
Inflammation-Related Changes in Mood Disorders and the Immunomodulatory Role of Lithium


Molecular and Cell Biology Unit, Poznan University of Medical Sciences, 61-701 Poznań, Poland
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(4), 1532; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041532
Submission received: 13 January 2021
/
Revised: 28 January 2021
/
Accepted: 1 February 2021
/
Published: 3 February 2021
(This article belongs to the Special Issue Molecular Mechanisms of Mood Stabilizers)
Abstract
:Mood disorders are chronic, recurrent diseases characterized by changes in mood and emotions. The most common are major depressive disorder (MDD) and bipolar disorder (BD). Molecular biology studies have indicated an involvement of the immune system in the pathogenesis of mood disorders, and showed their correlation with altered levels of inflammatory markers and energy metabolism. Previous reports, including meta-analyses, also suggested the role of microglia activation in the M1 polarized macrophages, reflecting the pro-inflammatory phenotype. Lithium is an effective mood stabilizer used to treat both manic and depressive episodes in bipolar disorder, and as an augmentation of the antidepressant treatment of depression with a multidimensional mode of action. This review aims to summarize the molecular studies regarding inflammation, microglia activation and energy metabolism changes in mood disorders. We also aimed to outline the impact of lithium on these changes and discuss its immunomodulatory effect in mood disorders.
1. Introduction
Mood disorders (also known as affective disorders) are chronic and recurrent psychiatric conditions affecting emotions, motivation, and energy. The alterations of affect caused by mood disorders impair the ability to cope with daily life, including performance at work or school. The wide spectrum of mood disorders can be classified by the severity and range of mood change. The most common include major depressive disorder and bipolar disorder [1,2].
Major depressive disorder (MDD) is characterized by depressed mood, anhedonia, and decreased energy, as well as problems with concentration, feelings of guilt, and in its severe episodes, suicidal thoughts and attempts. MDD affects about 163 million people worldwide [3], and the World Health Organization (WHO) ranked MDD as the single largest contributor to global disability [4].
Bipolar disorder (BD), previously known as manic depressive illness, is a chronic, multifactorial and biphasic condition characterized by episodes of elevated mood and increased motor efficiency—episodes of mania or hypomania—and episodes of depression expressed as low mood and decreased energy [5,6]. BD affects approximately 45 million people worldwide [3]. Due to an increased risk of suicide, and other comorbidities such as cardio- and cerebrovascular diseases, endocrine, and metabolic disorders, bipolar disorder shortens life expectancy by 11 years in females and 10 years in males [7,8].
2. Pathophysiology of Mood Disorders
Molecular biology studies have enabled rapid progress in elucidating the pathophysiology of mood disorders. Several theories were formulated, including an imbalance in monoaminergic neurotransmission, stress response, disturbances in the hypothalamic–pituitary–adrenal (HPA) axis, and neurotrophic pathways alterations. Changes were also observed within the immune system, not only in the central nervous system, but also in the periphery, including the production of inflammatory cytokines that lead to microglia activation. Such activation may also be induced as a result of alterations in mitochondrial activity and energy metabolism within the central nervous system.
2.1. Inflammation Theory
Inflammation is a response of the organism to harmful stimuli, and conditions such as physical injury or infection. The immune system triggers inflammation to maintain or restore the disturbed homeostasis. Inflammatory responses are mediated by cytokines, and this group includes interleukins (IL), chemokines, interferons (IFN), tumor necrosis factors (TNF), and lymphokines. Cytokines are produced by immune cells such as macrophages, T cells, B cells or mast cells, and non-immune cells such as endothelial cells, fibroblasts, and various stromal cells. The cytokines act through receptors modulating the immune responses by a self- (autocrine), locally (paracrine), or distally (endocrine) determined manner, either enhancing immune response or showing an anti-inflammatory effect by affecting various biological processes [9].
Dysfunction of the immune system and chronic inflammation have been implicated in the pathophysiology of numerous psychiatric disorders [10,11]. In 1990, Maes et al. [12] revealed a correlation between the cytokines level in blood and major depressive disorder. At the same time, Smith et al. [13] described the relationship between depression and pro-inflammatory cytokines produced by macrophages, which was the basis of the inflammatory theory that assumes the crucial role of immune cells present in the central nervous system (CNS)—resident macrophages called microglia [14]. The physiological microglia’s role is the active scanning of CNS and responding to tissue injury and infections. The microglia hypothesis of mood disorders describes the microglia’s over-activation in the brain regions controlling mood and cognition, e.g., amygdala, hippocampus and prefrontal cortex [15,16]. In bipolar disorder, immune disturbances underlie the disrupted white matter microstructure and functional connectivity. Higher inflammatory cytokines, higher macrophage/monocyte inflammatory activation patterns and reduced T cell activity correlate with white matter disruption [17,18,19]. High peripheral levels of pro-inflammatory cytokines, reflecting M1 macrophages, also correlate with poor antidepressant response [18]. Interestingly, a study by Benros et al. revealed that autoimmune and infectious diseases increases the risk of future mood disorder diagnosis [20]. Therefore, research on the role of microglia activation may be a promising direction to take in the studies on mood disorders pathogenesis.
2.2. Cytokines as Markers of Microglial Activation
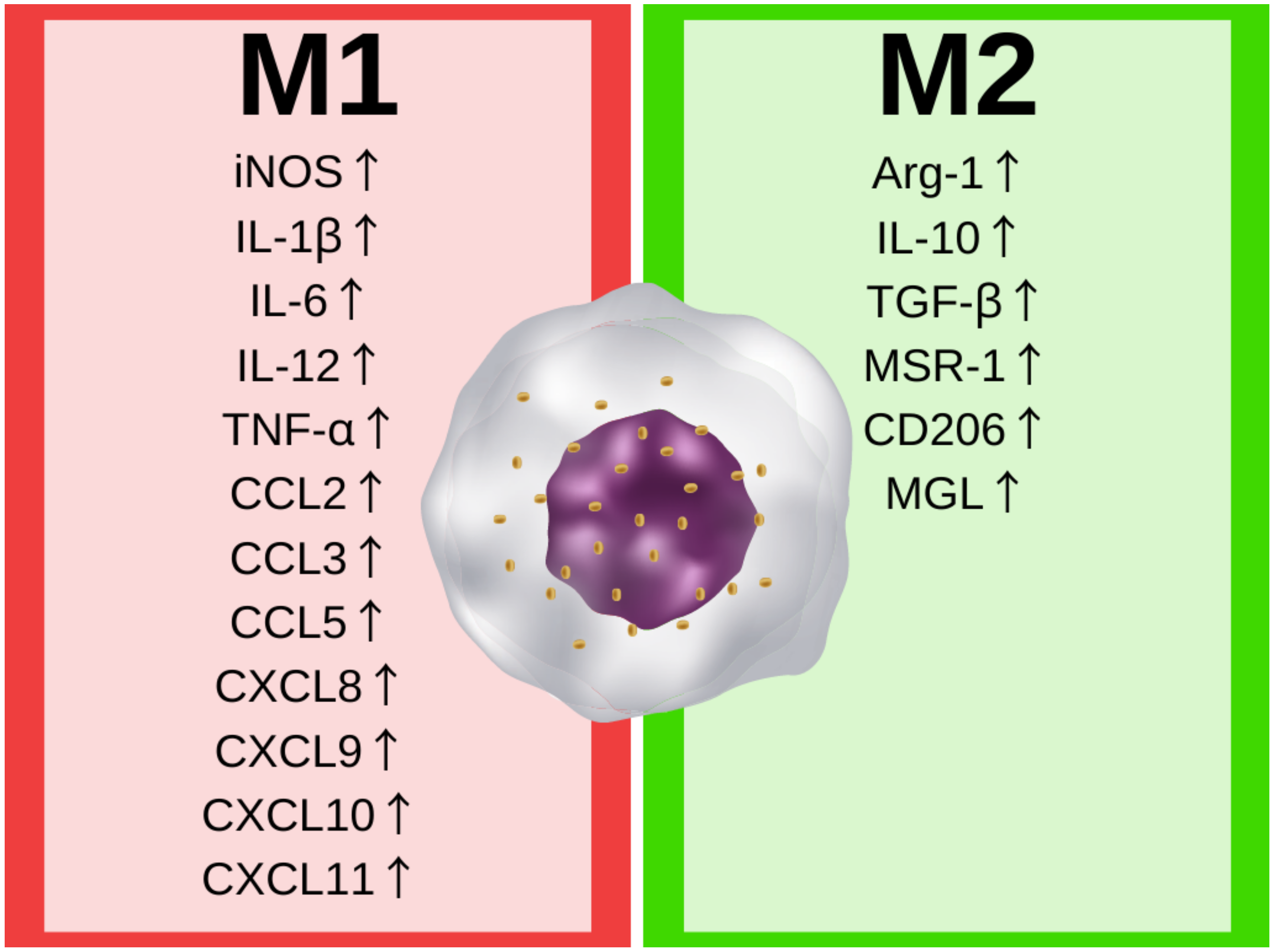
Macrophages and their counterparts are involved in the innate immune response in the central nervous system. The microglia—resident macrophages of the CNS—account for about 5–10% of the brain cells, and are the most common immune cells in the CNS [21]. The central physiological role of microglia is to maintain homeostasis by the induction and resolution of inflammation in response to exogenous (viral or bacterial) or endogenous (DNA, RNA, protein) signals by secreting several soluble factors, such as cytokines, chemokines, and neurotrophic factors, necessary for the CNS immune response and tissue repair processes [22,23]. The stimuli are recognized by pattern-recognition receptors (PRRs), the protein receptors on the microglia. The recognition of endogenous danger molecules—damage-associated molecular patterns (DAMPs)—or exogenous molecules frequently found in pathogens—pathogen-associated molecular patterns (PAMPs)—activates the microglia cells’ mechanism of adaption to stimuli, resulting in changes in their phenotype. The microglia phenotype can be generally assigned to the well-known M1/M2 macrophages paradigm. Classically activated (M1) macrophages are characterized by the secretion of pro-inflammatory cytokines and their response to the presence of a potential pathogen, while on the other hand, alternatively activated (M2) macrophages are involved in tissue repair and regeneration [22,24]. The two main phenotypes of macrophages are generally characterized by pathways regulating the metabolism of arginine. M1 macrophages upregulate inducible nitric oxide synthase (iNOS) expression, leading to L-arginine’s conversion to nitric oxide (NO) that can promote tissue injury at cytotoxic levels. Alternatively activated (or M2) macrophages upregulate arginase-1 (Arg-1), and metabolize L-arginine to L-citrulline, which results in the production of prolines and polyamines promoting tissue repair, proliferation, and the synthesis of extracellular matrix [22]. However, changes in arginine metabolism lead to important expression and secretion differences between phenotypes. The main differences in the secreted immune factors and expressed genes are shown in Figure 1.
