladenosine |
| METTL16 | Methyltransferase like 16 |
| METTL3 | Methyltransferase like 3 |
| NB | Naïve Bayes |
| NEK2 | Serine/threonine-protein kinase Nek2 |
| NN | Neural Network |
| NXF1 | Nuclear RNA export factor 1 |
| PHKA2 | Phosphorylase b kinase regulatory subunit alpha |
| PRDM16 | Histone-lysine N-methyltransferase PRDM16 |
| RABIF | Guanine nucleotide exchange factor MSS4 |
| RBM15 | RNA binding motif protein 15 |
| RF | Random Forest |
| SGD | Stochastic Gradient Descent |
| SMC4 | Structural maintenance of chromosomes protein 4 |
| SOCS2 | Suppressor of cytokine signaling 2 |
| SVM | Support Vector Machine |
| TAZ | Tafazzin |
| TBCD | Tubulin-specific chaperone D |
| TCGA | The Cancer Genome Atlas |
| TOP2A | DNA topoisomerase 2-alpha |
| UCK2 | Uridine-cytidine kinase 2 |
| WTAP | WT1 associated protein |
| YTHDC1 | YTH domain containing 1 |
| YTHDF1 | YTH N6-methyladenosine RNA binding protein 1 |
| YTHDF2 | YTH N6-methyladenosine RNA binding protein 2 |
| YTHDF3 | YTH N6-methyladenosine RNA binding protein 3 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.W.; Talati, C.; Kim, R. Hepatocellular carcinoma (HCC): Beyond sorafenib—Chemotherapy. J. Gastrointest. Oncol. 2017, 8, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.Y.; Song, J.; Liu, Y.; Song, C.X.; Yi, C. Mapping the epigenetic modifications of DNA and RNA. Protein. Cell 2020, 11, 792–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef] [PubMed]
- Haruehanroengra, P.; Zheng, Y.Y.; Zhou, Y.; Huang, Y.; Sheng, J. RNA modifications and cancer. RNA Biol. 2020, 17, 1560–1575. [Google Scholar] [CrossRef]
- Muthusamy, S. M6a mRNA methylation: A pleiotropic regulator of cancer. Gene 2020, 736, 144415. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Wu, J.; Liu, J.; Qin, Z.; Fan, H. N6-methyladenosine: A potential breakthrough for human cancer. Mol. Ther. Nucleic Acids 2020, 19, 804–813. [Google Scholar] [CrossRef]
- Wen, S.; Wei, Y.; Zen, C.; Xiong, W.; Niu, Y.; Zhao, Y. Long non-coding RNA NEAT1 promotes bone metastasis of prostate cancer through N6-methyladenosine. Mol. Cancer 2020, 19, 171. [Google Scholar] [CrossRef]
- Chang, G.; Shi, L.; Ye, Y.; Shi, H.; Zeng, L.; Tiwary, S.; Huse, J.T.; Huo, L.; Ma, L.; Ma, Y.; et al. YTHDF3 induces the translation of m6A-enriched gene transcripts to promote breast cancer brain metastasis. Cancer Cell 2020, 38, 857–871. [Google Scholar] [CrossRef]
- Geng, Y.; Guan, R.; Hong, W.; Huang, B.; Liu, P.; Guo, X.; Hu, S.; Yu, M.; Hou, B. Identification of m6A-related genes and m6A RNA methylation regulators in pancreatic cancer and their association with survival. Ann Transl. Med. 2020, 8, 387. [Google Scholar] [CrossRef]
- Su, Z.Z.; Zhang, Z.X.; Cao, L.; Ding, G.; Wang, Z.Z.; Xiao, W. Network pharmacological study on the active components of YinQiaoJieDu soft capsule. Chin. New Drugs J. 2017, 15, 1786–1791. [Google Scholar]
