What, When and How to Measure—Peripheral Biomarkers in Therapy of Huntington’s Disease


Abstract
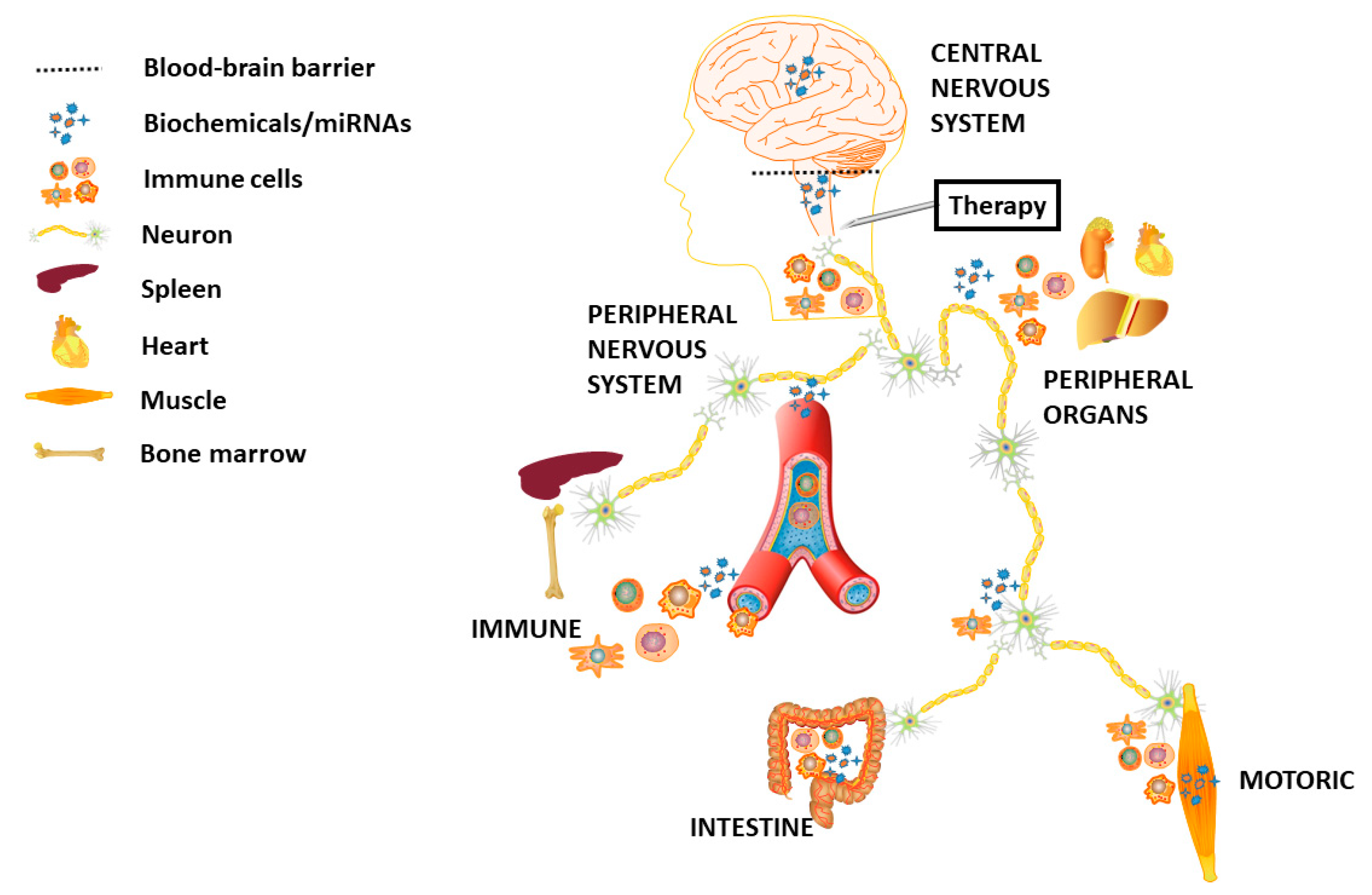
:1. Introduction
2. Disease-Modifying Strategies for HD
3. Biomarkers—General Information and Types Applicable for HD
4. HD Wet Biomarkers
4.1. Neurofilament Light Chain
4.2. Mutant Huntingtin
4.3. Oxidative Stress Markers
4.4. BDNF
4.5. Metabolic Markers
4.6. miRNAs
4.7. Immune System
4.8. Others
5. Microbiome

{kind=link}
| Blood Biomarkers | Type of Deregulation | References |
|---|---|---|
| NFL | up-regulation | [50,51,52] |
| mHTT | up-regulation | [64,65,66,67] |
| 8-OHdG | up-regulation no difference | [75,85,86,87] [86,90] |
| UA | up-regulation | [76] |
| Kynurenine | no difference up-regulation | [82] [83] |
| Tryptophan | down-regulation no difference | [82] [83,104] |
| Kynurenine/tryptophan ratio | up-regulation | [82,83] |
| BDNF | down-regulation (transcript only) | [95] |
| down-regulation | [40,96] | |
| no difference | [91,97,98] | |
| up-regulation | [99] | |
| 24OHC | down-regulation | [102] |
| Melatonin | no difference down-regulation | [138] [140] |
| GH | no difference up-regulation | [141,142] [141,151] |
| Metabolism of amino acids (valine, leucine and isoleucine) | changed | [107,108,109] |
| not changed | [108,110,111] | |
| Cholesterol profile | no difference down-regulation | [111,112,113] [97] |
| AT1R-AA | up-regulation | [136] |
| AGA | up-regulation | [137] |
| Serotonin N-acetylserotonin | down-regulation | [104] |
| up-regulation | [104] | |
| Cytokines | ||
| Eotaxin, MCP-1, MCP-4, MIP-1β | up-regulation | [125] |
| IL-4, IL-8, IL-10, TNFα, GM-CSF | up-regulation | [124] |
| MMP-9, VEGF, IL-18 | up-regulation | [152] |
| TGF-β1 | down-regulation | [134] |
| IL-6 | up-regulation | [132] |
| miRNAs | ||
| miR-34b | up-regulation | [120] |
| miR-10b-5p, miR-486-5p | up-regulation | [117] |
| miR-30d-5p, miR-877-5p, miR-425-5p, miR-223-3p, miR-223-5p, miR-222-3p, miR-338-3p, miR-130b-3p, miR-628-3p, miR-361-5p, miR-128, miR-22-5p, miR-942 | up-regulation | [121] |
| miR-9 | down-regulation | [119] |
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HD | Huntington’s disease |
| CNS | Central Nervous System |
| RNAi | RNA interreference |
| siRNA | Small interfering RNA |
| ASO | Anti-sense Oligonucleotide |
| CSF | Cerebrospinal fluid |
| PolyQ | Polyglutamine |
| BBB | Blood-brain barrier |
| NFL | Neurofilament light chain |
References
- Ghosh, R.; Tabrizi, S.J. Huntington disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 147, pp. 255–278. ISBN 978-0-444-63233-3. [Google Scholar]
- Paulson, H. Repeat expansion diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 147, pp. 105–123. ISBN 978-0-444-63233-3. [Google Scholar]
- Stoyas, C.A.; La Spada, A.R. The CAG–polyglutamine repeat diseases: A clinical, molecular, genetic, and pathophysiologic nosology. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 147, pp. 143–170. ISBN 978-0-444-63233-3. [Google Scholar]
- Chung, C.G.; Lee, H.; Lee, S.B. Mechanisms of Protein Toxicity in Neurodegenerative Diseases. Cell. Mol. Life Sci. 2018, 75, 3159–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-H.; Schilling, G.; Young, W.S.; Li, X.; Margolis, R.L.; Stine, O.C.; Wagster, M.V.; Abbott, M.H.; Franz, M.L.; Ranen, N.G.; et al. Huntington’s Disease Gene (IT15) Is Widely Expressed in Human and Rat Tissues. Neuron 1993, 11, 985–993. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, J.M.; Björkqvist, M.; Brundin, P. Beyond the Brain: Widespread Pathology in Huntington’s Disease. Lancet Neurol. 2009, 8, 765–774. [Google Scholar] [CrossRef]
- Bozzi, M.; Sciandra, F. Molecular Mechanisms Underlying Muscle Wasting in Huntington’s Disease. IJMS 2020, 21, 8314. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How Neuroinflammation Contributes to Neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Valadão, P.A.C.; Santos, K.B.S.; Ferreira e Vieira, T.H.; Macedo e Cordeiro, T.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s Disease: A Few New Twists on an Old Tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef]
- Stephen, C.D.; Hung, J.; Schifitto, G.; Hersch, S.M.; Rosas, H.D. Electrocardiogram Abnormalities Suggest Aberrant Cardiac Conduction in Huntington’s Disease: ECG Abnormalities in Huntington’s Disease. Mov. Disord. Clin. Pract. 2018, 5, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Cankar, K.; Melik, Z.; Kobal, J.; Starc, V. Evidence of Cardiac Electrical Remodeling in Patients with Huntington Disease. Brain Behav. 2018, 8, e01077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melik, Z.; Kobal, J.; Cankar, K.; Strucl, M. Microcirculation Response to Local Cooling in Patients with Huntington’s Disease. J. Neurol. 2012, 259, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [Green Version]
- Fiszer, A.; Krzyzosiak, W.J. Oligonucleotide-Based Strategies to Combat Polyglutamine Diseases. Nucleic Acids Res. 2014, 42, 6787–6810. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Wild, E.J.; Tabrizi, S.J. Therapies Targeting DNA and RNA in Huntington’s Disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, P.; Johnson, I.M.; Alli, S.; Dragatsis, I. Elimination of Huntingtin in the Adult Mouse Leads to Progressive Behavioral Deficits, Bilateral Thalamic Calcification, and Altered Brain Iron Homeostasis. PLoS Genet 2017, 13, e1006846. [Google Scholar] [CrossRef]