The classical activation of macrophages is induced by pathogens (by peptidoglycans, lipopolysaccharide, or flagellin on the surface of pathogens). The recognition of pathogens, or pathogen-like stimuli, induces the expression of pro-inflammatory interleukins, including, amongst others, IL-1β, IL-6, IL-12, TNF-α, CCL2 (or Monocyte Chemoattractant Protein 1 [MCP-1]), CCL3 (or Macrophage Inflammatory Protein 1-α [MIP-1-α]), CCL5, CXCL8 (or IL-8), CXCL9 CXCL10 and CXCL11 [22,25]. These cytokines lead to the further activation of the immune system, e.g., interleukin 12 stimulates T helper 1 (Th1) cell development and the secretion of IFN-γ that perpetuates the phenotype of M1 macrophages [24].
In contrast, the alternative activation of macrophages in the M2 phenotypes is caused by IL-4 produced by T helper 2 (Th2), prostaglandins, apoptotic cells, or IL-10, which upregulate Arg-1 expression and activity. Moreover, the M2 macrophages are characterized by the production of anti-inflammatory cytokines, e.g., IL-10 and transforming growth factor-beta (TGF-β). The M2 phenotype leads to a reduction in inflammation, and promotes tissue regeneration and elevated expressions of scavenger (MSR-1), mannose (CD206), and galactose-type (MGL) receptors. [24,26]. However, the M2 phenotype is not homogenous in the human body, and new conceptions defining the phenotype via several subtypes, depending on cell environment and activity, are receiving increasing attention [27].
Recent findings in the molecular biology and immunology of mood disorders have supported an association between the inflammatory responses of the immune system and macrophages’ phenotype switch upon microglial activation, as well as an increased risk of mood disorders. Studies showed altered levels of cytokines and chemokines associated with macrophage polarization in both MDD and BD measured in peripheral blood or the central nervous system (cerebrospinal fluid). The results of the studies analyzing the pro- and anti-inflammatory cytokines levels in mood disorders (major depressive disorder and bipolar disorder), either during manic or depressive episodes or in a euthymic state, are summarized in Table 1.
Despite a large number of studies and meta-analyses comparing the levels of inflammatory markers during the course of mood disorders, the data are still inconsistent. The reason for heterogeneity in these results may be the common use of a cross-sectional model instead of a prospective study, revealing the changes in the patient’s inflammatory markers’ shift, as well as the heterogeneity between the studies’ designs (e.g., different sample sizes, populations and ethnic groups, diagnostic criteria, disease duration or provided pharmacotherapy). Interestingly, some studies revealed the upregulation of both pro- and anti-inflammatory markers in differently polarized macrophages. Further comparison of the peripheral and central levels of pro- and anti-inflammatory cytokines in the context of the disease state, as well as normotymic drugs and comorbid disorders, seems essential to better understand the inflammation shifts in the course of the mood disorders.
The inconsistencies were also observed in brain samples and were recently summarized by a systematic review by Giridharan et al. [54]. They analyzed the data from brain inflammation studies evaluating microglia, astrocytes, cytokines, chemokines, adhesion molecules, and other inflammatory markers in postmortem BD brain samples. They reported that out of 51 studies in brain tissue, most of them confirmed neuroinflammation in BD postmortem samples. However, a large number of these studies did not evaluate the presence of infiltrating peripheral immune cells in the CNS parenchyma, cytokines levels, and microglia activation in the same postmortem brain sample. Moreover, the heterogeneity in postmortem samples regarding postmortem interval, studied brain area, age at diagnosis, the treatment, cause of death and comorbidities might underlie the observed inconsistencies. Therefore, more prospective studies, including precisely defined phenotypes, are necessary to elucidate how changing inflammatory marker levels influence the course of mood disorders. More in-depth research on the activated microglia phenotype would also be valuable in postmortem brain samples analyzed by cell type and brain region, to confirm the presence of neuroinflammation in the pathogenesis of mood disorders.
2.3. Energy Metabolism Markers of Microglia Activation
Mitochondria are intracellular organelles that regulate energy metabolism, the production of reactive oxygen species (ROS), cellular signaling, Ca2+ homeostasis, and apoptosis. Studies revealed that the mitochondrial dysfunction and damage play a crucial role in the development of neurodegenerative diseases, including amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease or multiple sclerosis, by enhancing innate and adaptive immune responses that result in neuroinflammation [55]. The dysfunction of mitochondria and impaired energy metabolism were also reported in the pathophysiology of mood disorders such as bipolar disorder and major depressive disorder [56,57,58,59]. The mitochondrial hypothesis suggests that changes in the cell energy metabolism activate inflammasome, increase cytokines production and further lead to apoptotic cell death. The changes underlying mitochondrial dysfunction in bipolar disorder include decreased ATP synthesis, disrupted electron transport chain (ETC), a switch from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, increased lactate levels and oxidative stress markers, and reduced antioxidant capacity (decreased glutathione peroxidase and superoxide dismutase) [60,61,62,63,64]. Mitochondrial dysfunction and increased lactate levels were also observed recently in an adolescent population of BD patients, suggesting that the impediment of mitochondrial oxidative phosphorylation and a shift towards anaerobic respiration and lactate production occur at the early stage of disease development [65]. These alterations further support the role of mitochondrial dysfunction leading to a shift towards anaerobic metabolism, and an associated elevated risk for cellular injury. These studies also suggested that, taking into account the similarities between mitochondria and bacteria, it is possible that the mitochondrial contents released to the cytoplasm of microglial cells are recognized as pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), which further activate pattern recognition receptor (PRR) signaling, thus enhancing inflammation [66].
