Targeting for Success: Demonstrating Proof-of-Concept with Mechanistic Early Phase Clinical Pharmacology Studies for Disease-Modification in Neurodegenerative Disorders


Abstract
:1. Introduction
2. Neurodegenerative Disease Mechanisms
3. Innovative Drug Development of Disease Modifying Treatments
4. Biomarkers
5. Early Phase Proof-of-Concept with Mechanistic Biomarkers
6. Reported Use and Classification of Early Clinical Phase Biomarkers
6.1. Target Occupancy
6.2. Target Activation
6.3. Physiological Response
6.4. Pathophysiological Response
6.5. Clinical Response
7. Biomarker Sources
8. Biomarker Selection, Development, and Validation
9. Limitations
10. Roadmap for Mechanistic, Data-Rich Early Phase Clinical Pharmacology Studies
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Gribkoff, V.K.; Kaczmarek, L.K. The need for new approaches in CNS drug discovery: Why drugs have failed, and what can be done to improve outcomes. Neuropharmacology 2017, 120, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J. Disease modification and Neuroprotection in neurodegenerative disorders. Transl. Neurodegener. 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Plascencia-Villa, G.; Perry, G. Status and future directions of clinical trials in Alzheimer’s disease. In International Review of Neurobiology; Academic Press Inc.: Cambridge, MA, USA, 2020; Volume 154, pp. 3–50. ISBN 9780128200766. [Google Scholar]
- Travessa, A.M.; Rodrigues, F.B.; Mestre, T.A.; Ferreira, J.J. Fifteen years of clinical trials in Huntington’s disease: A very low clinical drug development success rate. J. Huntingtons. Dis. 2017, 6, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.; Mitchell, J.; Lyon, M.; Moore, D. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). In Cochrane Database of Systematic Reviews; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2002. [Google Scholar]
- Hardiman, O.; van den Berg, L.H. Edaravone: A new treatment for ALS on the horizon? Lancet Neurol. 2017, 16, 490–491. [Google Scholar] [CrossRef] [Green Version]
- Maharshi, V.; Hasan, S. Nusinersen: The First Option beyond Supportive Care for Spinal Muscular Atrophy. Clin. Drug Investig. 2017, 37, 807–817. [Google Scholar] [CrossRef]
- Hodges, J.R.; Piguet, O. Progress and Challenges in Frontotemporal Dementia Research: A 20-Year Review. J. Alzheimer’s Dis. 2018, 62, 1467–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrar, M.A.; Park, S.B.; Vucic, S.; Carey, K.A.; Turner, B.J.; Gillingwater, T.H.; Swoboda, K.J.; Kiernan, M.C. Emerging therapies and challenges in spinal muscular atrophy. Ann. Neurol. 2017, 81, 355–368. [Google Scholar] [CrossRef]
- Ashizawa, T.; Öz, G.; Paulson, H.L. Spinocerebellar ataxias: Prospects and challenges for therapy development. Nat. Rev. Neurol. 2018, 14, 590–605. [Google Scholar] [CrossRef]
- Zeuner, K.E.; Schäffer, E.; Hopfner, F.; Brüggemann, N.; Berg, D. Progress of Pharmacological Approaches in Parkinson’s Disease. Clin. Pharmacol. Ther. 2019, 105, 1106–1120. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Dominey, T.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2020. J. Parkinsons. Dis. 2020, 10, 757–774. [Google Scholar] [CrossRef]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- McGhee, D.J.M.; Ritchie, C.W.; Zajicek, J.P.; Counsell, C.E. A review of clinical trial designs used to detect a disease-modifying effect of drug therapy in Alzheimer’s disease and Parkinson’s disease. BMC Neurol. 2016, 16, 92. [Google Scholar] [CrossRef] [Green Version]
- Henchcliffe, C.; Severt, W.L. Disease modification in Parkinson’s disease. Drugs Aging 2011, 28, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Obrocki, P.; Khatun, A.; Ness, D.; Senkevich, K.; Hanrieder, J.; Capraro, F.; Mattsson, N.; Andreasson, U.; Portelius, E.; Ashton, N.J.; et al. Perspectives in fluid biomarkers in neurodegeneration from the 2019 biomarkers in neurodegenerative diseases course—A joint PhD student course at University College London and University of Gothenburg. Alzheimer’s Res. Ther. 2020, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Bakkar, N.; Boehringer, A.; Bowser, R. Use of biomarkers in ALS drug development and clinical trials. Brain Res. 2015, 1607, 94–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Degroot, A. Biomarker-Guided Drug Development for Better Defined Early Patient Studies and Clinical Trial Efficiency. In Handbook of Behavioral Neuroscience; Elsevier B.V.: Amsterdam, The Netherlands, 2019; Volume 29, pp. 17–23. [Google Scholar]
- Beach, T.G. A Review of Biomarkers for Neurodegenerative Disease: Will They Swing Us Across the Valley? Neurol. Ther. 2017, 6, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.B. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp. Neurol. 2017, 298, 236–245. [Google Scholar] [CrossRef]
- Macaluso, M.; Krams, M.; Savitz, J.; Drevets, W.C.; Preskorn, S.H. New Approaches in Translational Medicine for Phase I Clinical Trials of CNS Drugs. In Handbook of Behavioral Neuroscience; Elsevier B.V.: Amsterdam, The Netherlands, 2019; Volume 29, pp. 81–91. [Google Scholar]
- Cohen, A.F.; Burggraaf, J.; Van Gerven, J.M.A.; Moerland, M.; Groeneveld, G.J. The use of biomarkers in human pharmacology (Phase I) studies. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 55–74. [Google Scholar] [CrossRef]
- Nagai, Y.; Eiko, M. Drug development for neurodegenerative diseases. In Neurodegenerative Disorders as Systemic Diseases; Springer Japan: Tokyo, Japan, 2015; pp. 183–216. ISBN 9784431545415. [Google Scholar]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Grievink, H.W.; Heuberger, J.A.A.C.; Huang, F.; Chaudhary, R.; Birkhoff, W.A.J.; Tonn, G.R.; Mosesova, S.; Erickson, R.; Moerland, M.; Haddick, P.C.G.; et al. DNL104, a Centrally Penetrant RIPK1 Inhibitor, Inhibits RIP1 Kinase Phosphorylation in a Randomized Phase I Ascending Dose Study in Healthy Volunteers. Clin. Pharmacol. Ther. 2020, 107, 406–414. [Google Scholar] [CrossRef]
- West, T.; Hu, Y.; Verghese, P.B.; Bateman, R.J.; Braunstein, J.B.; Fogelman, I.; Budur, K.; Florian, H.; Mendonca, N.; Holtzman, D.M. Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J. Prev. Alzheimer’s Dis. 2017, 4, 236–241. [Google Scholar]
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Sardi, S.P.; Cedarbaum, J.M.; Brundin, P. Targeted Therapies for Parkinson’s Disease: From Genetics to the Clinic. Mov. Disord. 2018, 33, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Visser, S.J.; Cohen, A.F.; Kenter, M.J.H. Integrating scientific considerations into R&D project valuation. Nat. Biotechnol. 2020, 38, 14–18. [Google Scholar] [PubMed]
- Cummings, J.; Fox, N. Defining Disease Modifying Therapy for Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2017, 4, 109–115. [Google Scholar]