- Kou, F.; Sun, H.; Wu, L.; Li, B.; Zhang, B.; Wang, X.; Yang, L. TOP2A promotes lung adenocarcinoma cells’ malignant progression and predicts poor prognosis in lung adenocarcinoma. J. Cancer 2020, 11, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Goodenow, D.; Emmanuel, F.; Berman, C.; Sahyouni, M.; Richardson, C. Bioflavonoids cause DNA double-strand breaks and chromosomal translocations through topoisomerase II-dependent and -independent mechanisms. Mutat. Res. 2020, 849, 503144. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Tsuda, M.; Bunch, H.; Sasanuma, H.; Austin, C.; Takeda, S. Type II DNA topoisomerases cause spontaneous double-strand breaks in genomic DNA. Genes 2019, 10, 868. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Li, T.; Sheng, M.; Yang, B. Exonuclease 1 (Exo1) participates in mammalian non-homologous end joining and contributes to drug resistance in ovarian cancer. Med. Sci. Monit. 2020, 26, e918751. [Google Scholar] [CrossRef]
- Hu, H.; Xu, H.; Lu, F.; Zhang, J.; Xu, L.; Xu, S.; Jiang, H.; Zeng, Q.; Chen, E.; He, Z. Exosome-derived miR-486–5p regulates cell cycle, proliferation and metastasis in lung adenocarcinoma via targeting NEK2. Front. Bioeng. Biotechnol. 2020, 8, 259. [Google Scholar] [CrossRef]
- Roberts, M.S.; Sahni, J.M.; Schrock, M.S.; Piemonte, K.M.; Weber-Bonk, K.L.; Seachrist, D.D.; Avril, S.; Anstine, L.J.; Singh, S.; Sizemore, S.T.; et al. LIN9 and NEK2 are core regulators of mitotic fidelity that can be therapeutically targeted to overcome taxane resistance. Cancer Res. 2020, 80, 1693–1706. [Google Scholar] [CrossRef] [Green Version]
- Deb, B.; Sengupta, P.; Sambath, J.; Kumar, P. Bioinformatics analysis of global proteomic and phosphoproteomic data sets revealed activation of NEK2 and AURKA in cancers. Biomolecules 2020, 10, 237. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar]
- Yin, H.; Que, R.; Liu, C.; Ji, W.; Sun, B.; Lin, X.; Zhang, Q.; Zhao, X.; Peng, Z.; Zhang, X.; et al. Survivin-targeted drug screening platform identifies a matrine derivative WM-127 as a potential therapeutics against hepatocellular carcinoma. Cancer Lett. 2018, 425, 54–64. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Yuan, J.; Zhang, J.; Han, F. The condensin subunits SMC2 and SMC4 interact for correct condensation and segregation of mitotic maize chromosomes. Plant J. 2020, 102, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Varghese, R.T.; Young, S.; Pham, L.; Liang, Y.; Pridham, K.J.; Guo, S.; Murphy, S.; Kelly, D.F.; Sheng, Z. Casein kinase 1 epsilon regulates glioblastoma cell survival. Sci. Rep. 2018, 8, 13621. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Takeda, K.; Mohan, N.; Shen, Y.; Jiang, J.; Rotstein, D.; Wu, W.J. Payload of T-DM1 binds to cell surface cytoskeleton-associated protein 5 to mediate cytotoxicity of hepatocytes. Oncotarget 2018, 9, 37200–37215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, N.H.; Park, J.E.; Hwang, J.W.; Myung, N.; Hwang, K.T.; Kim, Y.A.; Jang, C.Y.; Kim, Y.K. PRMT6-mediated H3R2me2a guides Aurora B to chromosome arms for proper chromosome segregation. Nat. Commun. 2020, 11, 612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.C.; Yeh, C.T.; Lin, K.H. Cancer stem cell functions in hepatocellular carcinoma and comprehensive therapeutic strategies. Cells 2020, 9, 1331. [Google Scholar] [CrossRef]
- Jayant, K.; Habib, N.; Huang, K.W.; Warwick, J.; Arasaradnam, R. Recent advances: The imbalance of immune cells and cytokines in the pathogenesis of hepatocellular carcinoma. Diagnostics 2020, 10, 338. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef]