The underlying cause of the disturbed energy metabolism and altered mitochondrial activity may be the microglia activation itself, which results in polarization towards either the M1 or the M2 phenotype. The M2 microglia (considered non-activated microglia) are characterized by low glucose consumption and oxidative phosphorylation for ATP synthesis. The pro-inflammatory M1 phenotype contributes to the switch from oxidative to glycolytic metabolism, with high glucose uptake leading to lactate, NO and citrulline production with activation of the penta phosphate pathway (PPP) [67,68]. These changes in energy metabolism increase pro-inflammatory interleukins levels and enhance the production of ROS that are harmful to all cellular components, further progressing the inflammation and energy metabolic changes.
Translocator protein (TSPO) is one of the mitochondrial neuroimmune markers of microglia activation. TSPO ligands are used in positron emission tomography (PET) imaging as a biomarker of microglia activation in neurodegenerative diseases [69,70,71,72]. Taking into account the neuroinflammatory theory of mood disorders that assumes that the activation of microglia contributes to disease phenotype, we suggest that this protein may also be an important marker of neuroinflammation in mood disorders. TSPO is an 18 kDa protein localized on the outer mitochondrial membrane. It is responsible for mitochondrial homeostasis by direct interaction with the voltage-dependent anion channel (VDAC) and the adenine nucleotide transporter (ANT), thus contributing to energy production, Ca2+ signaling, ROS generation and apoptosis [73,74]. Recent studies have described the effect of an elevated expression of TSPO and VDAC on both gene and protein levels in the group of BD and MDD patients, as compared to controls [40,75,76]. Scaini et al. indicated that the upregulation of TSPO-related proteins (TSPO and VDAC) is one of the causes of the inflammasome activation of the NLR family pyrin domain containing 3 (NLRP3), by increasing ROS production and Ca2+ dysregulation in bipolar disorder patients [75]. Inflammasome complexes are multi-domain, cytoplasmatic protein complexes composed of three components: a cytosolic pattern recognition receptors (PRR) (detecting molecules typical for the pathogenic activity), caspase-1, and an adaptor protein (responsible for caspase activation) [77]. Inflammasome complexes are activated by a wide range of stimuli, including, e.g., bacterial infections, extracellular ATP, but also mitochondrial content. This activation stimulates NF-κB signaling, which induces NLRP3 inflammasome complex assembly and activates caspase 1 [77]. Activated caspase 1 promotes the cleavage of pro-interleukins into mature interleukins (IL-1β and IL-18) [77,78], and their secretion, leading to chronic inflammation [75,79,80]. A study by Alcocer-Gómez et al. described the increased level of mitochondrial ROS and the increased gene expression of NLRP3 and caspase-1 in the MDD patients not treated with antidepressants [81]. Further in vivo studies by Pan et al. described the upregulation of NLRP3, NF-κB, caspase-1 and IL-1β gene expression after inducing depressive-like behavior via a chronic unpredictable mild stress protocol. These alterations in gene expression levels were restored after the administration of antidepressant treatment [82]. Further studies by Wong et al. showed that the inhibition of inflammasome signaling by caspase-1 knock-out reduced depression-like behavior in response to chronic restraint stress in animals, supporting the important role of NLRP3 inflammasome in the pathogenesis of depressive-like behavior [83].
Although changes in energy metabolism in microglia seem to be an important factor in the development of neuroinflammation, more studies are necessary to establish if these metabolic shifts are the cause or the result of the inflammation.
3. Lithium Influences Inflammation and Energy Metabolism
Lithium is the smallest monovalent cation used in the pharmacotherapy of psychiatric disorders. For over 70 years, lithium has been an efficient mood stabilizer with proven anti-manic and antidepressant properties [84]. Despite proven long-term efficacy in treating acute episodes of mania and depression, as well as prophylactic properties, the exact molecular mechanism of action is not fully understood. Despite the well-known modes of lithium action, including GSK-3β inhibition and the phosphatidylinositol pathway, previous studies have also indicated its immunomodulatory potential, suggesting that lithium may decrease the levels of pro-inflammatory cytokines in mood disorders, either in the periphery or in the central nervous system, and that these changes are related to its therapeutic efficacy [85,86,87].
The best-known mode of action for lithium is the inhibition of glycogen synthase kinase-3β (GSK-3β), ubiquitously expressed in the tissues and associated with inflammation. The GSK-3β can affect a variety of transcription factors, including NF-κB, which is crucial for innate immune response and regulates cytokines production [88]. The decreased transcriptional activity of NF-κB by GSK-3β inhibition—including lithium treatment—reduces the production of pro-inflammatory mediators such as IL-1β, IFN-γ, IL-6, and MCP-1, associated with M1 macrophages, and increases the production of anti-inflammatory cytokines [88,89,90]. Another potential downstream target of GSK-3β is a signal transducer and activator of transcription (STAT). A previous study showed that the inhibition of GSK-3β by lithium resulted in the reduced activation of STAT and the reduced secretion of pro-inflammatory cytokines [91,92]. Martin et al. [89] showed that the inhibition of GSK-3β upregulated anti-inflammatory IL-10 production and reduced pro-inflammatory cytokines, such as IL-1β, IL-6, TNF, IL-12, and IFNγ, associated with M1 macrophages by affecting Toll-like receptor-mediated production. The clinical study by Rapaport et al. reported decreased levels of inflammatory markers in serum in rapid cycling bipolar patients after 30 days of lithium treatment [86]. Boufidou et al. presented a study comparing the cytokine levels (IL-2, IL-6, IL-10 and IFN-gamma) in the peripheral blood of healthy volunteers and lithium-naive BD patients before and after lithium treatment [87]. Although they did not observe differences in the cytokine levels at the beginning of lithium treatment, after three months on lithium, the patients presented a significant decrease in all analyzed cytokines compared to controls. A study analyzing ex vivo interleukin IL-1β and IL-6 secretion in BD patient-derived monocytes stimulated with LPS showed that lithium treatment decreased the production of IL-6 compared to the non-treated control [93]. Recently, Wu et al. [94] analyzed immunophenotypes of BD patients and found that they had significantly higher percentages of total T cells, CD4+ T cells, activated B cells, and monocytes than healthy controls, whereas the treatment of patient-derived PBMCs with lithium in vitro increased the percentage of CD14+ monocytes and dendritic cells. The authors suggested that lithium plays an immunomodulatory role in CD14+ monocytes and dendritic cells [94]. A study from our center showed that long-term lithium decreased the expression of glial and pluripotency markers (in particular Oct-4, Sox-2, GFAP and Olig1) in peripheral blood, suggesting that lithium can reduce ongoing inflammatory processes in bipolar disorder [95].
The observations from clinical studies were also confirmed in vitro. The study by Nahman et al. [96] showed that pre-treatment of the rat primary glial cells with lithium suppressed LPS-induced inflammation, including reduced levels of M1 macrophages markers such as TNF-α, IL-1β and iNOS expression. Although the results indicated the anti-inflammatory potential of lithium, they were obtained by using concentrations higher than the therapeutic dose [96]. A study by Yuskaitis et al. [90] analyzed the influence of lithium and other GSK-3β inhibitors on microglia activation and migration, and they showed that lithium decreased microglial migration in mouse BV-2 cells as well as attenuating the injury-induced migration of microglia in situ in hippocampal mouse slices. Lithium also decreased inflammatory markers IL-6, NO and iNOS in microglial cells stimulated with LPS, showing the anti-inflammatory potential of lithium [90]. More recently, Dong et al. [97] confirmed that lithium treatment influenced microglial activation. The pretreatment of primary microglial cells with lithium followed by LPS stimulation inhibited LPS-induced microglial activation and pro-inflammatory cytokine production. This suppression resulted from LPS-induced toll-like receptor 4 (TLR4) downregulation and proceeded via inhibition of the PI3K/Akt/FoxO1 signaling pathway [97].
The anti-inflammatory effect of lithium was also confirmed in the recent animal study by Adams et al. [98]. They observed that chronic lithium treatment (12 weeks) decreased pro-inflammatory cytokines’ levels (IL-1β, IL-6 and RANTES) in plasma and the orbitofrontal cortex, leading to reduced motor impulsivity in rats. The authors suggested that improved impulse control deficits in patients treated with lithium may result from the reduced influence of pro-inflammatory signaling on neuronal activity.