- Atkinson, A.J.; Colburn, W.A.; DeGruttola, V.G.; DeMets, D.L.; Downing, G.J.; Hoth, D.F.; Oates, J.A.; Peck, C.C.; Schooley, R.T.; Spilker, B.A.; et al. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Amur, S.; Lavange, L.; Zineh, I.; Buckman-Garner, S.; Woodcock, J. Biomarker qualification: Toward a multiple stakeholder framework for biomarker development, regulatory acceptance, and utilization. Clin. Pharmacol. Ther. 2015, 98, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Verber, N.S.; Shepheard, S.R.; Sassani, M.; McDonough, H.E.; Moore, S.A.; Alix, J.J.P.; Wilkinson, I.D.; Jenkins, T.M.; Shaw, P.J. Biomarkers in motor neuron disease: A state of the art review. Front. Neurol. 2019, 10, 291. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Zetterberg, H. The Past and the Future of Alzheimer’s Disease Fluid Biomarkers. J. Alzheimer’s Dis. 2018, 62, 1125–1140. [Google Scholar] [CrossRef] [Green Version]
- Parnetti, L.; Gaetani, L.; Eusebi, P.; Paciotti, S.; Hansson, O.; El-Agnaf, O.; Mollenhauer, B.; Blennow, K.; Calabresi, P. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 2019, 18, 573–586. [Google Scholar] [CrossRef]
- Silajdzic, E.; Bjorkqvist, M. A critical evaluation of wet biomarkers for huntington’s disease: Current status and ways forward. J. Huntingtons. Dis. 2018, 7, 109–135. [Google Scholar] [CrossRef] [Green Version]
- Coarelli, G.; Brice, A.; Durr, A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view [version 1; referees: 3 approved]. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Cummings, J. The Role of Biomarkers in Alzheimer’s Disease Drug Development. In Advances in Experimental Medicine and Biology; Springer New York LLC: New York, NY, USA, 2019; Volume 1118, pp. 29–61. [Google Scholar]
- Danhof, M.; Alvan, G.; Dahl, S.G.; Kuhlmann, J.; Paintaud, G. Mechanism-based pharmacokinetic-pharmacodynamic modeling—A new classification of biomarkers. Pharm. Res. 2005, 22, 1432–1437. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve RD productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry; US Food and Drug Administration: Silver Spring, MD, USA, 2019. [Google Scholar]
- Committee for Medicinal Products for Human Use (CHMP). Guideline on the Clinical Investigation of Medicines for the Treatment of Alzheimer’s Disease; CHMP: London, UK, 2018. [Google Scholar]
- Committee for Medicinal Product for Human Use (CHMP). Guideline on Clinical Investigation of Medicinal Products for the Treatment of Amyotrophic Lateral Sclerosis (ALS); CHMP: London, UK, 2013. [Google Scholar]
- Cohen, A.F. Developing drug prototypes: Pharmacology replaces safety and tolerability? Nat. Rev. Drug Discov. 2010, 9, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Committee for Medicinal Products for Human Use (CHMP). Guideline on Strategies to Identify and Mitigate Risks for First-in-Human Clinical Trials with Investigational Medicinal Products; CHMP: London, UK, 2017. [Google Scholar]
- Ferrero, J.; Williams, L.; Stella, H.; Leitermann, K.; Mikulskis, A.; O’Gorman, J.; Sevigny, J. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401—A clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers’s Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, I.; Fujimoto, Y.; Yamada, M.; Abe, S.; Zhao, Q.; Cronenberger, C.; Togo, K.; Ishibashi, T.; Bednar, M.M.; Kupiec, J.W.; et al. Safety and pharmacokinetics of PF-04360365 following a single-dose intravenous infusion in Japanese subjects with mild-to-moderate Alzheimer’s disease: A multicenter, randomized, double-blind, placebo-controlled, dose-escalation study. Int. J. Clin. Pharmacol. Ther. 2013, 51, 911–923. [Google Scholar] [CrossRef]
- Landen, J.W.; Zhao, Q.; Cohen, S.; Borrie, M.; Woodward, M.; Billing, C.B.; Bales, K.; Alvey, C.; McCush, F.; Yang, J.; et al. Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate alzheimer disease: A phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin. Neuropharmacol. 2013, 36, 14–23. [Google Scholar] [CrossRef]
- Li, L.; Zhen, E.Y.; Decker, R.L.; Willis, B.A.; Waters, D.; Liu, P.; Hake, A.M.; Demattos, R.B.; Ayan-Oshodi, M. Pharmacokinetics and Pharmacodynamics of LY2599666, a PEG-Linked Antigen Binding Fragment that Targets Soluble Monomer Amyloid-β. J. Alzheimer’s Dis. 2019, 68, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Brashear, H.R. Pharmacokinetics, Pharmacodynamics, and Safety of Subcutaneous Bapineuzumab: A Single-Ascending-Dose Study in Patients with Mild to Moderate Alzheimer Disease. Clin. Pharmacol. Drug Dev. 2019, 8, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Umemura, K.; Ichimiya, Y.; Iseki, E.; Eto, K.; Miyakawa, K.; Kirino, E.; Shibata, N.; Baba, H.; Tsuchiwata, S. Safety and pharmacokinetics of bapineuzumab in a single ascending-dose study in Japanese patients with mild to moderate Alzheimer’s disease. Geriatr. Gerontol. Int. 2016, 16, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Leyhe, T.; Andreasen, N.; Simeoni, M.; Reich, A.; Von Arnim, C.A.F.; Tong, X.; Yeo, A.; Khan, S.; Loercher, A.; Chalker, M.; et al. Modulation of β-amyloid by a single dose of GSK933776 in patients with mild Alzheimer’s disease: A phase i study. Alzheimer’s Res. Ther. 2014, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Black, R.S.; Sperling, R.A.; Safirstein, B.; Motter, R.N.; Pallay, A.; Nichols, A.; Grundman, M. A single ascending dose study of bapineuzumab in patients with alzheimer disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K.; et al. An effector-reduced anti-β-amyloid (Aβ) antibody with unique Aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 2012, 32, 9677–9689. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Ichimiya, Y.; Shibata, N.; Nakajima, T.; Sudoh, S.; Tokuda, T.; Sujaku, T.; Yokokawa, S.; Hoshii, N.; Noguchi, H.; et al. Safety and tolerability of immune globulin intravenous (human), 10% solution in Japanese subjects with mild to moderate Alzheimer’s disease. Psychogeriatrics 2014, 14, 165–174. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Tirucherai, G.; Ahlijanian, M.K.; Kolaitis, G.; Bechtold, C.; Grundman, M. A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Seo, S.W.; Chang, J.W.; Lee, J., II; Kim, C.H.; Chin, J.; Choi, S.J.; Kwon, H.; Yun, H.J.; Lee, J.M.; et al. Stereotactic brain injection of human umbilical cord blood mesenchymal stem cells in patients with Alzheimer’s disease dementia: A phase 1 clinical trial. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2015, 1, 95–102. [Google Scholar] [CrossRef]
- Wahlberg, L.U.; Lind, G.; Almqvist, P.M.; Kusk, P.; Tornøe, J.; Juliusson, B.; Söderman, M.; Selldén, E.; Seiger, Å.; Eriksdotter-Jönhagen, M.; et al. Targeted delivery of nerve growth factor via encapsulated cell biodelivery in Alzheimer disease: A technology platform for restorative neurosurgery-Clinical article. J. Neurosurg. 2012, 117, 340–347. [Google Scholar] [CrossRef]
- Nolan, J.M.; Mulcahy, R.; Power, R.; Moran, R.; Howard, A.N. Nutritional Intervention to Prevent Alzheimer’s Disease: Potential Benefits of Xanthophyll Carotenoids and Omega-3 Fatty Acids Combined. J. Alzheimer’s Dis. 2018, 64, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafii, M.S.; Baumann, T.L.; Bakay, R.A.E.; Ostrove, J.M.; Siffert, J.; Fleisher, A.S.; Herzog, C.D.; Barba, D.; Pay, M.; Salmon, D.P.; et al. A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimers. Dement. 2014, 10, 571–581. [Google Scholar] [CrossRef]