- Zhong, L.; Liao, D.; Zhang, M.; Zeng, C.; Li, X.; Zhang, R.; Ma, H.; Kang, T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019, 442, 252–261. [Google Scholar] [CrossRef]
- Lin, Y.; Wei, X.; Jian, Z.; Zhang, X. METTL3 expression is associated with glycolysis metabolism and sensitivity to glycolytic stress in hepatocellular carcinoma. Cancer Med. 2020, 9, 2859–2867. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, F.; Peng, Y.; Fang, J.; Zhou, J. Identification of three m6A-related mRNAs signature and risk score for the prognostication of hepatocellular carcinoma. Cancer Med. 2020, 9, 1877–1889. [Google Scholar] [CrossRef]
- Li, J.; Zhu, L.; Shi, Y.; Liu, J.; Lin, L.; Chen, X. m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. Am. J. Transl. Res. 2019, 11, 6084–6092. [Google Scholar] [PubMed]
- Guo, X.; Li, K.; Jiang, W.; Hu, Y.; Xiao, W.; Huang, Y.; Feng, Y.; Pan, Q.; Wan, R. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol. Cancer 2020, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.M.; Huo, F.C.; Pei, D.S. Function and evolution of RNA N6-methyladenosine modification. Int. J. Biol. Sci. 2020, 16, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Mendel, M.; Chen, K.M.; Homolka, D.; Gos, P.; Pandey, R.R.; McCarthy, A.A.; Pillai, R.S. Methylation of structured RNA by the m6A writer METTL16 is essential for mouse embryonic development. Mol. Cell 2018, 71, 986–1000. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Gong, Y.; Shen, L.; Li, J.; Han, J.; Song, B.; Hu, L.; Wang, Q.; Wang, Z. Total Panax notoginseng saponin inhibits vascular smooth muscle cell proliferation and migration and intimal hyperplasia by regulating WTAP/p16 signals via m6A modulation. Biomed. Pharmacother. 2020, 124, 109935. [Google Scholar] [CrossRef]
- Juillard, F.; Mure, F.; Bazot, Q.; Manet, É.; Gruffat, H. Post-transcriptional regulation of Herpesvirus productive cycle genes expression: Importance of EB2 factor from Epstein-Barr virus (EBV) and its similar proteins. Virologie 2013, 17, 96–110. [Google Scholar]
- Lee, Y.; Choe, J.; Park, O.H.; Kim, Y.K. Molecular mechanisms driving mRNA degradation by m6A modification. Trends Genet. 2020, 36, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.J.; Zhu, G.Q.; Zhang, Q.W.; Zheng, K.I.; Chen, J.N.; Zhang, X.T.; Wang, Q.W.; Li, X.B. Survival-associated alternative messenger RNA splicing signatures in pancreatic ductal adenocarcinoma: A study based on RNA-sequencing data. DNA Cell Biol. 2019, 38, 1207–1222. [Google Scholar] [CrossRef]
- Huang, S.; Li, J.; Tam, N.L.; Sun, C.; Hou, Y.; Hughes, B.; Wang, Z.; Zhou, Q.; He, X.; Wu, L. Uridine-cytidine kinase 2 upregulation predicts poor prognosis of hepatocellular carcinoma and is associated with cancer aggressiveness. Mol. Carcinog. 2019, 58, 603–615. [Google Scholar] [CrossRef]
- Enomoto, M.; Tamori, A.; Murakami, Y.; Kawada, N. Interferon-α/β for treatment of chronic hepatitis C infection in the era of direct-acting antiviral agents. Hepatol. Res. 2014, 44, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.R.; Hafez, M.M.; Bahnassy, A.A.; Hassan, Z.K.; Mansour, T.; Kamal, M.M.; Khaled, H.M. Genetic profile of Egyptian hepatocellular-carcinoma associated with hepatitis C virus Genotype 4 by 15 K cDNA microarray: Preliminary study. BMC Res. Notes 2008, 1, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, I.; de la Fouchardiere, A.; Pissaloux, D.; Mully, T.W.; Garrido, M.C.; Vemula, S.S.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am. J. Surg. Pathol. 2015, 39, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiyin, H.; Na, N.; Han, X.; Fang, Y.; Wu, Y.; Lou, W.; Yang, X. BRSK2 induced by nutrient deprivation promotes Akt activity in pancreatic cancer via downregulation of mTOR activity. Oncotarget 2017, 8, 44669–44681. [Google Scholar] [CrossRef] [Green Version]