- Grondin, R.; Kaytor, M.D.; Ai, Y.; Nelson, P.T.; Thakker, D.R.; Heisel, J.; Weatherspoon, M.R.; Blum, J.L.; Burright, E.N.; Zhang, Z.; et al. Six-Month Partial Suppression of Huntingtin Is Well Tolerated in the Adult Rhesus Striatum. Brain 2012, 135, 1197–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-Nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Martier, R.; Konstantinova, P. Gene Therapy for Neurodegenerative Diseases: Slowing Down the Ticking Clock. Front. Neurosci. 2020, 14, 580179. [Google Scholar] [CrossRef]
- Smalley, E. First AAV Gene Therapy Poised for Landmark Approval. Nat. Biotechnol. 2017, 35, 998–999. [Google Scholar] [CrossRef] [PubMed]
- Alterman, J.F.; Godinho, B.M.D.C.; Hassler, M.R.; Ferguson, C.M.; Echeverria, D.; Sapp, E.; Haraszti, R.A.; Coles, A.H.; Conroy, F.; Miller, R.; et al. A Divalent SiRNA Chemical Scaffold for Potent and Sustained Modulation of Gene Expression throughout the Central Nervous System. Nat. Biotechnol. 2019, 37, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, J.; Corey, D.R. Allele-Selective Inhibition of Huntingtin Expression by Switching to an MiRNA-like RNAi Mechanism. Chem. Biol. 2010, 17, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Fiszer, A.; Mykowska, A.; Krzyzosiak, W.J. Inhibition of Mutant Huntingtin Expression by RNA Duplex Targeting Expanded CAG Repeats. Nucleic Acids Res. 2011, 39, 5578–5585. [Google Scholar] [CrossRef] [Green Version]
- Fiszer, A.; Olejniczak, M.; Galka-Marciniak, P.; Mykowska, A.; Krzyzosiak, W.J. Self-Duplexing CUG Repeats Selectively Inhibit Mutant Huntingtin Expression. Nucleic Acids Res. 2013, 41, 10426–10437. [Google Scholar] [CrossRef]
- Yu, D.; Pendergraff, H.; Liu, J.; Kordasiewicz, H.B.; Cleveland, D.W.; Swayze, E.E.; Lima, W.F.; Crooke, S.T.; Prakash, T.P.; Corey, D.R. Single-Stranded RNAs Use RNAi to Potently and Allele-Selectively Inhibit Mutant Huntingtin Expression. Cell 2012, 150, 895–908. [Google Scholar] [CrossRef] [Green Version]
- Ciesiolka, A.; Stroynowska-Czerwinska, A.; Joachimiak, P.; Ciolak, A.; Kozlowska, E.; Michalak, M.; Dabrowska, M.; Olejniczak, M.; Raczynska, K.D.; Zielinska, D.; et al. Artificial MiRNAs Targeting CAG Repeat Expansion in ORFs Cause Rapid Deadenylation and Translation Inhibition of Mutant Transcripts. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Fiszer, A.; Ellison-Klimontowicz, M.E.; Krzyzosiak, W.J. Silencing of Genes Responsible for PolyQ Diseases Using Chemically Modified Single-Stranded SiRNAs. Acta Biochim. Pol. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Fiszer, A.; Wroblewska, J.; Nowak, B.; Krzyzosiak, W. Mutant CAG Repeats Effectively Targeted by RNA Interference in SCA7 Cells. Genes 2016, 7, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotowska-Zimmer, A.; Ostrovska, Y.; Olejniczak, M. Universal RNAi Triggers for the Specific Inhibition of Mutant Huntingtin, Atrophin-1, Ataxin-3, and Ataxin-7 Expression. Mol. Ther. Nucleic Acids 2020, 19, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Liang, X.; Crooke, R.M.; Baker, B.F.; Geary, R.S. Antisense Drug Discovery and Development Technology Considered in a Pharmacological Context. Biochem. Pharmacol. 2020, 114196. [Google Scholar] [CrossRef] [PubMed]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained Therapeutic Reversal of Huntington’s Disease by Transient Repression of Huntingtin Synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, C.; Wood, M.J.A. Antisense Oligonucleotides: The next Frontier for Treatment of Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef]
- Stanek, L.M.; Yang, W.; Angus, S.; Sardi, P.S.; Hayden, M.R.; Hung, G.H.; Bennett, C.F.; Cheng, S.H.; Shihabuddin, L.S. Antisense Oligonucleotide-Mediated Correction of Transcriptional Dysregulation Is Correlated with Behavioral Benefits in the YAC128 Mouse Model of Huntington’s Disease. J. Huntingt. Dis. 2013, 2, 217–228. [Google Scholar] [CrossRef]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-Mediated Brain Drug Delivery: Overcoming Blood–Brain Barrier to Treat Neurodegenerative Diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allinson, J.L. Clinical Biomarker Validation. Bioanalysis 2018, 10, 957–968. [Google Scholar] [CrossRef]
- Johnson, E.B.; Gregory, S. Huntington’s disease: Brain imaging in Huntington’s disease. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2019; Volume 165, pp. 321–369. ISBN 978-0-12-818361-8. [Google Scholar]
- Squitieri, F.; Cannella, M.; Simonelli, M.; Sassone, J.; Martino, T.; Venditti, E.; Ciammola, A.; Colonnese, C.; Frati, L.; Ciarmiello, A. Distinct Brain Volume Changes Correlating with Clinical Stage, Disease Progression Rate, Mutation Size, and Age at Onset Prediction as Early Biomarkers of Brain Atrophy in Huntington’s Disease. CNS Neurosci. Ther. 2009, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, N.Z.; Barnes, J.; Frost, C.; Henley, S.M.D.; Wild, E.J.; Macdonald, K.; Barker, R.A.; Scahill, R.I.; Fox, N.C.; Tabrizi, S.J. Onset and Progression of Pathologic Atrophy in Huntington Disease: A Longitudinal MR Imaging Study. AJNR Am. J. Neuroradiol. 2010, 31, 1036–1041. [Google Scholar] [CrossRef] [Green Version]