- Eyjolfsdottir, H.; Eriksdotter, M.; Linderoth, B.; Lind, G.; Juliusson, B.; Kusk, P.; Almkvist, O.; Andreasen, N.; Blennow, K.; Ferreira, D.; et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimer’s Res. Ther. 2016, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Wang, P.N.; Chiu, M.J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.H.; De Fang, X.; Zhao, K.; Hung, C.H.; et al. UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604. [Google Scholar] [CrossRef]
- Lacosta, A.M.; Pascual-Lucas, M.; Pesini, P.; Casabona, D.; Pérez-Grijalba, V.; Marcos-Campos, I.; Sarasa, L.; Canudas, J.; Badi, H.; Monleón, I.; et al. Safety, tolerability and immunogenicity of an active anti-Aβ 40 vaccine (ABvac40) in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase i trial. Alzheimer’s Res. Ther. 2018, 10, 12. [Google Scholar] [CrossRef]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Zilka, N.; Kovacech, B.; Skrabana, R.; Vince-Kazmerova, Z.; Katina, S.; Fialova, L.; Prcina, M.; et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017, 16, 123–134. [Google Scholar] [CrossRef]
- Kutzsche, J.; Jürgens, D.; Willuweit, A.; Adermann, K.; Fuchs, C.; Simons, S.; Windisch, M.; Hümpel, M.; Rossberg, W.; Wolzt, M.; et al. Safety and pharmacokinetics of the orally available antiprionic compound PRI-002: A single and multiple ascending dose phase I study. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12001. [Google Scholar] [CrossRef]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood–brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 2018, 9, 2336. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.S.; Laxton, A.W.; Tang-Wai, D.F.; McAndrews, M.P.; Diaconescu, A.O.; Workman, C.I.; Lozano, A.M. Increased cerebral metabolism after 1 year of deep brain stimulation in Alzheimer disease. Arch. Neurol. 2012, 69, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Noreik, M.; Kuhn, J.; Hardenacke, K.; Lenartz, D.; Bauer, A.; Bührle, C.P.; Häussermann, P.; Hellmich, M.; Klosterkötter, J.; Wiltfang, J.; et al. Changes in nutritional status after deep brain stimulation of the nucleus basalis of Meynert in Alzheimer’s disease—Results of a phase I study. J. Nutr. Health Aging 2015, 19, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Laxton, A.W.; Tang-Wai, D.F.; McAndrews, M.P.; Zumsteg, D.; Wennberg, R.; Keren, R.; Wherrett, J.; Naglie, G.; Hamani, C.; Smith, G.S.; et al. A phase i trial of deep brain stimulation of memory circuits in Alzheimer’s disease. Ann. Neurol. 2010, 68, 521–534. [Google Scholar] [CrossRef]
- Scharre, D.W.; Weichart, E.; Nielson, D.; Zhang, J.; Agrawal, P.; Sederberg, P.B.; Knopp, M.V.; Rezai, A.R. Deep Brain Stimulation of Frontal Lobe Networks to Treat Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Family, N.; Maillet, E.L.; Williams, L.T.J.; Krediet, E.; Carhart-Harris, R.L.; Williams, T.M.; Nichols, C.D.; Goble, D.J.; Raz, S. Safety, tolerability, pharmacokinetics, and pharmacodynamics of low dose lysergic acid diethylamide (LSD) in healthy older volunteers. Psychopharmacology 2020, 237, 841–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maccecchini, M.L.; Chang, M.Y.; Pan, C.; John, V.; Zetterberg, H.; Greig, N.H. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-β peptide and τ levels: Target engagement, tolerabilityand pharmacokinetics in humans. J. Neurol. Neurosurg. Psychiatry 2012, 83, 894–902. [Google Scholar] [CrossRef]
- Brazier, D.; Perry, R.; Keane, J.; Barrett, K.; Elmaleh, D.R. Pharmacokinetics of Cromolyn and Ibuprofen in Healthy Elderly Volunteers. Clin. Drug Investig. 2017, 37, 1025–1034. [Google Scholar] [CrossRef]
- Forman, M.; Palcza, J.; Tseng, J.; Stone, J.A.; Walker, B.; Swearingen, D.; Troyer, M.D.; Dockendorf, M.F. Safety, Tolerability, and Pharmacokinetics of the β-Site Amyloid Precursor Protein-Cleaving Enzyme 1 Inhibitor Verubecestat (MK-8931) in Healthy Elderly Male and Female Subjects. Clin. Transl. Sci. 2019, 12, 545–555. [Google Scholar] [CrossRef]
- Chris Min, K.; Dockendorf, M.F.; Palcza, J.; Tseng, J.; Ma, L.; Stone, J.A.; Kleijn, H.J.; Hodsman, P.; Masuo, K.; Tanen, M.; et al. Pharmacokinetics and Pharmacodynamics of the BACE1 Inhibitor Verubecestat (MK-8931) in Healthy Japanese Adults: A Randomized, Placebo-Controlled Study. Clin. Pharmacol. Ther. 2019, 105, 1234–1243. [Google Scholar] [CrossRef]
- Timmers, M.; Streffer, J.R.; Russu, A.; Tominaga, Y.; Shimizu, H.; Shiraishi, A.; Tatikola, K.; Smekens, P.; Börjesson-Hanson, A.; Andreasen, N.; et al. Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: Randomized, double-blind, placebo-controlled study. Alzheimer’s Res. Ther. 2018, 10, 85. [Google Scholar] [CrossRef]
- Sakamoto, K.; Matsuki, S.; Matsuguma, K.; Yoshihara, T.; Uchida, N.; Azuma, F.; Russell, M.; Hughes, G.; Haeberlein, S.B.; Alexander, R.C.; et al. BACE1 Inhibitor Lanabecestat (AZD3293) in a Phase 1 Study of Healthy Japanese Subjects: Pharmacokinetics and Effects on Plasma and Cerebrospinal Fluid Aβ Peptides. J. Clin. Pharmacol. 2017, 57, 1460–1471. [Google Scholar] [CrossRef]
- Cebers, G.; Alexander, R.C.; Haeberlein, S.B.; Han, D.; Goldwater, R.; Ereshefsky, L.; Olsson, T.; Ye, N.; Rosen, L.; Russell, M.; et al. AZD3293: Pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 55, 1039–1053. [Google Scholar] [CrossRef]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS b-Amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8, ra150–ra363. [Google Scholar] [CrossRef] [PubMed]
- Timmers, M.; Van Broeck, B.; Ramael, S.; Slemmon, J.; De Waepenaert, K.; Russu, A.; Bogert, J.; Stieltjes, H.; Shaw, L.M.; Engelborghs, S.; et al. Profiling the dynamics of CSF and plasma Aβ reduction after treatment with JNJ-54861911, a potent oral BACE inhibitor. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, R.; Ahn, J.E.; Alexander, R.; Brodney, M.A.; He, P.; Leurent, C.; Mancuso, J.; Margolin, R.A.; Tankisheva, E.; Chen, D.; et al. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamic Effects of PF-06751979, a Potent and Selective Oral BACE1 Inhibitor: Results from Phase i Studies in Healthy Adults and Healthy Older Subjects. J. Alzheimer’s Dis. 2019, 71, 581–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulati, A.; Hornick, M.G.; Briyal, S.; Lavhale, M.S. A Novel Neuroregenerative Approach Using ET B Receptor Agonist, IRL-1620, to Treat CNS Disorders. Physiol. Res 2018, 67, 95–113. [Google Scholar] [CrossRef]
- Lues, I.; Weber, F.; Meyer, A.; Bühring, U.; Hoffmann, T.; Kühn-Wache, K.; Manhart, S.; Heiser, U.; Pokorny, R.; Chiesa, J.; et al. A phase 1 study to evaluate the safety and pharmacokinetics of PQ912, a glutaminyl cyclase inhibitor, in healthy subjects This work was previously published as abstracts presented at AAIC 2013 Conference Boston. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2015, 1, 182–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgievska, B.; Sandin, J.; Doherty, J.; Mörtberg, A.; Neelissen, J.; Andersson, A.; Gruber, S.; Nilsson, Y.; Schött, P.; Arvidsson, P.I.; et al. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J. Neurochem. 2013, 125, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Grundman, M.; Morgan, R.; Lickliter, J.D.; Schneider, L.S.; DeKosky, S.; Izzo, N.J.; Guttendorf, R.; Higgin, M.; Pribyl, J.; Mozzoni, K.; et al. A phase 1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a novel therapeutic candidate for Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 20–26. [Google Scholar] [CrossRef]