The anti-inflammatory potential of lithium was also considered in a recent study of the olanzapine (OLZ) and lithium co-treatment. Olanzapine is a second-generation antipsychotic drug with side effects, including metabolic alteration associated with low-grade systemic inflammation. The in vitro study by Fernandes et al. [99] used the activated RAW 267.7 macrophages treated with OLZ or OLZ and lithium, and evaluated the oxidation and inflammation at the gene and protein expression level. The macrophages treated only with OLZ showed increased proliferation and higher levels of oxidative markers and pro-inflammatory cytokines, with reduced anti-inflammatory IL-10 level. Co-treatment with lithium resulted in a significantly reduced level of oxidative and inflammatory markers, and an increased IL-10 level.
Despite studies describing the anti-inflammatory role of lithium, some pieces of evidence have showed also its pro-inflammatory action. Petersein et al. analyzed the blood collected from healthy subjects stimulated ex vivo with phytohemagglutinin (PHA), murine anti-human CD3 monoclonal antibody OKT3 and 5C3 monoclonal antibody (OKT3/5C3), as well as non-stimulated blood, followed by incubation with different psychotropic drugs combinations: lithium without antidepressant, co-treatment with lithium and antidepressant, antidepressant without lithium and no drug as a negative control. They showed that lithium alone or in combination with antidepressants increased several pro-inflammatory cytokines, including IL-1β, IL-6 and TNF-α, independently of stimulation [100]. However, it is not known if the same pro-inflammatory action would also be observed in the blood collected from mood disorder patients.
In regard to energy metabolism and mitochondrial dysfunction, the early in vitro study in cultured cerebellar granule neurons showed that lithium increased the mitochondrial calcium concentrations that desensitized mitochondria to depolarization [101]. Lithium was also described as a modulator of ETC and OXPHOS pathway activity, and enhanced the oxidative phosphorylation in human brain tissue [102]. A previous clinical study reported increased complex I activity and enhanced oxidative phosphorylation after lithium treatment, and this increase positively correlated with plasma lithium levels in BD patients [103].
On the other hand, the recent in vitro study analyzing the effects of different mood stabilizers on pig brain mitochondria showed no inhibitory effects of lithium on complex I- and II-linked respiration [104]. However, in cultured human neurons (SH-SY5Y), chronic lithium treatment (25–50 weeks) increased resistance to oxidative stress, and enhanced glucose consumption and glycolysis activity [105]. These results suggested the neuroprotective effect of chronic lithium exposure via the improved resistance of neurons to oxidative stress. A recent in vitro study performed in two rodent cell lines (murine microglial N9 and rat pheochromocytoma PC12) showed that lithium prevented ATP-induced cell death in PC12 neuronal cells, but in the N9 microglial cell line lithium treatment did not prevent the ATP-induced microglial switch to the M1 phenotype [106].
Previous preclinical studies showed that lithium reversed hyperactivity and risk-taking behavior after repeated injections of amphetamine [107,108], and thus was considered a validated model of mania in rats [109,110,111]. Therefore, studies using this model of mania confirmed that lithium prevented the amphetamine-induced inhibition of ETC complexes I, II, III, and IV in the hippocampus, striatum and prefrontal cortex. Lithium pre-treatment also attenuated hyperactive behavior, suggesting that restoring mitochondrial functioning improved the behavioral changes induced by amphetamine [112].
Recent clinical studies confirmed changes in OXPHOS and ETC activity. The co-expression network analysis of RNA-seq data from the whole blood of lithium-treated BD patients reported the significant over-representation of genes involved in mitochondrial functioning (specifically ETC and OXPHOS related genes), and the expression of these genes was lower in better lithium responders relative to poor lithium responders, thus indicating that lithium is involved in the regulation of these mitochondrial functioning pathways [113]. A recent study by Machado-Vieira et al. suggested that a shift from aerobic to anaerobic metabolism underlies mitochondrial dysfunction and energy metabolism alterations in bipolar disorder. Using proton magnetic resonance spectroscopy (H-MRS), they measured brain lactate in vivo in the cingulate cortex of BD patients during a depressive episode and after six weeks of lithium therapy to evaluate mitochondrial and metabolic dysfunction. They showed that during the depressive episode, lactate production increased and mitochondrial metabolism changed towards anaerobic glycolysis, whereas lithium therapy restored the lactate level in the brain and improving mitochondrial respiration [114]. Those results confirmed the important role of lithium in mood disorder therapy by suppressing the CNS neuroinflammation, not only as a potential anti-inflammatory agent, but also via its potential impact on the brain energy metabolism. Despite the long history of using lithium as a mood stabilizer, its influence on the immune system and energy metabolism is still not fully understood. Further studies considering its immunomodulatory potential may shed light on its role in the pathogenesis of mood disorders.
4. Anti-Manic and Antidepressant Properties of Anti-Inflammatory Drugs
The emerging evidence of neuroinflammation in the pathophysiology of mood disorders has led to several meta-analyses describing the efficacy of anti-inflammatory agents therapy for MDD and BD patients. A recent meta-analysis including 1610 MDD patients from 26 randomized controlled trials suggested the potential use of antidepressants in patients receiving non-steroidal anti-inflammatory drugs (NSAIDs) compared to the placebo group [115]. Moreover, subgroup analysis indicated a significant reduction in the severity of depressive symptoms in groups treated via monotherapy, as well as via adjunctive treatment with anti-inflammatory agents [115]. Another meta-analysis investigating the effect of adjunct treatment with NSAIDs, omega-3 fatty acids, N-acetylcysteine and pioglitazone on bipolar disorder indicated a significant effect on reducing depressive symptoms. However, these anti-inflammatory drugs were not effective in reducing mania symptoms after adjunctive anti-inflammatory therapy [116]. Another meta-analysis by Husain et al. [117] showed the beneficial effect of anti-inflammatory medications—with NSAIDs, N-acetyl cysteine, cytokine inhibitors or minocycline—on manic and depressive symptoms measured as post-treatment symptoms’ severity; however, the authors emphasized the small number of studies investigating the effect of the anti-inflammatory treatment of mania [117].
Based on the results of the presented meta-analyses, further studies analyzing the effects of anti-inflammatory agents on the course of mood disorders, particularly on the severity of manic episodes, seem necessary in order to better understand the role of inflammation in mood disorders.
5. Conclusions
Alterations of the pro- and anti-inflammatory markers underlie the changes in the immune system observed in mood disorders. Increased levels of M1-associated pro-inflammatory cytokines and chemokines indicate the potential role of microglia polarization as a pathological factor of mood disorders. The energy metabolic changes in mood disorders are another source of immune system alterations, leading to the state of neuroinflammation and microglial cells’ pro-inflammatory polarization. However, few studies so far have deeply investigated the energy metabolism-related pathway and its impact on microglia activation in the course of mood disorders. Lithium, a well-described and effective mood stabilizer, showed an immunomodulatory effect in previous studies. However, there is a lack of studies correlating the cytokines and metabolic-related genes levels with lithium treatment in the clinical setting. Therefore, more prospective studies evaluating the effects of lithium treatment, and the levels of neuroinflammatory and energy metabolism markers described above, may shed light on the molecular etiology and the course of mood disorders, as well as lithium’s mode of action.
Author Contributions
Conceptualization, K.S. and A.S.; writing—original draft preparation, K.S.; writing—review and editing, K.S. and A.S.; visualization, K.S.; supervision, A.S.; funding acquisition, A.S. All authors have read and agreed to the published version of the manuscript.