- Roussakis, A.-A.; Piccini, P. PET Imaging in Huntington’s Disease. JHD 2015, 4, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J.; et al. Detection of Huntington’s Disease Decades before Diagnosis: The Predict-HD Study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 874–880. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Scahill, R.I.; Durr, A.; Roos, R.A.; Leavitt, B.R.; Jones, R.; Landwehrmeyer, G.B.; Fox, N.C.; Johnson, H.; Hicks, S.L.; et al. Biological and Clinical Changes in Premanifest and Early Stage Huntington’s Disease in the TRACK-HD Study: The 12-Month Longitudinal Analysis. Lancet Neurol. 2011, 10, 31–42. [Google Scholar] [CrossRef]
- Disatnik, M.-H.; Joshi, A.U.; Saw, N.L.; Shamloo, M.; Leavitt, B.R.; Qi, X.; Mochly-Rosen, D. Potential Biomarkers to Follow the Progression and Treatment Response of Huntington’s Disease. J. Exp. Med. 2016, 213, 2655–2669. [Google Scholar] [CrossRef] [Green Version]
- Drouet, V.; Perrin, V.; Hassig, R.; Dufour, N.; Auregan, G.; Alves, S.; Bonvento, G.; Brouillet, E.; Luthi-Carter, R.; Hantraye, P.; et al. Sustained Effects of Nonallele-Specific Huntingtin Silencing. Ann. Neurol. 2009, 65, 276–285. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Cleeter, M.W.J.; Xuereb, J.; Taanman, J.W.; Cooper, J.M.; Schapira, A.H.V. Biochemical Abnormalities and Excitotoxicity in Huntington’s Disease Brain. Ann. Neurol. 1999, 45, 25–32. [Google Scholar] [CrossRef]
- Killoran, A. Biomarkers for Huntington’s Disease: A Brief Overview. J. Rare Dis. Res. Treat. 2016, 1, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Palermo, G.; Mazzucchi, S.; Della Vecchia, A.; Siciliano, G.; Bonuccelli, U.; Azuar, C.; Ceravolo, R.; Lista, S.; Hampel, H.; Baldacci, F. Different Clinical Contexts of Use of Blood Neurofilament Light Chain Protein in the Spectrum of Neurodegenerative Diseases. Mol. Neurobiol. 2020, 57, 4667–4691. [Google Scholar] [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Scahill, R.I.; Tabrizi, S.J.; Zetterberg, H.; Langbehn, D.; et al. Neurofilament Light Protein in Blood as a Potential Biomarker of Neurodegeneration in Huntington’s Disease: A Retrospective Cohort Analysis. Lancet Neurol. 2017, 16, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Byrne, L.M.; Rodrigues, F.B.; Johnson, E.B.; Wijeratne, P.A.; De Vita, E.; Alexander, D.C.; Palermo, G.; Czech, C.; Schobel, S.; Scahill, R.I.; et al. Evaluation of Mutant Huntingtin and Neurofilament Proteins as Potential Markers in Huntington’s Disease. Sci. Transl. Med. 2018, 10, eaat7108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.B.; Byrne, L.M.; Gregory, S.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.; Zetterberg, H.; Tabrizi, S.J.; et al. Neurofilament Light Protein in Blood Predicts Regional Atrophy in Huntington Disease. Neurology 2018, 90, e717–e723. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s Disease: Current Status and Prospects for the Future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [Green Version]
- Andersson, E.; Janelidze, S.; Lampinen, B.; Nilsson, M.; Leuzy, A.; Stomrud, E.; Blennow, K.; Zetterberg, H.; Hansson, O. Blood and Cerebrospinal Fluid Neurofilament Light Differentially Detect Neurodegeneration in Early Alzheimer’s Disease. Neurobiol. Aging 2020, 95, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Li, C.-H.; Yang, K.-C.; Lin, F.-J.; Wu, C.-C.; Chieh, J.-J.; Chiu, M.-J. Blood NfL: A Biomarker for Disease Severity and Progression in Parkinson Disease. Neurology 2019, 93, e1104–e1111. [Google Scholar] [CrossRef]
- Li, Q.-F.; Dong, Y.; Yang, L.; Xie, J.-J.; Ma, Y.; Du, Y.-C.; Cheng, H.-L.; Ni, W.; Wu, Z.-Y. Neurofilament Light Chain Is a Promising Serum Biomarker in Spinocerebellar Ataxia Type 3. Mol. Neurodegener. 2019, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament Light Chain as a Biomarker in Neurological Disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef]
- Forgrave, L.M.; Ma, M.; Best, J.R.; DeMarco, M.L. The Diagnostic Performance of Neurofilament Light Chain in CSF and Blood for Alzheimer’s Disease, Frontotemporal Dementia, and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-analysis. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2019, 11, 730–743. [Google Scholar] [CrossRef]
- Kuhle, J.; Barro, C.; Andreasson, U.; Derfuss, T.; Lindberg, R.; Sandelius, Å.; Liman, V.; Norgren, N.; Blennow, K.; Zetterberg, H. Comparison of Three Analytical Platforms for Quantification of the Neurofilament Light Chain in Blood Samples: ELISA, Electrochemiluminescence Immunoassay and Simoa. Clin. Chem. Lab. Med. 2016, 54. [Google Scholar] [CrossRef]
- Baldacci, F.; Mazzucchi, S.; Della Vecchia, A.; Giampietri, L.; Giannini, N.; Koronyo-Hamaoui, M.; Ceravolo, R.; Siciliano, G.; Bonuccelli, U.; Elahi, F.M.; et al. The Path to Biomarker-Based Diagnostic Criteria for the Spectrum of Neurodegenerative Diseases. Expert Rev. Mol. Diagn. 2020, 20, 421–441. [Google Scholar] [CrossRef]