- Ahn, J.E.; Carrieri, C.; Dela Cruz, F.; Fullerton, T.; Hajos-Korcsok, E.; He, P.; Kantaridis, C.; Leurent, C.; Liu, R.; Mancuso, J.; et al. Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF-06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies. Clin. Pharmacol. Ther. 2020, 107, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Logovinsky, V.; Schuck, E.; Kaplow, J.; Chang, M.K.; Miyagawa, T.; Wong, N.; Ferry, J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel γ-secretase modulator, E2212, in healthy human subjects. J. Clin. Pharmacol. 2014, 54, 528–536. [Google Scholar] [CrossRef]
- Tsai, R.M.; Miller, Z.; Koestler, M.; Rojas, J.C.; Ljubenkov, P.A.; Rosen, H.J.; Rabinovici, G.D.; Fagan, A.M.; Cobigo, Y.; Brown, J.A.; et al. Reactions to Multiple Ascending Doses of the Microtubule Stabilizer TPI-287 in Patients with Alzheimer Disease, Progressive Supranuclear Palsy, and Corticobasal Syndrome: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 215–224. [Google Scholar] [CrossRef]
- Duma, C.; Kopyov, O.; Kopyov, A.; Berman, M.; Lander, E.; Elam, M.; Arata, M.; Weiland, D.; Cannell, R.; Caraway, C.; et al. Human intracerebroventricular (ICV) injection of autologous, non-engineered, adipose-derived stromal vascular fraction (ADSVF) for neurodegenerative disorders: Results of a 3-year phase 1 study of 113 injections in 31 patients. Mol. Biol. Rep. 2019, 46, 5257–5272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meininger, V.; Pradat, P.F.; Corse, A.; Al-Sarraj, S.; Brooks, B.R.; Caress, J.B.; Cudkowicz, M.; Kolb, S.J.; Lange, D.; Leigh, P.N.; et al. Safety, pharmacokinetic, and functional effects of the Nogo-A monoclonal antibody in amyotrophic lateral sclerosis: A randomized, first-in-human clinical trial. PLoS ONE 2014, 9, e97803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Oh, K.-W.; Moon, C.; Kim, H.Y.; Oh, S.; Park, J.; Lee, J.H.; Chang, I.Y.; Kim, K.S.; Kim, S.H. Phase I Trial of Repeated Intrathecal Autologous Bone Marrow-Derived Mesenchymal Stromal Cells in Amyotrophic Lateral Sclerosis. Stem Cells Transl. Med. 2015, 4, 590–597. [Google Scholar] [CrossRef]
- Mazzini, L.; Ferrero, I.; Luparello, V.; Rustichelli, D.; Gunetti, M.; Mareschi, K.; Testa, L.; Stecco, A.; Tarletti, R.; Miglioretti, M.; et al. Mesenchymal stem cell transplantation in amyotrophic lateral sclerosis: A Phase I clinical trial. Exp. Neurol. 2010, 223, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Blanquer, M.; Moraleda, J.M.; Iniesta, F.; Gómez-Espuch, J.; Meca-Lallana, J.; Villaverde, R.; Pérez-Espejo, M.Á.; Ruíz-López, F.J.; Santos, J.M.G.; Bleda, P.; et al. Neurotrophic bone marrow cellular nests prevent spinal motoneuron degeneration in amyotrophic lateral sclerosis patients: A pilot safety study. Stem Cells 2012, 30, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Petrou, P.; Gothelf, Y.; Argov, Z.; Gotkine, M.; Levy, Y.S.; Kassis, I.; Vaknin-Dembinsky, A.; Ben-Hur, T.; Offen, D.; Abramsky, O.; et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis. JAMA Neurol. 2016, 73, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Staff, N.P.; Madigan, N.N.; Morris, J.; Jentoft, M.; Sorenson, E.J.; Butler, G.; Gastineau, D.; Dietz, A.; Windebank, A.J. Safety of intrathecal autologous adipose-derived mesenchymal stromal cells in patients with ALS. Neurology 2016, 87, 2230–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syková, E.; Rychmach, P.; Drahorádová, I.; Konrádová, Š.; Růžičková, K.; Voříšek, I.; Forostyak, S.; Homola, A.; Bojar, M. Transplantation of mesenchymal stromal cells in patients with amyotrophic lateral sclerosis: Results of phase I/IIa clinical trial. Cell Transplant. 2017, 26, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Mazzini, L.; Gelati, M.; Profico, D.C.; Sgaravizzi, G.; Projetti Pensi, M.; Muzi, G.; Ricciolini, C.; Rota Nodari, L.; Carletti, S.; Giorgi, C.; et al. Human neural stem cell transplantation in ALS: Initial results from a phase I trial. J. Transl. Med. 2015, 13, 17. [Google Scholar] [CrossRef]
- Feldman, E.L.; Boulis, N.M.; Hur, J.; Johe, K.; Rutkove, S.B.; Federici, T.; Polak, M.; Bordeau, J.; Sakowski, S.A.; Glass, J.D. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: Phase 1 trial outcomes. Ann. Neurol. 2014, 75, 363–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geijo-Barrientos, E.; Pastore-Olmedo, C.; De Mingo, P.; Blanquer, M.; Gómez Espuch, J.; Iniesta, F.; Iniesta, N.G.; García-Hernández, A.; Martín-Estefanía, C.; Barrios, L.; et al. Intramuscular Injection of Bone Marrow Stem Cells in Amyotrophic Lateral Sclerosis Patients: A Randomized Clinical Trial. Front. Neurosci. 2020, 14, 195. [Google Scholar] [CrossRef]
- Thonhoff, J.R.; Beers, D.R.; Zhao, W.; Pleitez, M.; Simpson, E.P.; Berry, J.D.; Cudkowicz, M.E.; Appel, S.H. Expanded autologous regulatory T-lymphocyte infusions in ALS A phase I, first-in-human study. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabavi, S.M.; Arab, L.; Jarooghi, N.; Bolurieh, T.; Abbasi, F.; Mardpour, S.; Azimyian, V.; Moeininia, F.; Maroufizadeh, S.; Sanjari, L.; et al. Safety, feasibility of intravenous and intrathecal injection of autologous bone marrow derived mesenchymal stromal cells in patients with amyotrophic lateral sclerosis: An open label phase I clinical trial. Cell J. 2019, 20, 592–598. [Google Scholar]
- Riley, J.; Glass, J.; Feldman, E.L.; Polak, M.; Bordeau, J.; Federici, T.; Johe, K.; Boulis, N.M. Intraspinal stem cell transplantation in amyotrophic lateral sclerosis: A phase I trial, cervical microinjection, and final surgical safety outcomes. Neurosurgery 2014, 74, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Glass, J.D.; Boulis, N.M.; Johe, K.; Rutkove, S.B.; Federici, T.; Polak, M.; Kelly, C.; Feldman, E.L. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: Results of a phase I trial in 12 patients. Stem Cells 2012, 30, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Sufit, R.L.; Ajroud-Driss, S.; Casey, P.; Kessler, J.A. Open label study to assess the safety of VM202 in subjects with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 269–278. [Google Scholar] [CrossRef]
- Chiò, A.; Mora, G.; La Bella, V.; Caponnetto, C.; Mancardi, G.; Sabatelli, M.; Siciliano, G.; Silani, V.; Corbo, M.; Moglia, C.; et al. Repeated courses of granulocyte colony-stimulating factor in amyotrophic lateral sclerosis: Clinical and biological results from a prospective multicenter study. Muscle Nerve 2011, 43, 189–195. [Google Scholar] [CrossRef]
- Warita, H.; Kato, M.; Asada, R.; Yamashita, A.; Hayata, D.; Adachi, K.; Aoki, M. Safety, Tolerability, and Pharmacodynamics of Intrathecal Injection of Recombinant Human HGF (KP-100) in Subjects With Amyotrophic Lateral Sclerosis: A Phase I Trial. J. Clin. Pharmacol. 2019, 59, 677–687. [Google Scholar] [CrossRef]
- Berry, J.D.; Shefner, J.M.; Conwit, R.; Schoenfeld, D.; Keroack, M.; Felsenstein, D.; Krivickas, L.; David, W.S.; Vriesendorp, F.; Pestronk, A.; et al. Design and Initial Results of a Multi-Phase Randomized Trial of Ceftriaxone in Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e61177. [Google Scholar] [CrossRef] [PubMed]