Funding
The study was funded by The National Science Centre in Poland, grant number 2016/21/B/NZ5/00148.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Phillips, M.L.; Kupfer, D.J. Bipolar disorder 2—Bipolar disorder diagnosis: Challenges and future directions. Lancet 2013, 381, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Chesney, E.; Goodwin, G.M.; Fazel, S. Risks of all-cause and suicide mortality in mental disorders: A meta-review. World Psychiatry 2014, 13, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates; CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2017; pp. 1–24. [Google Scholar]
- Grande, I.; Berk, M.; Birmaher, B.; Vieta, E. Bipolar disorder. Lancet 2016, 387, 1561–1572. [Google Scholar] [CrossRef]
- Vieta, E.; Berk, M.; Schulze, T.G.; Calabrese, J.R.; Gao, K.; Miskowiak, K.W.; Grande, I. Bipolar disorders. Nat. Rev. Dis. Prim. 2018, 4, 18008. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Muzina, D.J.; Kemp, D.E.; Blank, D.; Woldeyohannes, H.O.; Lofchy, J.; Soczynska, J.K.; Banik, S.; Konarski, J.Z. Bipolar disorder and suicide: Research synthesis and clinical translation. Curr. Psychiatry Rep. 2008, 10, 66–72. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Konarski, J.Z.; Yatham, L.N. Comorbidity in bipolar disorder: A framework for rational treatment selection. Hum. Psychopharmacol. 2004, 19, 369–386. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.A.; Thomsen, C. The role of the innate immune system in psychiatric disorders. Mol. Cell. Neurosci. 2013, 53, 52–62. [Google Scholar] [CrossRef]
- Stuart, M.J.; Baune, B.T. Chemokines and chemokine receptors in mood disorders, schizophrenia, and cognitive impairment: A systematic review of biomarker studies. Neurosci. Biobehav. Rev. 2014, 42, 93–115. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; Suy, E.; Vandervorst, C.; De Jonckheere, C.; Raus, J. Immune disturbances during major depression: Upregulated expression of interleukin-2 receptors. Neuropsychobiology 1990, 24, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.S. The macrophage theory of depression. Med. Hypotheses 1991, 35, 298–306. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stertz, L.; Magalhães, P.V.S.S.; Kapczinski, F. Is bipolar disorder an inflammatory condition? the relevance of microglial activation. Curr. Opin. Psychiatry 2013, 26, 19–26. [Google Scholar] [CrossRef]
- Frick, L.R.; Williams, K.; Pittenger, C. Microglial dysregulation in psychiatric disease. Clin. Dev. Immunol. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Becking, K.; Haarman, B.C.M.; Grosse, L.; Nolen, W.A.; Claes, S.; Arolt, V.; Schoevers, R.A.; Drexhage, H.A. The circulating levels of CD4+ t helper cells are higher in bipolar disorder as compared to major depressive disorder. J. Neuroimmunol. 2018, 319, 28–36. [Google Scholar] [CrossRef]
- Benedetti, F.; Poletti, S.; Hoogenboezem, T.A.; Locatelli, C.; de Wit, H.; Wijkhuijs, A.J.M.; Colombo, C.; Drexhage, H.A. Higher baseline proinflammatory cytokines mark poor antidepressant response in bipolar disorder. J. Clin. Psychiatry 2017, 78, e986–e993. [Google Scholar] [CrossRef]
- Snijders, G.; Schiweck, C.; Mesman, E.; Grosse, L.; De Wit, H.; Nolen, W.A.; Drexhage, H.A.; Hillegers, M.H.J. A dynamic course of T cell defects in individuals at risk for mood disorders. Brain Behav. Immun. 2016, 58, 11–17. [Google Scholar] [CrossRef]
- Benros, M.E.; Waltoft, B.L.; Nordentoft, M.; Ostergaard, S.D.; Eaton, W.W.; Krogh, J.; Mortensen, P.B. Autoimmune diseases and severe infections as risk factors for mood disorders a nationwide study. JAMA Psychiatry 2013, 70, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.U.; De Vellis, J. Microglia in health and disease. J. Neurosci. Res. 2005, 81, 302–313. [Google Scholar] [CrossRef]
- Mills, C.D.; Ley, K. M1 and M2 macrophages: The chicken and the egg of immunity. J. Innate Immun. 2014, 6, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ka, M.B.; Daumas, A.; Textoris, J.; Mege, J.-L. Phenotypic diversity and emerging new tools to study macrophage activation in bacterial infectious diseases. Front. Immunol. 2014, 5, 500. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.K.; Needham, D. Targeted drug delivery. Expert Opin. Ther. Pat. 1999, 9, 1499–1513. [Google Scholar] [CrossRef]
- Roszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2015, 2015, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Brunoni, A.R.; Supasitthumrong, T.; Teixeira, A.L.; Vieira, E.L.; Gattaz, W.F.; Benseñor, I.M.; Lotufo, P.A.; Lafer, B.; Berk, M.; Carvalho, A.F.; et al. Differences in the immune-inflammatory profiles of unipolar and bipolar depression. J. Affect. Disord. 2020, 262, 8–15. [Google Scholar] [CrossRef]
- Fiedorowicz, J.G.; Prossin, A.R.; Johnson, C.P.; Christensen, G.E.; Magnotta, V.A.; Wemmie, J.A. Peripheral inflammation during abnormal mood states in bipolar i disorder. J. Affect. Disord. 2015, 187, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Modabbernia, A.; Taslimi, S.; Brietzke, E.; Ashrafi, M. Cytokine alterations in bipolar disorder: A meta-analysis of 30 studies. Biol. Psychiatry 2013, 74, 15–25. [Google Scholar] [CrossRef]
- Miklowitz, D.J.; Portnoff, L.C.; Armstrong, C.C.; Keenan-Miller, D.; Breen, E.C.; Muscatell, K.A.; Eisenberger, N.I.; Irwin, M.R. Inflammatory cytokines and nuclear factor-kappa B activation in adolescents with bipolar and major depressive disorders. Psychiatry Res. 2016, 241, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Oskooei, V.K.; Omrani, M.D.; Taheri, M. Dysregulation of cytokine coding genes in peripheral blood of bipolar patients. J. Affect. Disord. 2019, 256, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Domínguez, A.; Hernández, M.E.; Berlanga, C.; Gutiérrez-Mora, D.; Moreno, J.; Heinze, G.; Pavón, L. Immune variations in bipolar disorder: Phasic differences. Bipolar Disord. 2007, 9, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ho, R.C.M.; Mak, A. Interleukin (IL)-6, tumour necrosis factor alpha (TNF-α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: A meta-analysis and meta-regression. J. Affect. Disord. 2012, 139, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.A.; Freitas, T.H.; Maes, M.; de Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N.; et al. Peripheral cytokine and chemokine alterations in depression: A meta-analysis of 82 studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Cassano, P.; Bui, E.; Rogers, A.H.; Walton, Z.E.; Ross, R.; Zeng, M.; Nadal-Vicens, M.; Mischoulon, D.; Baker, A.W.; Keshaviah, A.; et al. Inflammatory cytokines in major depressive disorder: A case-control study. Aust. N. Z. J. Psychiatry 2017, 51, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Munkholm, K.; Braüner, J.V.; Kessing, L.V.; Vinberg, M. Cytokines in bipolar disorder vs. healthy control subjects: A systematic review and meta-analysis. J. Psychiatr. Res. 2013, 47, 1119–1133. [Google Scholar] [CrossRef]
- Enache, D.; Pariante, C.M.; Mondelli, V. Markers of central inflammation in major depressive disorder: A systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav. Immun. 2019, 81, 24–40. [Google Scholar] [CrossRef]