- Wild, E.J.; Boggio, R.; Langbehn, D.; Robertson, N.; Haider, S.; Miller, J.R.C.; Zetterberg, H.; Leavitt, B.R.; Kuhn, R.; Tabrizi, S.J.; et al. Quantification of Mutant Huntingtin Protein in Cerebrospinal Fluid from Huntington’s Disease Patients. J. Clin. Investig. 2015, 125, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Fodale, V.; Boggio, R.; Daldin, M.; Cariulo, C.; Spiezia, M.C.; Byrne, L.M.; Leavitt, B.R.; Wild, E.J.; Macdonald, D.; Weiss, A.; et al. Validation of Ultrasensitive Mutant Huntingtin Detection in Human Cerebrospinal Fluid by Single Molecule Counting Immunoassay. JHD 2017, 6, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southwell, A.L.; Smith, S.E.P.; Davis, T.R.; Caron, N.S.; Villanueva, E.B.; Xie, Y.; Collins, J.A.; Li Ye, M.; Sturrock, A.; Leavitt, B.R.; et al. Ultrasensitive Measurement of Huntingtin Protein in Cerebrospinal Fluid Demonstrates Increase with Huntington Disease Stage and Decrease Following Brain Huntingtin Suppression. Sci. Rep. 2015, 5, 12166. [Google Scholar] [CrossRef]
- Moscovitch-Lopatin, M.; Weiss, A.; Rosas, H.D.; Ritch, J.; Doros, G.; Kegel, K.B.; Difiglia, M.; Kuhn, R.; Bilbe, G.; Paganetti, P.; et al. Optimization of an HTRF Assay for the Detection of Soluble Mutant Huntingtin in Human Buffy Coats: A Potential Biomarker in Blood for Huntington Disease. PLoS Curr. 2010, 2, RRN1205. [Google Scholar] [CrossRef] [PubMed]
- Moscovitch-Lopatin, M.; Goodman, R.E.; Eberly, S.; Ritch, J.J.; Rosas, H.D.; Matson, S.; Matson, W.; Oakes, D.; Young, A.B.; Shoulson, I.; et al. HTRF Analysis of Soluble Huntingtin in PHAROS PBMCs. Neurology 2013, 81, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.; Träger, U.; Wild, E.J.; Grueninger, S.; Farmer, R.; Landles, C.; Scahill, R.I.; Lahiri, N.; Haider, S.; Macdonald, D.; et al. Mutant Huntingtin Fragmentation in Immune Cells Tracks Huntington’s Disease Progression. J. Clin. Investig. 2012, 122, 3731–3736. [Google Scholar] [CrossRef]
- Hensman Moss, D.J.; Robertson, N.; Farmer, R.; Scahill, R.I.; Haider, S.; Tessari, M.A.; Flynn, G.; Fischer, D.F.; Wild, E.J.; Macdonald, D.; et al. Quantification of Huntingtin Protein Species in Huntington’s Disease Patient Leukocytes Using Optimised Electrochemiluminescence Immunoassays. PLoS ONE 2017, 12, e0189891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corey-Bloom, J.; Gluhm, S.; Herndon, A.; Haque, A.S.; Park, S.; Gilbert, P.E. Benton Judgment of Line Orientation (JoLO) Test: A Brief and Useful Measure for Assessing Visuospatial Abilities in Manifest, but Not Premanifest, Huntington’s Disease. JHD 2016, 5, 91–96. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Haque, A.S.; Park, S.; Nathan, A.S.; Baker, R.W.; Thomas, E.A. Salivary Levels of Total Huntingtin Are Elevated in Huntington’s Disease Patients. Sci. Rep. 2018, 8, 7371. [Google Scholar] [CrossRef]
- Silajdžić, E.; Björkqvist, M. A Critical Evaluation of Wet Biomarkers for Huntington’s Disease: Current Status and Ways Forward. JHD 2018, 7, 109–135. [Google Scholar] [CrossRef] [Green Version]
- Baldo, B.; Sajjad, M.U.; Cheong, R.Y.; Bigarreau, J.; Vijayvargia, R.; McLean, C.; Perrier, A.L.; Seong, I.S.; Halliday, G.; Petersén, Å.; et al. Quantification of Total and Mutant Huntingtin Protein Levels in Biospecimens Using a Novel AlphaLISA Assay. eNeuro 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Corey-Bloom, J.; Fischer, R.S.; Kim, A.; Snell, C.; Parkin, G.M.; Granger, D.A.; Granger, S.W.; Thomas, E.A. Levels of Interleukin-6 in Saliva, but Not Plasma, Correlate with Clinical Metrics in Huntington’s Disease Patients and Healthy Control Subjects. IJMS 2020, 21, 6363. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Winderickx, J.; Franssens, V.; Liu, B. A Mitochondria-Associated Oxidative Stress Perspective on Huntington’s Disease. Front. Mol. Neurosci. 2018, 11, 329. [Google Scholar] [CrossRef]
- Tang, Q.; Liu, H.; Shi, X.-J.; Cheng, Y. Blood Oxidative Stress Marker Aberrations in Patients with Huntington’s Disease: A Meta-Analysis Study. Oxidative Med. Cell. Longev. 2020, 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-M.; Wu, Y.-R.; Cheng, M.-L.; Liu, J.-L.; Lee, Y.-M.; Lee, P.-W.; Soong, B.-W.; Chiu, D.T.-Y. Increased Oxidative Damage and Mitochondrial Abnormalities in the Peripheral Blood of Huntington’s Disease Patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Corey-Bloom, J.; Haque, A.; Aboufadel, S.; Snell, C.; Fischer, R.S.; Granger, S.W.; Granger, D.A.; Thomas, E.A. Uric Acid as a Potential Peripheral Biomarker for Disease Features in Huntington’s Patients. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotty, G.F.; Ascherio, A.; Schwarzschild, M.A. Targeting Urate to Reduce Oxidative Stress in Parkinson Disease. Exp. Neurol. 2017, 298, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Zhou, B.; Chen, Y.-H.; Ma, Z.-L.; Gou, Y.; Zhang, C.-L.; Yu, W.-F.; Jiao, L. Serum Uric Acid Levels in Patients with Parkinson’s Disease: A Meta-Analysis. PLoS ONE 2017, 12, e0173731. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.F.; Kumar, P.; Bahado-Singh, R.O.; Robinson, A.; Mann, D.; Green, B.D. Novel Metabolite Biomarkers of Huntington’s Disease As Detected by High-Resolution Mass Spectrometry. J. Proteome Res. 2016, 15, 1592–1601. [Google Scholar] [CrossRef]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The Kynurenine Pathway Modulates Neurodegeneration in a Drosophila Model of Huntington’s Disease. Curr. Biol. 2011, 21, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Möller, T. Neuroinflammation in Huntington’s Disease. J. Neural. Transm. 2010, 117, 1001–1008. [Google Scholar] [CrossRef]