- Bozik, M.E.; Mather, J.L.; Kramer, W.G.; Gribkoff, V.K.; Ingersoll, E.W. Safety, tolerability, and pharmacokinetics of KNS-760704 (dexpramipexole) in healthy adult subjects. J. Clin. Pharmacol. 2011, 51, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Zhang, R.; Block, G.; Katz, J.; Barohn, R.; Kasarskis, E.; Forshew, D.; Gopalakrishnan, V.; Mcgrath, M.S. NP001 regulation of macrophage activation markers in ALS: A phase I clinical and biomarker study. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, D.J.; Shahbazi, M.; Silani, V.; Ludolph, A.C.; Weishaupt, J.H.; Ajroud-Driss, S.; Fields, K.G.; Remanan, R.; Appel, S.H.; Morelli, C.; et al. Pyrimethamine significantly lowers cerebrospinal fluid Cu/Zn superoxide dismutase in amyotrophic lateral sclerosis patients with SOD1 mutations. Ann. Neurol. 2017, 81, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, D.J.; Andersen, P.M.; Remanan, R.; Marklund, S.; Benjamin, D. Pyrimethamine decreases levels of SOD1 in leukocytes and cerebrospinal fluid of ALS patients: A phase i pilot study. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.D.; Miller, R.G.; Bradley, W.G.; Moore, D.H.; Saperstein, D.S.; Flynn, L.E.; Katz, J.S.; Forshew, D.A.; Metcalf, J.S.; Banack, S.A.; et al. Phase I clinical trial of safety of L-serine for ALS patients. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 107–111. [Google Scholar] [CrossRef]
- Atassi, N.; Ratai, E.M.; Greenblatt, D.J.; Pulley, D.; Zhao, Y.; Bombardier, J.; Wallace, S.; Eckenrode, J.; Cudkowicz, M.; Dibernardo, A. A phase I, pharmacokinetic, dosage escalation study of creatine monohydrate in subjects with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, E.J.; Guo, S.; Benson, M.D.; Booten, S.; Freier, S.; Hughes, S.G.; Kim, T.W.; Jesse Kwoh, T.; Matson, J.; Norris, D.; et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid 2016, 23, 148–157. [Google Scholar] [CrossRef]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Soragni, E.; Miao, W.; Iudicello, M.; Jacoby, D.; De Mercanti, S.; Clerico, M.; Longo, F.; Piga, A.; Ku, S.; Campau, E.; et al. Epigenetic Therapy for Friedreich ataxia. Ann. Neurol. 2014, 76, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Libri, V.; Yandim, C.; Athanasopoulos, S.; Loyse, N.; Natisvili, T.; Law, P.P.; Chan, P.K.; Mohammad, T.; Mauri, M.; Tam, K.T.; et al. Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich’s ataxia: An exploratory, open-label, dose-escalation study. Lancet 2014, 384, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Zesiewicz, T.; Heerinckx, F.; De Jager, R.; Omidvar, O.; Kilpatrick, M.; Shaw, J.; Shchepinov, M.S. Randomized, clinical trial of RT001: Early signals of efficacy in Friedreich’s ataxia. Mov. Disord. 2018, 33, 1000–1005. [Google Scholar] [CrossRef]
- Sha, S.J.; Miller, Z.A.; Min, S.W.; Zhou, Y.; Brown, J.; Mitic, L.L.; Karydas, A.; Koestler, M.; Tsai, R.; Corbetta-Rastelli, C.; et al. An 8-week, open-label, dose-finding study of nimodipine for the treatment of progranulin insufficiency from GRN gene mutations. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.T.R.; Mahuran, D.J.; Sathe, S.; Kolodny, E.H.; Rigat, B.A.; Raiman, J.A.; Tropak, M.B. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Mol. Genet. Metab. 2011, 102, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Diemen, M.P.J.; Hart, E.P.; Abbruscato, A.; Mead, L.; Beelen, I.; Bergheanu, S.C.; Hameeteman, P.W.; Coppen, E.; Winder, J.Y.; Moerland, M.; et al. Safety, Pharmacokinetics and Pharmacodynamics of SBT-020 in Patients with Early Stage Huntington’s Disease, a two-part study. Br. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Guy, J.; Feuer, W.J.; Davis, J.L.; Porciatti, V.; Gonzalez, P.J.; Koilkonda, R.D.; Yuan, H.; Hauswirth, W.W.; Lam, B.L. Gene Therapy for Leber Hereditary Optic Neuropathy: Low- and Medium-Dose Visual Results. Ophthalmology 2017, 124, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Feuer, W.J.; Schiffman, J.C.; Davis, J.L.; Porciatti, V.; Gonzalez, P.; Koilkonda, R.D.; Yuan, H.; Lalwani, A.; Lam, B.L.; Guy, J. Gene Therapy for Leber Hereditary Optic Neuropathy: Initial Results. Ophthalmology 2016, 123, 558–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaGanke, C.; Samkoff, L.; Edwards, K.; Henson, L.J.; Repovic, P.; Lynch, S.; Stone, L.; Mattson, D.; Galluzzi, A.; Fisher, T.L.; et al. Safety/tolerability of the anti-semaphorin 4D antibody VX15/2503 in a randomized phase 1 trial. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.A.; Imrey, P.B.; Planchon, S.M.; Bermel, R.A.; Fisher, E.; Fox, R.J.; Bar-Or, A.; Sharp, S.L.; Skaramagas, T.T.; Jagodnik, P.; et al. Pilot trial of intravenous autologous culture-expanded mesenchymal stem cell transplantation in multiple sclerosis. Mult. Scler. J. 2018, 24, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karussis, D.; Karageorgiou, C.; Vaknin-Dembinsky, A.; Gowda-Kurkalli, B.; Gomori, J.M.; Kassis, I.; Bulte, J.W.M.; Petrou, P.; Ben-Hur, T.; Abramsky, O.; et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 1187–1194. [Google Scholar] [CrossRef]
- Ziemssen, T.; Tumani, H.; Sehr, T.; Thomas, K.; Paul, F.; Richter, N.; Samara, E.; Spiegelstein, O.; Sorani, E.; Bar-Ilan, O.; et al. Safety and in vivo immune assessment of escalating doses of oral laquinimod in patients with RRMS. J. Neuroinflamm. 2017, 14, 172. [Google Scholar] [CrossRef]
- Kosa, P.; Wu, T.; Phillips, J.; Leinonen, M.; Masvekar, R.; Komori, M.; Wichman, A.; Sandford, M.; Bielekova, B. Idebenone does not inhibit disability progression in primary progressive MS. Mult. Scler. Relat. Disord. 2020, 45, 102434. [Google Scholar] [CrossRef]
- Singer, W.; Dietz, A.B.; Zeller, A.D.; Gehrking, T.L.; Schmelzer, J.D.; Schmeichel, A.M.; Gehrking, J.A.; Suarez, M.D.; Sletten, D.M.; Minota Pacheco, K.V.; et al. Intrathecal administration of autologous mesenchymal stem cells in multiple system atrophy. Neurology 2019, 93, E77–E87. [Google Scholar] [CrossRef] [PubMed]
- Meissner, W.G.; Traon, A.P.L.; Foubert-Samier, A.; Galabova, G.; Galitzky, M.; Kutzelnigg, A.; Laurens, B.; Lührs, P.; Medori, R.; Péran, P.; et al. A Phase 1 Randomized Trial of Specific Active α-Synuclein Immunotherapies PD01A and PD03A in Multiple System Atrophy. Mov. Disord. 2020, 35, 1957–1965. [Google Scholar] [CrossRef]
- Selden, N.R.; Al-Uzri, A.; Huhn, S.L.; Koch, T.K.; Sikora, D.M.; Nguyen-Driver, M.D.; Guillaume, D.J.; Koh, J.L.; Gultekin, S.H.; Anderson, J.C.; et al. Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis. J. Neurosurg. Pediatr. 2013, 11, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Grover, A.; Hammon, K.; Hart, G.; Slasor, P.; Cherukuri, A.; Ajayi, T.; Jacoby, D.; Schulz, A.; Specchio, N.; et al. Clinical Pharmacokinetics and Pharmacodynamics of Cerliponase Alfa, Enzyme Replacement Therapy for CLN2 Disease by Intracerebroventricular Administration. Clin. Transl. Sci. 2020. [Google Scholar] [CrossRef]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A.; et al. Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: A non-randomised, open-label, phase 1–2 trial. Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef] [Green Version]