- Rowland, T.; Perry, B.I.; Upthegrove, R.; Barnes, N.; Chatterjee, J.; Gallacher, D.; Marwaha, S. Neurotrophins, cytokines, oxidative stress mediators and mood state in bipolar disorder: Systematic review and meta-analyses. Br. J. Psychiatry 2018, 213, 514–525. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.M.; Scully, P.; Scott, L.V.; Dinan, T.G. Cytokine profiles in bipolar affective disorder: Focus on acutely ill patients. J. Affect. Disord. 2006, 90, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, V.; Tuglu, C.; Gorgulu, Y.; Kunduracilar, H.; Uyanik, M.S. Assessment of cytokine levels and hs-CRP in bipolar I disorder before and after treatment. Psychiatry Res. 2015, 228, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Jung, H.G.; Myint, A.M.; Kim, H.; Park, S.H. Imbalance between pro-inflammatory and anti-inflammatory cytokines in bipolar disorder. J. Affect. Disord. 2007, 104, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.-M.; Su, T.-P.; Li, C.-T.; Tsai, S.-J.; Chen, M.-H.; Tu, P.-C.; Chiou, W.-F. Comparison of pro-inflammatory cytokines among patients with bipolar disorder and unipolar depression and normal controls. Bipolar Disord. 2015, 17, 269–277. [Google Scholar] [CrossRef]
- Munkholm, K.; Vinberg, M.; Vedel Kessing, L. Cytokines in bipolar disorder: A systematic review and meta-analysis. J. Affect. Disord. 2013, 144, 16–27. [Google Scholar] [CrossRef]
- Leighton, S.P.; Nerurkar, L.; Krishnadas, R.; Johnman, C.; Graham, G.J.; Cavanagh, J. Chemokines in depression in health and in inflammatory illness: A systematic review and meta-analysis. Mol. Psychiatry 2018, 23, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Drexhage, R.C.; Hoogenboezem, T.H.; Versnel, M.A.; Berghout, A.; Nolen, W.A.; Drexhage, H.A. The activation of monocyte and T cell networks in patients with bipolar disorder. Brain Behav. Immun. 2011, 25, 1206–1213. [Google Scholar] [CrossRef]
- Isgren, A.; Sellgren, C.; Ekman, C.J.; Holmén-Larsson, J.; Blennow, K.; Zetterberg, H.; Jakobsson, J.; Landén, M. Markers of neuroinflammation and neuronal injury in bipolar disorder: Relation to prospective clinical outcomes. Brain Behav. Immun. 2017, 65, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, P.; Bellani, M.; Isola, M.; Bergami, A.; Marinelli, V.; Dusi, N.; Rambaldelli, G.; Tansella, M.; Maria Finardi, A.; Martino, G.; et al. Increased M1/decreased M2 signature and signs of Th1/Th2 shift in chronic patients with bipolar disorder, but not in those with schizophrenia. Transl. Psychiatry 2014, 4, e406-e406. [Google Scholar] [CrossRef] [Green Version]
- Brietzke, E.; Kauer-Sant’Anna, M.; Teixeira, A.L.; Kapczinski, F. Abnormalities in serum chemokine levels in euthymic patients with bipolar disorder. Brain Behav. Immun. 2009, 23, 1079–1082. [Google Scholar] [CrossRef]
- Barbosa, I.G.; Rocha, N.P.; Bauer, M.E.; De Miranda, A.S.; Huguet, R.B.; Reis, H.J.; Zunszain, P.A.; Horowitz, M.A.; Pariante, C.M.; Teixeira, A.L. Chemokines in bipolar disorder: Trait or state? Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Parisi, M.M.; Colombo, R.; Becker, M.; Fries, G.; Ascoli, B.M.; Géa, L.P.; Kauer-Sant’anna, M.; Kapczinski, F.; Klamt, F.; et al. Depression and mania induce pro-inflammatory activation of macrophages following application of serum from individuals with bipolar disorder. Clin. Psychopharmacol. Neurosci. 2018, 16, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayana, P.; Colpo, G.D.; Simões, L.R.; Giridharan, V.V.; Teixeira, A.L.; Quevedo, J.; Barichello, T. A systemic review of evidence for the role of inflammatory biomarkers in bipolar patients. J. Psychiatr. Res. 2017, 92, 160–182. [Google Scholar] [CrossRef] [PubMed]
- Garabadu, D.; Agrawal, N.; Sharma, A.; Sharma, S. Mitochondrial metabolism: A common link between neuroinflammation and neurodegeneration. Behav. Pharmacol. 2019, 30, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Cikankova, T.; Sigitova, E.; Zverova, M.; Fisar, Z.; Raboch, J.; Hroudova, J. Mitochondrial Dysfunctions in Bipolar Disorder: Effect of the Disease and Pharmacotherapy. CNS Neurol. Disord. Drug Targets 2016, 16, 176–186. [Google Scholar] [CrossRef]
- Kato, T. The role of mitochondrial dysfunction in bipolar disorder. Drug News Perspect. 2006, 19, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; Romay-Tallon, R.; Brymer, K.J.; Caruncho, H.J.; Kalynchuk, L.E. Mitochondria and mood: Mitochondrial dysfunction as a key player in the manifestation of depression. Front. Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef]
- Culmsee, C.; Michels, S.; Scheu, S.; Arolt, V.; Dannlowski, U.; Alferink, J. Mitochondria, microglia, and the immune system—How are they linked in affective disorders? Front. Psychiatry 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Konradi, C.; Eaton, M.; MacDonald, M.L.; Walsh, J.; Benes, F.M.; Heckers, S. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch. Gen. Psychiatry 2004, 61, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Valvassori, S.S.; Bavaresco, D.V.; Feier, G.; Cechinel-Recco, K.; Steckert, A.V.; Varela, R.B.; Borges, C.; Carvalho-Silva, M.; Gomes, L.M.; Streck, E.L.; et al. Increased oxidative stress in the mitochondria isolated from lymphocytes of bipolar disorder patients during depressive episodes. Psychiatry Res. 2018, 264, 192–201. [Google Scholar] [CrossRef]
- Frey, B.N.; Stanley, J.A.; Nery, F.G.; Serap Monkul, E.; Nicoletti, M.A.; Chen, H.H.; Hatch, J.P.; Caetano, S.C.; Ortiz, O.; Kapczinski, F.; et al. Abnormal cellular energy and phospholipid metabolism in the left dorsolateral prefrontal cortex of medication-free individuals with bipolar disorder: An in vivo 1 H MRS study. Bipolar Disord. Suppl. 2007, 9, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.; Renshaw, P.F. Mitochondrial dysfunction in bipolar disorder: Evidence from magnetic resonance spectroscopy research. Mol. Psychiatry 2005, 10, 900–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Dimick, M.K.; Sultan, A.; Duong, A.; Park, S.S.; El Soufi El Sabbagh, D.; Goldstein, B.I.; Andreazza, A.C. Peripheral biomarkers of mitochondrial dysfunction in adolescents with bipolar disorder. J. Psychiatr. Res. 2020, 123, 187–193. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An organelle of bacterial origin controlling inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Gimeno-Bayón, J.; López-López, A.; Rodríguez, M.J.; Mahy, N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J. Neurosci. Res. 2014, 92, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Sucksdorff, M.; Matilainen, M.; Tuisku, J.; Polvinen, E.; Vuorimaa, A.; Rokka, J.; Nylund, M.; Rissanen, E.; Airas, L. Brain TSPO-PET predicts later disease progression independent of relapses in multiple sclerosis. Brain 2020, 143. [Google Scholar] [CrossRef]
- Lavisse, S.; Goutal, S.; Wimberley, C.; Tonietto, M.; Bottlaender, M.; Gervais, P.; Kuhnast, B.; Peyronneau, M.-A.; Barret, O.; Lagarde, J.; et al. Increased microglial activation in patients with Parkinson disease using [18F]-DPA714 TSPO PET imaging. Parkinsonism Relat. Disord. 2021, 82, 29–36. [Google Scholar] [CrossRef]
- Hellwig, S.; Domschke, K. Update on PET imaging biomarkers in the diagnosis of neuropsychiatric disorders. Curr. Opin. Neurol. 2019, 32, 539–547. [Google Scholar] [CrossRef]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent developments in TSPO PET imaging as a biomarker of neuroinflammation in neurodegenerative disorders. Int. J. Mol. Sci. 2019, 20, 3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatliff, J.; Campanella, M. The 18 kDa translocator protein (TSPO): A new perspective in mitochondrial biology. Curr. Mol. Med. 2012, 12, 356–368. [Google Scholar] [CrossRef]