- Forrest, C.M.; Mackay, G.M.; Stoy, N.; Spiden, S.L.; Taylor, R.; Stone, T.W.; Darlington, L.G. Blood Levels of Kynurenines, Interleukin-23 and Soluble Human Leucocyte Antigen-G at Different Stages of Huntington’s Disease. J. Neurochem. 2010, 112, 112–122. [Google Scholar] [CrossRef]
- Stoy, N.; Mackay, G.M.; Forrest, C.M.; Christofides, J.; Egerton, M.; Stone, T.W.; Darlington, L.G. Tryptophan Metabolism and Oxidative Stress in Patients with Huntington’s Disease. J. Neurochem. 2005, 93, 611–623. [Google Scholar] [CrossRef]
- Zeun, P.; Scahill, R.I.; Tabrizi, S.J.; Wild, E.J. Fluid and Imaging Biomarkers for Huntington’s Disease. Mol. Cell. Neurosci. 2019, 97, 67–80. [Google Scholar] [CrossRef]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in Huntington Disease Is Safe, Tolerable, Bioavailable in Brain and Reduces Serum 8OH2′dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Long, J.D.; Matson, W.R.; Juhl, A.R.; Leavitt, B.R.; Paulsen, J.S. 8OHdG as a Marker for Huntington Disease Progression. Neurobiol. Dis. 2012, 46, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Túnez, I.; Sánchez-López, F.; Agüera, E.; Fernández-Bolaños, R.; Sánchez, F.M.; Tasset-Cuevas, I. Important Role of Oxidative Stress Biomarkers in Huntington’s Disease. J. Med. Chem. 2011, 54, 5602–5606. [Google Scholar] [CrossRef] [PubMed]
- Biglan, K.M.; Dorsey, E.R.; Evans, R.V.V.; Ross, C.A.; Hersch, S.; Shoulson, I.; Matson, W.; Kieburtz, K. Plasma 8-Hydroxy-2′-Deoxyguanosine Levels in Huntington Disease and Healthy Controls Treated with Coenzyme Q10. J. Huntingt. Dis. 2012, 1, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Langbehn, D.R.; Leavitt, B.R.; Roos, R.A.; Durr, A.; Craufurd, D.; Kennard, C.; Hicks, S.L.; Fox, N.C.; Scahill, R.I.; et al. Biological and Clinical Manifestations of Huntington’s Disease in the Longitudinal TRACK-HD Study: Cross-Sectional Analysis of Baseline Data. Lancet Neurol. 2009, 8, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Borowsky, B.; Warner, J.; Leavitt, B.R.; Tabrizi, S.J.; Roos, R.A.C.; Durr, A.; Becker, C.; Sampaio, C.; Tobin, A.J.; Schulman, H. 8OHdG Is Not a Biomarker for Huntington Disease State or Progression. Neurology 2013, 80, 1934–1941. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, A.; Corey-Bloom, J.; Thomas, E.A.; Desplats, P. Evaluation of Biochemical and Epigenetic Measures of Peripheral Brain-Derived Neurotrophic Factor (BDNF) as a Biomarker in Huntington’s Disease Patients. Front. Mol. Neurosci. 2020, 12, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyileten, C.; Sharif, L.; Wicik, Z.; Jakubik, D.; Jarosz-Popek, J.; Soplinska, A.; Postula, M.; Czlonkowska, A.; Kaplon-Cieslicka, A.; Mirowska-Guzel, D. The Relation of the Brain-Derived Neurotrophic Factor with MicroRNAs in Neurodegenerative Diseases and Ischemic Stroke. Mol. Neurobiol. 2021, 58, 329–347. [Google Scholar] [CrossRef]
- Yu, C.; Li, C.H.; Chen, S.; Yoo, H.; Qin, X.; Park, H. Decreased BDNF Release in Cortical Neurons of a Knock-in Mouse Model of Huntington’s Disease. Sci. Rep. 2018, 8, 16976. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Role of Brain-Derived Neurotrophic Factor in Huntington’s Disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Krzysztoń-Russjan, J.; Zielonka, D.; Jackiewicz, J.; Kuśmirek, S.; Bubko, I.; Klimberg, A.; Marcinkowski, J.T.; Anuszewska, E.L. A Study of Molecular Changes Relating to Energy Metabolism and Cellular Stress in People with Huntington’s Disease: Looking for Biomarkers. J. Bioenerg. Biomembr. 2013, 45, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Ciammola, A.; Sassone, J.; Cannella, M.; Calza, S.; Poletti, B.; Frati, L.; Squitieri, F.; Silani, V. Low Brain-Derived Neurotrophic Factor (BDNF) Levels in Serum of Huntington’s Disease Patients. Am. J. Med. Genet. 2007, 144B, 574–577. [Google Scholar] [CrossRef]
- Wang, R.; Ross, C.A.; Cai, H.; Cong, W.-N.; Daimon, C.M.; Carlson, O.D.; Egan, J.M.; Siddiqui, S.; Maudsley, S.; Martin, B. Metabolic and Hormonal Signatures in Pre-Manifest and Manifest Huntington’s Disease Patients. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccato, C.; Marullo, M.; Vitali, B.; Tarditi, A.; Mariotti, C.; Valenza, M.; Lahiri, N.; Wild, E.J.; Sassone, J.; Ciammola, A.; et al. Brain-Derived Neurotrophic Factor in Patients with Huntington’s Disease. PLoS ONE 2011, 6, e22966. [Google Scholar] [CrossRef] [Green Version]
- Betti, L.; Palego, L.; Unti, E. Brain-Derived Neurotrophic Factor (BDNF) and Serotonin Transporter (SERT) in Platelets of Patients with Mild Huntington’s Disease: Relationships with Social Cognition Symptoms. Arch. Ital. De Biol. 2018, 27–39. [Google Scholar] [CrossRef]