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; et al. Randomized phase I clinical trial of anti–α-synuclein antibody BIIB054. Mov. Disord. 2019, 34, 1154–1163. [Google Scholar] [CrossRef]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti--Synuclein Monoclonal Antibody, in Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti–α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Van Horne, C.G.; Quintero, J.E.; Gurwell, J.A.; Wagner, R.P.; Slevin, J.T.; Gerhardt, G.A. Implantation of autologous peripheral nerve grafts into the substantia nigra of subjects with idiopathic Parkinson’s disease treated with bilateral STN DBS: A report of safety and feasibility. J. Neurosurg. 2016, 126, 1140–1147. [Google Scholar] [CrossRef] [Green Version]
- Christine, C.W.; Bankiewicz, K.S.; Van Laar, A.D.; Richardson, R.M.; Ravina, B.; Kells, A.P.; Boot, B.; Martin, A.J.; Nutt, J.; Thompson, M.E.; et al. Magnetic resonance imaging–guided phase 1 trial of putaminal AADC gene therapy for Parkinson’s disease. Ann. Neurol. 2019, 85, 704–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittermeyer, G.; Christine, C.W.; Rosenbluth, K.H.; Baker, S.L.; Starr, P.; Larson, P.; Kaplan, P.L.; Forsayeth, J.; Aminoff, M.J.; Bankiewicz, K.S. Long-term evaluation of a phase 1 study of AADC gene therapy for parkinson’s disease. Hum. Gene Ther. 2011, 23, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, S.I.; Fujimoto, K.I.; Kato, S.; Mizukami, H.; Asari, S.; Ikeguchi, K.; Kawakami, T.; Urabe, M.; Kume, A.; Sato, T.; et al. A phase i study of aromatic l-amino acid decarboxylase gene therapy for parkinson’s disease. Mol. Ther. 2010, 18, 1731–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palfi, S.; Gurruchaga, J.M.; Scott Ralph, G.; Lepetit, H.; Lavisse, S.; Buttery, P.C.; Watts, C.; Miskin, J.; Kelleher, M.; Deeley, S.; et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 2014, 383, 1138–1146. [Google Scholar] [CrossRef]
- Tsai, S.T.; Chu, S.C.; Liu, S.H.; Pang, C.Y.; Hou, T.W.; Lin, S.Z.; Chen, S.Y. Neuroprotection of granulocyte colony-stimulating factor for early stage Parkinson’s disease. Cell Transplant. 2017, 26, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendelman, H.E.; Zhang, Y.; Santamaria, P.; Olson, K.E.; Schutt, C.R.; Bhatti, D.; Shetty, B.L.D.; Lu, Y.; Estes, K.A.; Standaert, D.G.; et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. NPJ Park. Dis. 2017, 3, 10. [Google Scholar] [CrossRef]
- Paul, G.; Zachrisson, O.; Varrone, A.; Almqvist, P.; Jerling, M.; Lind, G.; Rehncrona, S.; Linderoth, B.; Bjartmarz, H.; Shafer, L.L.; et al. Safety and tolerability of intracerebroventricular PDGF-BB in Parkinson’s disease patients. J. Clin. Investig. 2015, 125, 1339–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef]
- Charles, D.; Tolleson, C.; Davis, T.L.; Gill, C.E.; Molinari, A.L.; Bliton, M.J.; Tramontana, M.G.; Salomon, R.M.; Kao, C.; Wang, L.; et al. Pilot study assessing the feasibility of applying bilateral subthalamic nucleus deep brain stimulation in very early stage Parkinson’s disease: Study design and rationale. J. Parkinsons. Dis. 2012, 2, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Peterschmitt, M.J.; Crawford, N.P.S.; Gaemers, S.J.M.; Ji, A.J.; Sharma, J.; Pham, T.T. Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Oral Venglustat in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 10, 86–98. [Google Scholar] [CrossRef]
- Jucaite, A.; Svenningsson, P.; Rinne, J.O.; Cselényi, Z.; Varnäs, K.; Johnström, P.; Amini, N.; Kirjavainen, A.; Helin, S.; Minkwitz, M.; et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: A PET study in Parkinson’s disease. Brain 2015, 138, 2687–2700. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Shi, A.; Pang, H.; Xue, W.; Li, Y.; Cao, G.; Yan, B.; Dong, F.; Li, K.; Xiao, W.; et al. Safety, tolerability, and pharmacokinetics of a single ascending dose of baicalein chewable tablets in healthy subjects. J. Ethnopharmacol. 2014, 156, 210–215. [Google Scholar] [CrossRef]
- Mischley, L.K.; Leverenz, J.B.; Lau, R.C.; Polissar, N.L.; Neradilek, M.B.; Samii, A.; Standish, L.J. A randomized, double-blind phase I/IIa study of intranasal glutathione in Parkinson’s disease. Mov. Disord. 2015, 30, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Boxer, A.L.; Qureshi, I.; Ahlijanian, M.; Grundman, M.; Golbe, L.I.; Litvan, I.; Honig, L.S.; Tuite, P.; McFarland, N.R.; O’Suilleabhain, P.; et al. Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: A randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol. 2019, 18, 549–558. [Google Scholar] [CrossRef]
- Canesi, M.; Giordano, R.; Lazzari, L.; Isalberti, M.; Isaias, I.U.; Benti, R.; Rampini, P.; Marotta, G.; Colombo, A.; Cereda, E.; et al. Finding a new therapeutic approach for no-option Parkinsonisms: Mesenchymal stromal cells for progressive supranuclear palsy. J. Transl. Med. 2016, 14, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VandeVrede, L.; Dale, M.L.; Fields, S.; Frank, M.; Hare, E.; Heuer, H.W.; Keith, K.; Koestler, M.; Ljubenkov, P.A.; McDermott, D.; et al. Open-Label Phase 1 Futility Studies of Salsalate and Young Plasma in Progressive Supranuclear Palsy. Mov. Disord. Clin. Pract. 2020, 7, 440–447. [Google Scholar] [CrossRef]
- Tsai, Y.A.; Liu, R.S.; Lirng, J.F.; Yang, B.H.; Chang, C.H.; Wang, Y.C.; Wu, Y.S.; Ho, J.H.C.; Lee, O.K.; Soong, B.W. Treatment of spinocerebellar ataxia with mesenchymal stem cells: A phase I/IIa clinical study. Cell Transplant. 2017, 26, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Santos-Morales, O.; Díaz-Machado, A.; Jiménez-Rodríguez, D.; Pomares-Iturralde, Y.; Festary-Casanovas, T.; González-Delgado, C.A.; Pérez-Rodríguez, S.; Alfonso-Muñoz, E.; Viada-González, C.; Piedra-Sierra, P.; et al. Nasal administration of the neuroprotective candidate NeuroEPO to healthy volunteers: A randomized, parallel, open-label safety study. BMC Neurol. 2017, 17, 129. [Google Scholar] [CrossRef] [Green Version]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN Rx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Kletzl, H.; Marquet, A.; Günther, A.; Tang, W.; Heuberger, J.; Groeneveld, G.J.; Birkhoff, W.; Mercuri, E.; Lochmüller, H.; Wood, C.; et al. The oral splicing modifier RG7800 increases full length survival of motor neuron 2 mRNA and survival of motor neuron protein: Results from trials in healthy adults and patients with spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Sturm, S.; Günther, A.; Jaber, B.; Jordan, P.; Al Kotbi, N.; Parkar, N.; Cleary, Y.; Frances, N.; Bergauer, T.; Heinig, K.; et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br. J. Clin. Pharmacol. 2019, 85, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Stewart, J.J.; Green, C.L.; Jones, N.; Liang, M.; Xu, Y.; Wilkins, D.E.C.; Moulard, M.; Czechowska, K.; Lanham, D.; McCloskey, T.W.; et al. Role of receptor occupancy assays by flow cytometry in drug development. Cytom. B Clin. Cytom. 2016, 90, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Muller, P.Y.; Brennan, F.R. Safety assessment and dose selection for first-in-human clinical trials with immunomodulatory monoclonal antibodies. Clin. Pharmacol. Ther. 2009, 85, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Ritter, A.; Zhong, K. Clinical Trials for Disease-Modifying Therapies in Alzheimer’s Disease: A Primer, Lessons Learned, and a Blueprint for the Future. J. Alzheimer’s Dis. 2018, 64, S3–S22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cudkowicz, M.E.; Titus, S.; Kearney, M.; Yu, H.; Sherman, A.; Schoenfeld, D.; Hayden, D.; Shui, A.; Brooks, B.; Conwit, R.; et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: A multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014, 13, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Haché, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children with Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J. Child Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef] [Green Version]