- Kundu, A.; Nam, H.; Shelar, S.; Chandrashekar, D.S.; Brinkley, G.; Karki, S.; Mitchell, T.; Livi, C.B.; Buckhaults, P.; Kirkman, R.; et al. PRDM16 suppresses HIF-targeted gene expression in kidney cancer. J. Exp. Med. 2020, 217, e20191005. [Google Scholar] [CrossRef] [Green Version]
- Tian, G.; Cowan, N.J. Tubulin-specific chaperones: Components of a molecular machine that assembles the α/β heterodimer. Methods Cell Biol. 2013, 115, 155–171. [Google Scholar]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m6A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef]
- Dai, D.; Wang, H.; Zhu, L.; Jin, H.; Wang, X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, K.; Cai, J.; Zhang, M.; Zhang, X.; Xiong, X.; Meng, H.; Xu, X.; Huang, Z.; Peng, J.; et al. Landscape and regulation of m6A and m6Am methylome across human and mouse tissues. Mol. Cell 2020, 77, 426–440. [Google Scholar] [CrossRef]
- Lan, Q.; Liu, P.Y.; Haase, J.; Bell, J.L.; Hüttelmaier, S.; Liu, T. The critical role of RNA m6A methylation in cancer. Cancer Res. 2019, 79, 1285–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Nie, P.; Peng, D.; He, Z.; Liu, M.; Xie, Y.; Miao, Y.; Zuo, Z.; Ren, J. m6AVar: A database of functional variants involved in m6A modification. Nucleic Acids Res. 2018, 46, D139–D145. [Google Scholar] [CrossRef] [PubMed]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, C.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A.; et al. Orange: Data mining toolbox in python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | AUC | CA | F1 | Precision | Recall |
|---|---|---|---|---|---|
| RF | 0.70 | 0.67 | 0.63 | 0.64 | 0.67 |
| LG | 0.65 | 0.70 | 0.64 | 0.67 | 0.70 |
| NB | 0.64 | 0.65 | 0.65 | 0.64 | 0.65 |
| NN | 0.64 | 0.70 | 0.65 | 0.67 | 0.70 |
| SVM | 0.64 | 0.69 | 0.63 | 0.66 | 0.69 |
| KNN | 0.62 | 0.66 | 0.61 | 0.61 | 0.66 |
| SGD | 0.59 | 0.64 | 0.64 | 0.64 | 0.64 |
| DT | 0.57 | 0.68 | 0.64 | 0.64 | 0.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Qi, D.; Zhu, B.; Ye, X. Analysis of m6A RNA Methylation-Related Genes in Liver Hepatocellular Carcinoma and Their Correlation with Survival. Int. J. Mol. Sci. 2021, 22, 1474. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031474
Li Y, Qi D, Zhu B, Ye X. Analysis of m6A RNA Methylation-Related Genes in Liver Hepatocellular Carcinoma and Their Correlation with Survival. International Journal of Molecular Sciences. 2021; 22(3):1474. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031474
Chicago/Turabian StyleLi, Yong, Dandan Qi, Baoli Zhu, and Xin Ye. 2021. "Analysis of m6A RNA Methylation-Related Genes in Liver Hepatocellular Carcinoma and Their Correlation with Survival" International Journal of Molecular Sciences 22, no. 3: 1474. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031474