- Xie, Y.; Hayden, M.R.; Xu, B. BDNF Overexpression in the Forebrain Rescues Huntington’s Disease Phenotypes in YAC128 Mice. J. Neurosci. 2010, 30, 14708–14718. [Google Scholar] [CrossRef]
- Canals, J.M. Brain-Derived Neurotrophic Factor Regulates the Onset and Severity of Motor Dysfunction Associated with Enkephalinergic Neuronal Degeneration in Huntington’s Disease. J. Neurosci. 2004, 24, 7727–7739. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Mariotti, C.; Nanetti, L.; Salvatore, E.; Squitieri, F.; Bentivoglio, A.R.; Bandettini del Poggio, M.; Piacentini, S.; Monza, D.; Valenza, M.; et al. Whole Body Cholesterol Metabolism Is Impaired in Huntington’s Disease. Neurosci. Lett. 2011, 494, 245–249. [Google Scholar] [CrossRef]
- Leoni, V.; Long, J.D.; Mills, J.A.; Di Donato, S.; Paulsen, J.S. Plasma 24S-Hydroxycholesterol Correlation with Markers of Huntington Disease Progression. Neurobiol. Dis. 2013, 55, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas, H.D.; Doros, G.; Bhasin, S.; Thomas, B.; Gevorkian, S.; Malarick, K.; Matson, W.; Hersch, S.M. A Systems-level “Misunderstanding”: The Plasma Metabolome in Huntington’s Disease. Ann. Clin. Transl. Neurol. 2015, 2, 756–768. [Google Scholar] [CrossRef]
- Cheng, M.-L.; Chang, K.-H.; Wu, Y.-R.; Chen, C.-M. Metabolic Disturbances in Plasma as Biomarkers for Huntington’s Disease. J. Nutr. Biochem. 2016, 31, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Benaich, S.; Rabier, D.; Durr, A. Validation of Plasma Branched Chain Amino Acids as Biomarkers in Huntington Disease. Arch. Neurol. 2011, 68. [Google Scholar] [CrossRef] [Green Version]
- Underwood, B.R.; Broadhurst, D.; Dunn, W.B.; Ellis, D.I.; Michell, A.W.; Vacher, C.; Mosedale, D.E.; Kell, D.B.; Barker, R.A.; Grainger, D.J.; et al. Huntington Disease Patients and Transgenic Mice Have Similar Pro-Catabolic Serum Metabolite Profiles. Brain 2006, 129, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, N.A.; Onkenhout, W.; Kerstens, H.J.; Roos, R.A.C. Cystathionine Levels in Patients With Huntington Disease. PLoS Curr. 2015. [Google Scholar] [CrossRef]
- Mastrokolias, A.; Pool, R.; Mina, E.; Hettne, K.M.; van Duijn, E.; van der Mast, R.C.; van Ommen, G.; ‘t Hoen, P.A.C.; Prehn, C.; Adamski, J.; et al. Integration of Targeted Metabolomics and Transcriptomics Identifies Deregulation of Phosphatidylcholine Metabolism in Huntington’s Disease Peripheral Blood Samples. Metabolomics 2016, 12, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, B.M.; Kłaczkow, G.; Jaworska, M.; Krzysztoń-Russjan, J.; Anuszewska, E.L.; Zielonka, D.; Klimberg, A.; Marcinkowski, J.T. Huntington’ Disease-Imbalance of Amino Acid Levels in Plasma of Patients and Mutation Carriers. Ann. Agric. Environ. Med. 2013, 20, 779–783. [Google Scholar]
- Ciancarelli, I.; De Amicis, D.; Di Massimo, C.; Sandrini, G.; Pistarini, C.; Carolei, A.; Tozzi Ciancarelli, M.G. Influence of Intensive Multifunctional Neuro-Rehabilitation on Neuronal Oxidative Damage in Patients with Huntington’s Disease. Funct. Neurol. 2015. [Google Scholar] [CrossRef]
- Nambron, R.; Silajdžić, E.; Kalliolia, E.; Ottolenghi, C.; Hindmarsh, P.; Hill, N.R.; Costelloe, S.J.; Martin, N.G.; Positano, V.; Watt, H.C.; et al. A Metabolic Study of Huntington’s Disease. PLoS ONE 2016, 11, e0146480. [Google Scholar] [CrossRef] [Green Version]
- Süssmuth, S.D.; Müller, V.M.; Geitner, C.; Landwehrmeyer, G.B.; Iff, S.; Gemperli, A.; Orth, M. Fat-Free Mass and Its Predictors in Huntington’s Disease. J. Neurol. 2015, 262, 1533–1540. [Google Scholar] [CrossRef]
- Sarko, D.K.; McKinney, C.E. Exosomes: Origins and Therapeutic Potential for Neurodegenerative Disease. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; How Huang, K.; Jen Lee, M.; Galas, D.J.; Wang, K. The MicroRNA Spectrum in 12 Body Fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Juźwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. MicroRNA Dysregulation in Neurodegenerative Diseases: A Systematic Review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef] [PubMed]
- Hoss, A.G.; Lagomarsino, V.N.; Frank, S.; Hadzi, T.C.; Myers, R.H.; Latourelle, J.C. Study of Plasma-Derived MiRNAs Mimic Differences in Huntington’s Disease Brain: Plasma-Derived MiRNAS Mimic Differences in HD Brain. Mov. Disord. 2015, 30, 1961–1964. [Google Scholar] [CrossRef] [Green Version]
- Hoss, A.G.; Labadorf, A.; Latourelle, J.C.; Kartha, V.K.; Hadzi, T.C.; Gusella, J.F.; MacDonald, M.E.; Chen, J.F.; Akbarian, S.; Weng, Z.; et al. miR-10b-5p expression in Huntington’s disease brain relates to age of onset and the extent of striatal involvement. BMC Med. Genom. 2015, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.-H.; Wu, Y.-R.; Chen, C.-M. Down-Regulation of MiR-9* in the Peripheral Leukocytes of Huntington’s Disease Patients. Orphanet J. Rare Dis. 2017, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Gaughwin, P.M.; Ciesla, M.; Lahiri, N.; Tabrizi, S.J.; Brundin, P.; Björkqvist, M. Hsa-MiR-34b Is a Plasma-Stable MicroRNA That Is Elevated in Pre-Manifest Huntington’s Disease. Hum. Mol. Genet. 2011, 20, 2225–2237. [Google Scholar] [CrossRef]