- de Lange, E.C. The mastermind approach to CNS drug therapy: Translational prediction of human brain distribution, target site kinetics, and therapeutic effects. Fluids Barriers CNS 2013, 10, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, A.; Varrone, A.; Gulyás, B.; Salvadori, P.; Gee, A.; Windhorst, A.; Vercouillie, J.; Bormans, G.; Lammertsma, A.A.; Halldin, C. Guidelines to PET measurements of the target occupancy in the brain for drug development. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2255–2262. [Google Scholar] [CrossRef]
- Johnström, P.; Bergman, L.; Varnäs, K.; Malmquist, J.; Halldin, C.; Farde, L. Development of rapid multistep carbon-11 radiosynthesis of the myeloperoxidase inhibitor AZD3241 to assess brain exposure by PET microdosing. Nucl. Med. Biol. 2015, 42, 555–560. [Google Scholar] [CrossRef]
- Liang, M.; Schwickart, M.; Schneider, A.K.; Vainshtein, I.; Del Nagro, C.; Standifer, N.; Roskos, L.K. Receptor occupancy assessment by flow cytometry as a pharmacodynamic biomarker in biopharmaceutical development. Cytom. Part B Clin. Cytom. 2016, 90, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Dzamko, N.; Deak, M.; Hentati, F.; Reith, A.D.; Prescott, A.R.; Alessi, D.R.; Nichols, R.J. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem. J. 2010, 430, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Coimbra, J.R.M.; Marques, D.F.F.; Baptista, S.J.; Pereira, C.M.F.; Moreira, P.I.; Dinis, T.C.P.; Santos, A.E.; Salvador, J.A.R. Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front. Chem. 2018, 6, 178. [Google Scholar] [CrossRef] [Green Version]
- West, A.B.; Cookson, M.R. Identification of Bona Fide LRRK2 Kinase Substrates. Mov. Disord. 2016, 31, 1140–1141. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 2016, 5, e12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, N.D.; Scheltens, P. Treating Alzheimer’s disease with monoclonal antibodies: Current status and outlook for the future. Alzheimer’s Res. Ther. 2013, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Bowman, L.; Landray, M.; Peto, R. The Magic of Randomization versus the Myth of Real-World Evidence. N. Engl. J. Med. 2020, 382, 674–678. [Google Scholar] [CrossRef]
- Lipsmeier, F.; Taylor, K.I.; Kilchenmann, T.; Wolf, D.; Scotland, A.; Schjodt-Eriksen, J.; Cheng, W.Y.; Fernandez-Garcia, I.; Siebourg-Polster, J.; Jin, L.; et al. Evaluation of smartphone-based testing to generate exploratory outcome measures in a phase 1 Parkinson’s disease clinical trial. Mov. Disord. 2018, 33, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Htike, T.T.; Mishra, S.; Kumar, S.; Padmanabhan, P.; Gulyás, B. Peripheral Biomarkers for Early Detection of Alzheimer’s and Parkinson’s Diseases. Mol. Neurobiol. 2019, 56, 2256–2277. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Howden, A.J.M.; Sarhan, A.R.; Lis, P.; Ito, G.; Martinez, T.N.; Brockmann, K.; Gasser, T.; Alessi, D.R.; Sammler, E.M. Interrogating Parkinson’s disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils. Biochem. J. 2018, 475, 23–44. [Google Scholar] [CrossRef] [Green Version]
- Seol, W.; Kim, H.; Son, I. Urinary Biomarkers for Neurodegenerative Diseases. Exp. Neurobiol. 2020, 29, 325–333. [Google Scholar] [CrossRef]
- Shen, J.; Swift, B.; Mamelok, R.; Pine, S.; Sinclair, J.; Attar, M. Design and Conduct Considerations for First-in-Human Trials. Clin. Transl. Sci. 2019, 12, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Sattler, R.; Ayukawa, Y.; Coddington, L.; Sawa, A.; Block, D.; Chipkin, R.; Rothstein, J.D. Human nasal olfactory epithelium as a dynamic marker for CNS therapy development. Exp. Neurol. 2011, 232, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teves, J.M.Y.; Bhargava, V.; Kirwan, K.R.; Corenblum, M.J.; Justiniano, R.; Wondrak, G.T.; Anandhan, A.; Flores, A.J.; Schipper, D.A.; Khalpey, Z.; et al. Parkinson’s Disease Skin Fibroblasts Display Signature Alterations in Growth, Redox Homeostasis, Mitochondrial Function, and Autophagy. Front. Neurosci. 2018, 11, 737. [Google Scholar] [CrossRef] [Green Version]
- Tanis, K.Q.; Podtelezhnikov, A.A.; Blackman, S.C.; Hing, J.; Railkar, R.A.; Lunceford, J.; Klappenbach, J.A.; Wei, B.; Harman, A.; Camargo, L.M.; et al. An Accessible Pharmacodynamic Transcriptional Biomarker for Notch Target Engagement. Clin. Pharmacol. Ther. 2016, 99, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Suhy, J.; Chiao, P.; Chen, T.; Klein, G.; Purcell, D.; Oh, J.; Verma, A.; Sampat, M.; Barakos, J. Amyloid PET screening for enrichment of early-stage Alzheimer disease clinical trials: Experience in a phase 1b clinical trial. Alzheimer Dis. Assoc. Disord. 2016, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yee, L.M.; Lively, T.G.; McShane, L.M. Biomarkers in early-phase trials: Fundamental issues. Bioanalysis 2018, 10, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Zhu, Y.; Hsiao-Nakamoto, J.; Tang, X.; Dugas, J.C.; Moscovitch-Lopatin, M.; Glass, J.D.; Brown, R.H., Jr.; Ladha, S.S.; Lacomis, D.; et al. Longitudinal biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, M.; Rey, F.; Fantini, V.; Pansarasa, O.; Di Giulio, A.M.; Carelli, S.; Cereda, C. From neuronal differentiation of iPSCs to 3D neuro-organoids: Modelling and therapy of neurodegenerative diseases. Int. J. Mol. Sci. 2018, 19, 3972. [Google Scholar] [CrossRef] [Green Version]
- Houghton, R.; Chamberlain, J. Conference Report: Analytical challenges in the qualification and validation of pharmacodynamic biomarkers. In Bioanalysis; Future Science Ltd.: London, UK, 2011; Volume 3, pp. 945–948. [Google Scholar]
- Mullard, A. News in Brief: Innovative antidepressants arrive; Anti-amyloid failures stack up as Alzheimer antibody flops. Nat. Rev. Drug Discov. 2019, 18, 2019. [Google Scholar]
- van den Brink, W.J.; van den Berg, D.J.; Bonsel, F.E.M.; Hartman, R.; Wong, Y.C.; van der Graaf, P.H.; de Lange, E.C.M. Fingerprints of CNS drug effects: A plasma neuroendocrine reflection of D2 receptor activation using multi-biomarker pharmacokinetic/pharmacodynamic modelling. Br. J. Pharmacol. 2018, 175, 3832–3843. [Google Scholar] [CrossRef] [Green Version]
- US Food and Drug Administration. Early Alzheimer’s Disease: Developing Drugs for Treatment Guidance for Industry; US Food and Drug Administration: Silver Spring, MD, USA, 2018. [Google Scholar]



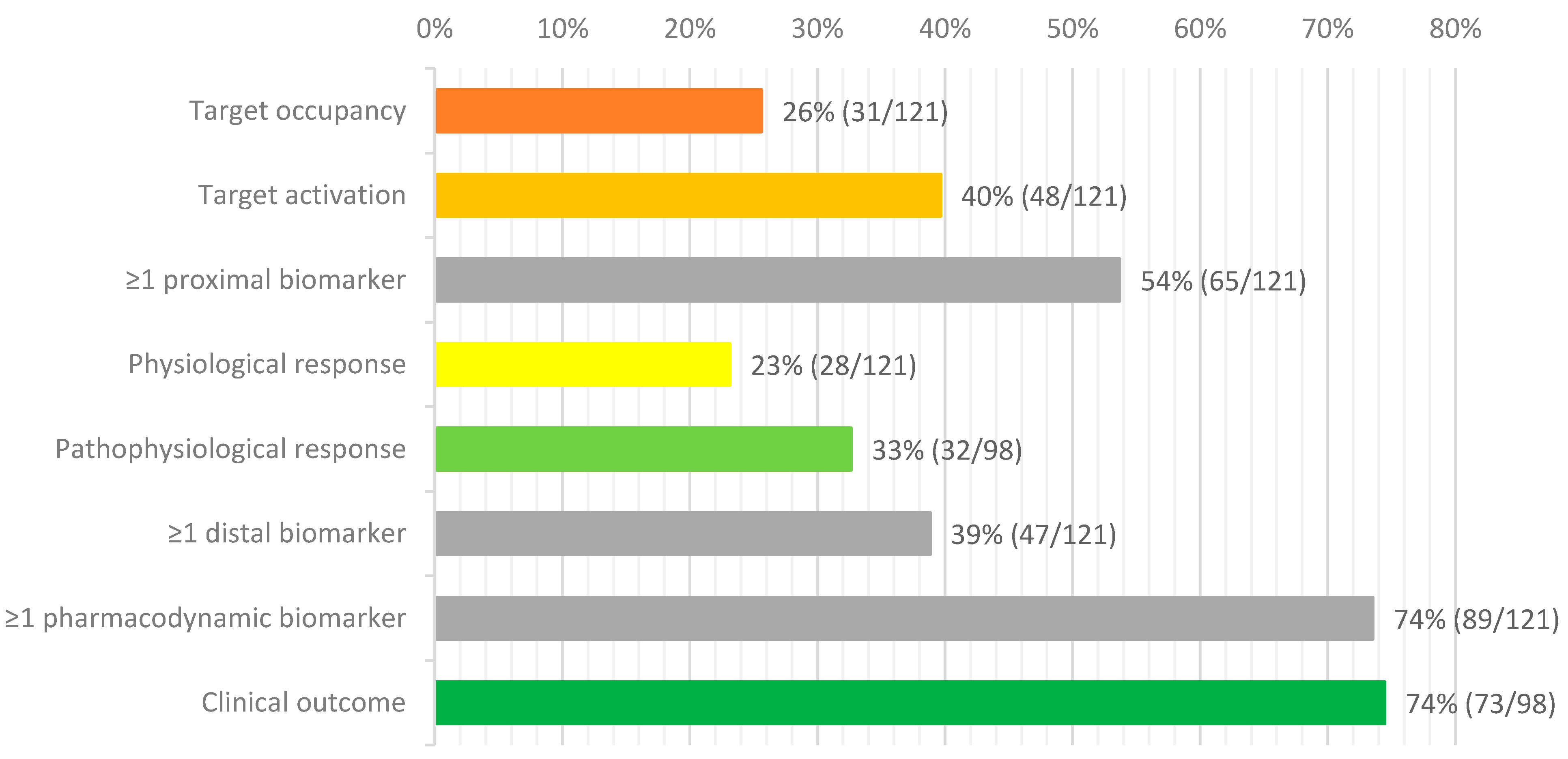{kind=link}
{kind=link}
| Biomarker Category | Use in Drug Development | Examples from NND DMT Drug Development |
|---|---|---|