- Bader, S.; Wolf, L.; Milenkovic, V.M.; Gruber, M.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. Differential effects of TSPO ligands on mitochondrial function in mouse microglia cells. Psychoneuroendocrinology 2019, 106, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Barichello, T.; Fries, G.R.; Kennon, E.A.; Andrews, T.; Nix, B.R.; Zunta-Soares, G.; Valvassori, S.S.; Soares, J.C.; Quevedo, J. TSPO upregulation in bipolar disorder and concomitant downregulation of mitophagic proteins and NLRP3 in fl ammasome activation. Neuropsychopharmacology 2018, 44, 1–9. [Google Scholar] [CrossRef]
- Setiawan, E.; Attwells, S.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Xu, C.; Sharma, S.; Kish, S.; Houle, S.; et al. Association of translocator protein total distribution volume with duration of untreated major depressive disorder: A cross-sectional study. Lancet Psychiatry 2018, 5, 339–347. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [Green Version]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction, oxidative stress, and neuroinflammation: Intertwined roads to neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Alcocer-Gómez, E.; de Miguel, M.; Casas-Barquero, N.; Núñez-Vasco, J.; Sánchez-Alcazar, J.A.; Fernández-Rodríguez, A.; Cordero, M.D. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav. Immun. 2014, 36, 111–117. [Google Scholar] [CrossRef]
- Pan, Y.; Chen, X.Y.; Zhang, Q.Y.; Kong, L.D. Microglial NLRP3 inflammasome activation mediates IL-1β-related inflammation in prefrontal cortex of depressive rats. Brain Behav. Immun. 2014, 41, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.L.; Inserra, A.; Lewis, M.D.; Mastronardi, C.A.; Leong, L.; Choo, J.; Kentish, S.; Xie, P.; Morrison, M.; Wesselingh, S.L.; et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol. Psychiatry 2016, 21, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Cade, J.F. Lithium salts in the treatment of psychotic excitement. Med. J. Aust. 1949, 2, 349–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Jope, R.S. Inflammation and lithium: Clues to mechanisms contributing to suicide-linked traits. Transl. Psychiatry 2014, 4, e488. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, M.H.; Guylai, L.; Whybrow, P. Immune parameters in rapid cycling bipolar patients before and after lithium treatment. J. Psychiatr. Res. 1999, 33, 335–340. [Google Scholar] [CrossRef]
- Boufidou, F.; Nikolaou, C.; Alevizos, B.; Liappas, I.A.; Christodoulou, G.N. Cytokine production in bipolar affective disorder patients under lithium treatment. J. Affect. Disord. 2004, 82, 309–313. [Google Scholar] [CrossRef]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor—Mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009, 21, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Jope, R.S. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflamm. 2009, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Rowse, A.L.; Naves, R.; Cashman, K.S.; McGuire, D.J.; Mbana, T.; Raman, C.; de Sarno, P. Lithium controls central nervous system autoimmunity through modulation of IFN-γ signaling. PLoS ONE 2012, 7, e52658. [Google Scholar] [CrossRef] [PubMed]
- Knijff, E.M.; Nadine, B.M.; Kupka, R.W.; De Wit, H.J.; Ruwhof, C.; Akkerhuis, G.W.; Nolen, W.A.; Drexhage, H.A. An imbalance in the production of IL-1β and IL-6 by monocytes of bipolar patients: Restoration by lithium treatment. Bipolar Disord. 2007, 9, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.N.; Lee, C.S.; Wu, B.J.; Sun, H.J.; Chang, C.H.; Chen, C.Y.; Chen, C.K.; Wu, L.S.H.; Cheng, A.T.A. Immunophenotypes associated with bipolar disorder and lithium treatment. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Ferensztajn-Rochowiak, E.; Kucharska-Mazur, J.; Tarnowski, M.; Samochowiec, J.; Ratajczak, M.Z.; Rybakowski, J.K. Stem cells, pluripotency and glial cell markers in peripheral blood of bipolar patients on long-term lithium treatment. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 80, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Nahman, S.; Belmaker, R.H.R.; Azab, A.N. Effects of lithium on lipopolysaccharide-induced inflammation in rat primary glia cells. Innate Immun. 2012, 18, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhang, X.; Dai, X.; Lu, S.; Gui, B.; Jin, W.; Zhang, S.; Zhang, S.; Qian, Y. Lithium ameliorates lipopolysaccharide-induced microglial activation via inhibition of toll-like receptor 4 expression by activating the PI3K/Akt/FoxO1 pathway. J. Neuroinflamm. 2014, 11, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, W.K.; Levesque, D.L.; Cocker, P.J.; Kaur, S.; Bodnar, T.S.; Young, A.H.; Winstanley, C.A. Decreased motor impulsivity following chronic lithium treatment in male rats is associated with reduced levels of pro-inflammatory cytokines in the orbitofrontal cortex. Brain Behav. Immun. 2020, 89, 339–349. [Google Scholar] [CrossRef]
- Fernandes, M.S.; Barbisan, F.; Azzolin, V.F.; Do Prado-Lima, P.A.S.; Teixeira, C.F.; Da Cruz Jung, I.E.; Assmann, C.E.; Riffel, R.T.; Duarte, M.M.M.F.; Aguiar-Ribeiro, E.M.; et al. Lithium is able to minimize olanzapine oxidative-inflammatory induction on macrophage cells. PLoS ONE 2019, 14, 1–16. [Google Scholar] [CrossRef]
- Petersein, C.; Sack, U.; Mergl, R.; Schönherr, J.; Schmidt, F.M.; Lichtblau, N.; Kirkby, K.C.; Bauer, K.; Himmerich, H. Impact of lithium alone and in combination with antidepressants on cytokine production in vitro. J. Neural Transm. 2015, 122, 109–122. [Google Scholar] [CrossRef]
- Cebers, G.; Zhivotovsky, B.; Ankarcrona, M.; Liljequist, S. AMPA neurotoxicity in cultured cerebellar granule neurons: Mode of cell death. Brain Res. Bull. 1997, 43, 393–403. [Google Scholar] [CrossRef]
- Maurer, I.C.; Schippel, P.; Volz, H.P. Lithium-induced enhancement of mitochondrial oxidative phosphorylation in human brain tissue. Bipolar Disord. 2009, 11, 515–522. [Google Scholar] [CrossRef]
- De Sousa, R.T.; Streck, E.L.; Zanetti, M.V.; Ferreira, G.K.; Diniz, B.S.; Brunoni, A.R.; Busatto, G.F.; Gattaz, W.F.; Machado-Vieira, R. Lithium increases leukocyte mitochondrial complex I activity in bipolar disorder during depressive episodes. Psychopharmacology (Berl.) 2015, 232, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Cikánková, T.; Fišar, Z.; Hroudová, J. In vitro effects of antidepressants and mood-stabilizing drugs on cell energy metabolism. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Nciri, R.; Desmoulin, F.; Allagui, M.S.; Murat, J.-C.; Feki, A.E.; Vincent, C.; Croute, F. Neuroprotective effects of chronic exposure of SH-SY5Y to low lithium concentration involve glycolysis stimulation, extracellular pyruvate accumulation and resistance to oxidative stress. Int. J. Neuropsychopharmacol. 2013, 16, 365–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubert, C.; Andrejew, R.; Figueiro, F.; Bergamin, L.; Kapczinski, F.; da Silva Magalhães, P.V.; Battastini, A.M.O. Lithium-induced neuroprotective activity in neuronal and microglial cells: A purinergic perspective. Psychiatry Res. 2021, 295, 113562. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, Y.; Tan, H.; Bharti, V.; Che, Y.; Wang, J.F. Chronic treatment with mood stabilizer lithium inhibits amphetamine-induced risk-taking manic-like behaviors. Neurosci. Lett. 2015, 603, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Valvassori, S.S.; Tonin, P.T.; Varela, R.B.; Carvalho, A.F.; Mariot, E.; Amboni, R.T.; Bianchini, G.; Andersen, M.L.; Quevedo, J. Lithium modulates the production of peripheral and cerebral cytokines in an animal model of mania induced by dextroamphetamine. Bipolar Disord. 2015, 17, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Valvassori, S.S.; Tonin, P.T.; Dal-Pont, G.C.; Varela, R.B.; Cararo, J.H.; Garcia, A.F.; Gava, F.F.; Menegas, S.; Soares, J.C.; Quevedo, J. Coadministration of lithium and celecoxib reverses manic-like behavior and decreases oxidative stress in a dopaminergic model of mania induced in rats. Transl. Psychiatry 2019, 9. [Google Scholar] [CrossRef]