- Díez-Planelles, C.; Sánchez-Lozano, P.; Crespo, M.C.; Gil-Zamorano, J.; Ribacoba, R.; González, N.; Suárez, E.; Martínez-Descals, A.; Martínez-Camblor, P.; Álvarez, V.; et al. Circulating MicroRNAs in Huntington’s Disease: Emerging Mediators in Metabolic Impairment. Pharmacol. Res. 2016, 108, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating MicroRNAs as Stable Blood-Based Markers for Cancer Detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Dalrymple, A.; Wild, E.J.; Joubert, R.; Sathasivam, K.; Bjo, M.; Bates, G.P.; Leavitt, B.R.; Keir, G.; Ward, M.; Tabrizi, S.J. Proteomic Profiling of Plasma in Huntington’s Disease Reveals Neuroinflammatory Activation and Biomarker Candidates. J. Proteome Res. 2007, 8, 2833–2840. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A Novel Pathogenic Pathway of Immune Activation Detectable before Clinical Onset in Huntington’s Disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, E.; Magnusson, A.; Lahiri, N.; Krus, U.; Orth, M.; Tabrizi, S.J.; Björkqvist, M. Abnormal Peripheral Chemokine Profile in Huntington’s Disease. PLoS Curr. 2011, 3, RRN1231. [Google Scholar] [CrossRef]
- Kwan, W.; Magnusson, A.; Chou, A.; Adame, A.; Carson, M.J.; Kohsaka, S.; Masliah, E.; Moller, T.; Ransohoff, R.; Tabrizi, S.J.; et al. Bone Marrow Transplantation Confers Modest Benefits in Mouse Models of Huntington’s Disease. J. Neurosci. 2012, 32, 133–142. [Google Scholar] [CrossRef]
- Simard, A.R.; Rivest, S. Bone Marrow Stem Cells Have the Ability to Populate the Entire Central Nervous System into Fully Differentiated Parenchymal Microglia. FASEB J. 2004, 18, 998–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M.; Perry, V.H. Microglial Physiology: Unique Stimuli, Specialized Responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M.V. Local Self-Renewal Can Sustain CNS Microglia Maintenance and Function throughout Adult Life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Lane, T.E.; Carson, M.; Bergmann, C.; Wyss-Coray, T. (Eds.) Central Nervous System Diseases and Inflammation; Springer: New York, NY, USA, 2008; ISBN 978-0-387-73893-2. [Google Scholar]
- Kwan, W.; Träger, U.; Davalos, D.; Chou, A.; Bouchard, J.; Andre, R.; Miller, A.; Weiss, A.; Giorgini, F.; Cheah, C.; et al. Mutant Huntingtin Impairs Immune Cell Migration in Huntington Disease. J. Clin. Investig. 2012, 122, 4737–4747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Träger, U.; Andre, R.; Magnusson-Lind, A.; Miller, J.R.C.; Connolly, C.; Weiss, A.; Grueninger, S.; Silajdžić, E.; Smith, D.L.; Leavitt, B.R.; et al. Characterisation of Immune Cell Function in Fragment and Full-Length Huntington’s Disease Mouse Models. Neurobiol. Dis. 2015, 73, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, A.; Caria, M.; Arata, L.; Di Geronimo, L.; Canonica, G.W.; Hugh Fudenberg, H. Evidence of T-Lymphocyte Functional Impairment in Huntington’s Disease. Clin. Immunol. Immunopathol. 1986, 39, 121–130. [Google Scholar] [CrossRef]
- Di Pardo, A.; Alberti, S.; Maglione, V.; Amico, E.; Cortes, E.P.; Elifani, F.; Battaglia, G.; Busceti, C.L.; Nicoletti, F.; Vonsattel, J.P.G.; et al. Changes of Peripheral TGF-Β1 Depend on Monocytes-Derived Macrophages in Huntington Disease. Mol. Brain 2013, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Dobson, L.; Träger, U.; Farmer, R.; Hayardeny, L.; Loupe, P.; Hayden, M.R.; Tabrizi, S.J. Laquinimod Dampens Hyperactive Cytokine Production in Huntington’s Disease Patient Myeloid Cells. J. Neurochem. 2016, 137, 782–794. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-H.; Heidecke, H.; Schröder, A.; Paul, F.; Wachter, R.; Hoffmann, R.; Ellrichmann, G.; Dragun, D.; Waschbisch, A.; Stegbauer, J.; et al. Increase of Angiotensin II Type 1 Receptor Auto-Antibodies in Huntington’s Disease. Mol. Neurodegener. 2014, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushara, K.O.; Nance, M.; Gomez, C.M. Antigliadin Antibodies in Huntington’s Disease. Neurology 2004, 62, 132–133. [Google Scholar] [CrossRef]
- Aziz, N.A.; Pijl, H.; Frölich, M.; Schröder-van der Elst, J.P.; Bent, C.; Roelfsema, F.; Roos, R.A.C. Delayed Onset of the Diurnal Melatonin Rise in Patients with Huntington’s Disease. J. Neurol. 2009, 256, 1961–1965. [Google Scholar] [CrossRef] [Green Version]
- Herzog-Krzywoszanska, R.; Krzywoszanski, L. Sleep Disorders in Huntington’s Disease. Front. Psychiatry 2019, 10, 221. [Google Scholar] [CrossRef] [Green Version]
- Kalliolia, E.; Silajdžić, E.; Nambron, R.; Hill, N.R.; Doshi, A.; Frost, C.; Watt, H.; Hindmarsh, P.; Björkqvist, M.; Warner, T.T. Plasma Melatonin Is Reduced in Huntington’s Disease. Mov. Disord. 2014, 29, 1511–1515. [Google Scholar] [CrossRef] [Green Version]
- Kalliolia, E.; Silajdžić, E.; Nambron, R.; Costelloe, S.J.; Martin, N.G.; Hill, N.R.; Frost, C.; Watt, H.C.; Hindmarsh, P.; Björkqvist, M.; et al. A 24-Hour Study of the Hypothalamo-Pituitary Axes in Huntington’s Disease. PLoS ONE 2015, 10, e0138848. [Google Scholar] [CrossRef] [PubMed]
- Popovic, V.; Svetel, M.; Djurovic, M.; Petrovic, S.; Doknic, M.; Pekic, S.; Miljic, D.; Milic, N.; Glodic, J.; Dieguez, C.; et al. Circulating and Cerebrospinal Fluid Ghrelin and Leptin: Potential Role in Altered Body Weight in Huntington’s Disease. Eur. J. Endocrinol. 2004, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Muñoz, M.E.; Arrieta, M.-C.; Ramer-Tait, A.E.; Walter, J. A Critical Assessment of the “Sterile Womb” and “in Utero Colonization” Hypotheses: Implications for Research on the Pioneer Infant Microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The Gut Microbiota Influences Blood-Brain Barrier Permeability in Mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [Green Version]
- Alonso, R.; Pisa, D.; Carrasco, L. Brain Microbiota in Huntington’s Disease Patients. Front. Microbiol. 2019, 10, 2622. [Google Scholar] [CrossRef]
- Wasser, C.I.; Mercieca, E.-C.; Kong, G.; Hannan, A.J.; McKeown, S.J.; Glikmann-Johnston, Y.; Stout, J.C. Gut Dysbiosis in Huntington’s Disease: Associations among Gut Microbiota, Cognitive Performance and Clinical Outcomes. Brain Commun. 2020, 2, fcaa110. [Google Scholar] [CrossRef]
- Kong, G.; Cao, K.-A.L.; Judd, L.M.; Li, S.; Renoir, T.; Hannan, A.J. Microbiome Profiling Reveals Gut Dysbiosis in a Transgenic Mouse Model of Huntington’s Disease. Neurobiol. Dis. 2020, 135, 104268. [Google Scholar] [CrossRef]
- Stan, T.L.; Soylu-Kucharz, R.; Burleigh, S.; Prykhodko, O.; Cao, L.; Franke, N.; Sjögren, M.; Haikal, C.; Hållenius, F.; Björkqvist, M. Increased Intestinal Permeability and Gut Dysbiosis in the R6/2 Mouse Model of Huntington’s Disease. Sci. Rep. 2020, 10, 18270. [Google Scholar] [CrossRef]
- Fang, P.; Kazmi, S.A.; Jameson, K.G.; Hsiao, E.Y. The Microbiome as a Modifier of Neurodegenerative Disease Risk. Cell Host Microbe 2020, 28, 201–222. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host Microbiota Constantly Control Maturation and Function of Microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.; Moutereau, S.; Durr, A.; Krystkowiak, P.; Azulay, J.-P.; Tranchant, C.; Broussolle, E.; Morin, F.; Bachoud-Lévi, A.-C.; Maison, P. Neuroendocrine Disturbances in Huntington’s Disease. PLoS ONE 2009, 4, e4962. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.-H.; Wu, Y.-R.; Chen, Y.-C.; Chen, C.-M. Plasma Inflammatory Biomarkers for Huntington’s Disease Patients and Mouse Model. Brain Behav. Immun. 2015, 44, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. IJMS 2020, 21, 9338. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, A.; Turnu, L.; Porro, B.; Squellerio, I.; Cavalca, V.; Tremoli, E.; Di Minno, M.N.D. 8-Hydroxy-2-Deoxyguanosine Levels and Cardiovascular Disease: A Systematic Review and Meta-Analysis of the Literature. Antioxid. Redox Signal. 2016, 24, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, D.; Ramos, K.S.; Baks, L.; Wiersma, M.; Lanters, E.A.H.; Bogers, A.J.J.C.; de Groot, N.M.S.; Brundel, B.J.J.M. Blood-Based 8-Hydroxy-2′-Deoxyguanosine Level: A Potential Diagnostic Biomarker for Atrial Fibrillation. Heart Rhythm. 2021, 18, 271–277. [Google Scholar] [CrossRef]
- Appl, T.; Kaltenbach, L.; Lo, D.C.; Terstappen, G.C. Targeting Mutant Huntingtin for the Development of Disease-Modifying Therapy. Drug Discov. Today 2012, 17, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the Proteostasis Network in Huntington’s Disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Fellmann, C.; Gowen, B.G.; Lin, P.-C.; Doudna, J.A.; Corn, J.E. Cornerstones of CRISPR–Cas in Drug Discovery and Therapy. Nat. Rev. Drug Discov. 2017, 16, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.-L.; Sánchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for Efficient Noninvasive Gene Delivery to the Central and Peripheral Nervous Systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-Responsive Gut Commensal Modulates TH17 Axis and Disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef] [PubMed]

| Company | Technology/Strategy | Molecule | Clinical Phase |
|---|---|---|---|
| Ionis Pharmaceuticals/Roche | ASO/Non-allele-selective | RG6042 | I/II—completed III—scheduled for completion in 2023 |
| Wave Life Sciences/Takeda | ASO/Allele-selective | WVE-120101, WVE-120102 | I/II—scheduled for completion in 2020 |
| UniQure Biopharma | RNAi/Non-allele-selective | rAAV5-miHTT (AMT-130) | I/II—scheduled for completion in 2022 |
| Voyager/Sanofi-Genzyme | RNAi/Non-allele-selective | AAV-shRNA (VY-HTT01) | - |
| Spark Therapeutics | RNAi/Non-allele-selective | AAV-shRNA | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Przybyl, L.; Wozna-Wysocka, M.; Kozlowska, E.; Fiszer, A. What, When and How to Measure—Peripheral Biomarkers in Therapy of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 1561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041561
Przybyl L, Wozna-Wysocka M, Kozlowska E, Fiszer A. What, When and How to Measure—Peripheral Biomarkers in Therapy of Huntington’s Disease. International Journal of Molecular Sciences. 2021; 22(4):1561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041561
Chicago/Turabian StylePrzybyl, Lukasz, Magdalena Wozna-Wysocka, Emilia Kozlowska, and Agnieszka Fiszer. 2021. "What, When and How to Measure—Peripheral Biomarkers in Therapy of Huntington’s Disease" International Journal of Molecular Sciences 22, no. 4: 1561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041561