- Menegas, S.; Dal-Pont, G.C.; Cararo, J.H.; Varela, R.B.; Aguiar-Geraldo, J.M.; Possamai-Della, T.; Andersen, M.L.; Quevedo, J.; Valvassori, S.S. Efficacy of folic acid as an adjunct to lithium therapy on manic-like behaviors, oxidative stress and inflammatory parameters in an animal model of mania. Metab. Brain Dis. 2020, 35, 413–425. [Google Scholar] [CrossRef]
- Szczepankiewicz, D.; Celichowski, P.; Kołodziejski, P.A.; Pruszyńska-Oszmałek, E.; Sassek, M.; Zakowicz, P.; Banach, E.; Langwiński, W.; Sakrajda, K.; Nowakowska, J.; et al. Transcriptome changes in three brain regions during chronic lithium administration in the rat models of mania and depression. Int. J. Mol. Sci. 2021, 22, 1148. [Google Scholar] [CrossRef]
- Valvassori, S.S.; Rezin, G.T.; Ferreira, C.L.; Moretti, M.; Gonçalves, C.L.; Cardoso, M.R.; Streck, E.L.; Kapczinski, F.; Quevedo, J. Effects of mood stabilizers on mitochondrial respiratory chain activity in brain of rats treated with d-amphetamine. J. Psychiatr. Res. 2010, 44, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Stacey, D.; Schubert, K.O.; Clark, S.R.; Amare, A.T.; Milanesi, E.; Maj, C.; Leckband, S.G.; Shekhtman, T.; Kelsoe, J.R.; Gurwitz, D.; et al. A gene co-expression module implicating the mitochondrial electron transport chain is associated with long-term response to lithium treatment in bipolar affective disorder. Transl. Psychiatry 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado-Vieira, R.; Zanetti, M.V.; Otaduy, M.C.; De Sousa, R.T.; Soeiro-De-Souza, M.G.; Costa, A.C.; Carvalho, A.F.; Leite, C.C.; Busatto, G.F.; Zarate, C.A.; et al. Increased brain lactate during depressive episodes and reversal effects by lithium monotherapy in drug-naive bipolar disorder: A 3-T 1H-MRS study. J. Clin. Psychopharmacol. 2017, 37, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Guo, W.; Feng, Y.; Deng, H.; Li, G.; Nie, H.; Guo, G.; Yu, H.; Ma, Y.; Wang, J.; et al. Efficacy and safety of anti-inflammatory agents for the treatment of major depressive disorder: A systematic review and meta-analysis of randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, J.D.; Kakar, R.; Berk, M.; Kessing, L.V.; Vinberg, M.; Baune, B.T.; Mansur, R.B.; Brietzke, E.; Goldstein, B.I.; Mcintyre, R.S. Anti-inflammatory agents in the treatment of bipolar depression: A systematic review and meta-analysis. Bipolar Disord. 2016, 18, 89–101. [Google Scholar] [CrossRef]
- Husain, M.I.; Strawbridge, R.; Stokes, P.R.A.; Young, A.H. Anti-inflammatory treatments for mood disorders: Systematic review and meta-analysis. J. Psychopharmacol. 2017, 31, 1137–1148. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Characteristic molecular markers elevated in M1 and M2 phenotype. iNOS—inducible nitric oxide synthase; IL-1β—interleukin 1 beta; IL-6—interleukin 6; IL-12—interleukin 12; TNF-α—tumor necrosis factor alpha; CCL2—C-C motif chemokine ligand 2 or monocyte chemoattractant protein1; CCL3—C-C motif chemokine ligand 3 or macrophage inflammatory protein 1-alpha; CCL5—C-C motif chemokine ligand 5; CXCL8—C-X-C motif chemokine ligand 8 or interleukin 8; CXCL9—C-X-C motif chemokine ligand 9; CXCL10—C-X-C motif chemokine ligand 10; CXCL11–C-X-C motif chemokine ligand 11; Arg-1—arginase-1; IL-10—interleukin 10; TGF-β—transforming growth factor beta; MSR-1—macrophage scavenger receptor 1; CD206—mannose receptor (or cluster of differentiation 206); MGL—galactose-type lectin.
Figure 1.
Characteristic molecular markers elevated in M1 and M2 phenotype. iNOS—inducible nitric oxide synthase; IL-1β—interleukin 1 beta; IL-6—interleukin 6; IL-12—interleukin 12; TNF-α—tumor necrosis factor alpha; CCL2—C-C motif chemokine ligand 2 or monocyte chemoattractant protein1; CCL3—C-C motif chemokine ligand 3 or macrophage inflammatory protein 1-alpha; CCL5—C-C motif chemokine ligand 5; CXCL8—C-X-C motif chemokine ligand 8 or interleukin 8; CXCL9—C-X-C motif chemokine ligand 9; CXCL10—C-X-C motif chemokine ligand 10; CXCL11–C-X-C motif chemokine ligand 11; Arg-1—arginase-1; IL-10—interleukin 10; TGF-β—transforming growth factor beta; MSR-1—macrophage scavenger receptor 1; CD206—mannose receptor (or cluster of differentiation 206); MGL—galactose-type lectin.


{kind=link}
Table 1.
Changes in pro- and anti-inflammatory interleukins in mood disorders.
| Interleukin | Major Depressive Disorder | Bipolar Disorder | ||
|---|---|---|---|---|
| Depression | Euthymia | Mania | ||
| Interleukin 1 beta | ↑ IL-1β [28] | ↑ IL-1β [29] | ↑ IL-1β [30,31,32,33] | ↓ IL-1β [29,34] |
| ↔ IL-1β [35,36,37,38] | ↔IL-1β [34] | ↔ IL-1β [39] | ||
| Interleukin 6 | ↑ IL-6 [32,35,36,37,40] | ↑IL-6 [29,34] | ↑ IL-6 [32,33,41] | ↑IL-6 [29,32,41,42,43,44] |
| ↔IL-6 [38,39] | ↓IL-6 [34] | |||
| Soluble form of interleukin 6 receptor | ↑sIL-6R [45] | N/A | ↑sIL-6R [30] | N/A |
| Tumor necrosis factor alpha | ↑TNF-α [35,36,37,40] | ↑TNF-α [29,34,42] | ↑TNF-α [30,33] | ↑TNF-α [29,30,41,42,43,44,46] |
| ↔TNF-α [31,38] | ||||
| C-C motif chemokine ligand 2 | ↑CCL2 [37,47] | ↑CCL2 [29] | ↑CCL2 [45,48,49] | ↑CCL2 [29] |
| ↔CCL2 [30] | ↔CCL2 [30,38] | ↔CCL2 [30] | ||
| ↓CCL2 [50] | ||||
| C-C motif chemokine ligand 3 | ↔CCL3 [37,47] | N/A | ↑CCL3 [50] | N/A |
| ↔CCL3 [51] | ||||
| C-C motif chemokine ligand 5 | ↔CCL5 [37,47] | N/A | N/A | N/A |
| C-X-C motif chemokine ligand 8 | ↑CXCL8 [47] | ↑CXCL8 [42] | ↑CXCL8 [33,42] | ↑CXCL8 [42] |
| ↔ CXCL8 [30] | ||||
| ↓CXCL8 [52] | ||||
| C-X-C motif chemokine ligand 9 | ↔ CXCL9 [47] | ↓CXCL9 [53] | ↑CXCL9 [52] | ↓CXCL9 [53] |
| ↔CXCL9 [53] | ||||
| Interferon gamma | ↔IFN-γ [35,36] | N/A | ↑IFN-γ [33] | ↑IFN-γ [43] |
| ↓IFN-γ [37] | ↔IFN-γ [39,41] | |||
| Interleukin 4 | ↔IL-4 [35,36] | ↑IL-4 [29] | ↑IL-4 [39,50] | ↑IL-4 [29,34] |
| ↔IL-4 [38,41] | ↔IL-4 [41] ↓IL-4 [44] | |||
| Transforming growth factor beta | N/A | ↑TGF-β [53] | ↑TGF-β [33] | ↑TGF-β [53] |
| ↔TGF-β [53] | ||||
| ↓TGF-β [50] | ||||
↑—a significant increase in the interleukin level; ↓—a significant decrease in the interleukin level; ↔—no significant changes in the interleukin level. N/A—data not available.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sakrajda, K.; Szczepankiewicz, A. Inflammation-Related Changes in Mood Disorders and the Immunomodulatory Role of Lithium. Int. J. Mol. Sci. 2021, 22, 1532. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041532
AMA Style
Sakrajda K, Szczepankiewicz A. Inflammation-Related Changes in Mood Disorders and the Immunomodulatory Role of Lithium. International Journal of Molecular Sciences. 2021; 22(4):1532. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041532
Chicago/Turabian StyleSakrajda, Kosma, and Aleksandra Szczepankiewicz. 2021. "Inflammation-Related Changes in Mood Disorders and the Immunomodulatory Role of Lithium" International Journal of Molecular Sciences 22, no. 4: 1532. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041532
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.