| Response | Pharmacodynamic biomarker as indicator of intended drug activity | CSF total amyloid-β and fragments in response to amyloid-β antibody treatments |
| ||
| Efficacy response marker as a surrogate for a clinical endpoint | Braak staging with tau PET as a surrogate biomarker for clinical AD (though no validated surrogate biomarkers are available yet for NDDs). | |
| Diagnostic | Patient selection | GBA1 gene mutation in PD patients SOD1 gene mutation in ALS patients |
| Predictive | Patient stratification Trial enrichment via inclusion criteria | Tau PET to identify AD patients more likely to respond to anti-tau therapies |
| Prognostic | Patient stratification Trial enrichment with patients likely to have disease | Percentage of weight loss at baseline for life expectancy and disease progression in ALS patients |
| Safety | Detect AEs and off-target drug responses | MRI for structural changes (including tumor or syrinx formation) within the brain after stem cell transplantation for ALS |
| Indication | Drug Category | Drug Target | Trials Reporting Mechanistic Biomarker | Peripheral Biomarkers | Central Biomarkers | Types of Biomarkers | Study Population | References |
|---|---|---|---|---|---|---|---|---|
| AD | Antibody | Amyloid β | 10/11 (91%) | Plasma total Aβ and Aβ fragments (Aβ1-x, Aβ1-40, Aβ1-42, Aβ3–42, Aβ1–38, Aβ18–35) | CSF Aβ species (Aβ1-x, Aβ1-40, Aβ1-42), t-tau, and p-tau181 | Target occupancy and pathophysiological response | HVs and patients | [53,54,55,56,57,58,59,60,61,62,63] |
| Tau protein | 1/1 (100%) | - | CSF N-terminal tau, mid-domain tau, Aβ40, and Aβ42 | Target occupancy and physiological response | HVs | [64] | ||
| Cell therapy | Cytotropic factors, anti-inflammatory, neurogenesis | 1/1 (100%) | - | CSF Aβ, t-tau and p-tau; PiB-PET changes in parenchymal amyloid deposition; FDG-PET metabolic changes | (patho)physiological response | Patients | [65] | |
| Nerve growth factor | 0/1 (0%) | - | - | - | Patients | [66] | ||
| Dietary | Xanthophyll Carotenoids, Omega-3 Fatty Acids | 0/1 (0%) | - | - | - | Patients | [67] | |
| Gene therapy | Nerve growth factor | 1/1 (100%) | - | PET brain glucose metabolism (post-mortem brain autopsy gene-mediated NGF expression and bioactivity) | Physiological response (and post-mortem target occupancy and activation) | Patients | [68] | |
| Growth factor | Nerve growth factor | 1/1 (100%) | - | MRI for implant position; CSF Aβ1–42, t-tau, p-tau181, NfL, glial fibrillary acidic protein (GFAP), AChE and choline acetyltransferase (ChAT) activity and protein levels | Target occupancy, activation and (patho)physiological response | Patients | [69] | |
| Immunotherapy | Amyloid β | 3/3 (100%) | Plasma anti-Aβ40 antibodies, Aβ peptides (Aβ40, Aβ42) and cytokines (IL-6, TNF-α, IL-1β, MCP-1, IL-2, sIL-2R); Serum antibody titres (Aβ IgM, Aβ IgG), Aβ1–40, AβX–40, Aβ1–42; In Vitro lymphocyte proliferation and cytokine production; PBMC β-specific and Qβ-specific responses of T-cells | CSF antibody titres, AβX–40, AβX–42, Aβ1–42, AβN–42, t-tau, p-tau181; MRI brain volumetric assessment | Target activation and (patho)physiological response | Patients | [70,71,72] | |
| Tau protein | 1/1 (100%) | IgG and IgM titre anti-vaccin peptide, anti-KLH antibody titre, anti-pathological-tau antibody titre; Lymphocyte immunoprofiling | - | Target activation and physiological response | Patients | [73] | ||
| Peptide | Amyloid β | 0/1 (0%) | - | - | - | HVs | [74] | |
| Focused ultrasound with injected microbubbles | BBB-opening to amyloid β and tau | 1/1 (100%) | - | PET BBB opening and amyloid β deposition | Target occupancy and pathophysiological response | Patients | [75] | |
| DBS | Cerebral glucose metabolism | 3/4 (75%) | - | PET cerebral glucose metabolism | Physiological response | Patients | [76,77,78,79] | |
| Small molecule | 5-HT2A receptor | 0/1 (0%) | - | - | - | HVs | [80] | |
| Amyloid precursor protein (APP) synthesis | 1/1 (100%) | - | CSF sAPPα, sAPPβ, t-tau, p-tau, Aβ42 and inflammatory markers (complement 3, factor H, MCP-1, YKL-40, sCD14) | Target activation and (patho)physiological response | Patients | [81] | ||
| Amyloid production and associated inflammatory response | 0/1 (0%) | - | - | - | HVs | [82] | ||
| BACE1 | 7/8 (89%) | Plasma total Aβ and Aβ fragments (Aβ1–37, Aβ1–38, Aβ1–40, Aβ1–42, Aβx-40), total sAPP and fragments (sAPPα, sAPPβ) | CSF total Aβ and fragments (Aβx-38, Aβx-40, Aβx-42, Aβ1–37, Aβ1–38, Aβ1–40, Aβ1–42), total sAPP and fragments (sAPPα, sAPPβ), BACE1, t-tau, p-tau181 | Target occupancy, activation and pathophysiological response | HVs and patients | [83,84,85,86,87,88,89,90] | ||
| ET(B) receptor | 0/1 (0%) | - | - | - | HVs | [91] | ||
| Glutaminyl cyclase (QC) | 1/1 (100%) | Serum QC activity | CSF QC activity | Target occupancy and activation | HVs | [92] | ||
| Glycogen synthase kinase-3β (GSK3β) | 1/1 (100%) | Lymphocyte GS phosphorylation | - | Target occupancy | HVs | [93] | ||
| Sigma-2 receptor complex | 0/1 (0%) | - | - | - | HVs | [94] | ||
| γ-secretase | 2/2 (100%) | Plasma Aβx–42 | CSF total Aβ and Aβ fragments (Aβ42, Aβ40, Aβ37, Aβ38) | Target activation | HVs | [95,96] | ||
| RIPK1 inhibitor * | 1/1 (100%) | PBMCs reduction of pS166 RIPK1 | - | Target occupancy and activation | HVs | [30] | ||
| Microtubule stabilization | 1/1 (100%) | - | CSF NfL, t-tau, p-tau, Aβ42, YKL-40 | Pathophysiological response | Patients | [97] | ||
| Cell therapy | Neuroprotective effects | 1/1 (100%) | - | CSF t-tau, p-tau, Aß42 | Pathophysiological response | Patients | [98] | |
| Overall use of mechanistic biomarkers in early phase AD trials | 37/47 (79%) | |||||||
| ALS | Antibody | Neurite outgrowth inhibitor Nogo-A | 1/1 (100%) | Muscle biopsy Nogo-A RNA and protein expression; Plasma Nogo-A protein gamma sarcoglycan; EMG (MUNE) | - | Target occupancy and activation | Patients | [99] |
| Antisense Oligonucleotide | SOD1 | 2/2 (100%) | Plasma p-NfH, NfL | CSF SOD1, p-NfH, NfL | Target activation and pathophysiological response | Patients | [33,100] | |
| Cell therapy | Neurotrophic growth factors and cytokines secretion, immunomodulation and cell proliferation or replacement | 5/13 (38%) | MRI muscle volume CD4 + CD25 + FOXP3 + Tregs, proliferation of autologous responder T lymphocytes; EMG of TA muscles (CMAP, FD, SMUP, MUNE, MUNIX, MUSIX); EIM | CSF cytokines (TGF-b1, TGF-b2, TGF-b3, IL-6, IL-10, MCP-1) | (patho)physiological response | Patients | [101,102,103,104,105,106,107,108,109,110,111,112,113] | |
| Gene therapy | Hepatocyte growth factor | 1/1 (100%) | Serum HGF; Muscle circumference | - | Target activation and pathophysiological response | Patients | [114] | |
| Growth factor | Granulocyte colony-stimulating factor | 1/1 (100%) | Blood cell counts, CD34 + cells, serum cytokines/chemokines (IL-1b, IL-1ra, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 (p70), IL-13, IL-15, IL-17, eotaxin, bFGF, FGF-2, TGF-a, G-CSF, GM-CSF, IFN-γ, IP-10, MCP-1, MIP- 1a, MIP-1b, PDGF-BB, RANTES, TNF-a, VEGF) | CSF BMC presence, cytokines/chemokines (IL-1b, IL-1ra, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 (p70), IL-13, IL-15, IL-17, eotaxin, bFGF, FGF-2, TGF-a, G-CSF, GM-CSF, IFN-γ, IP-10, MCP-1, MIP- 1a, MIP-1b, PDGF-BB, RANTES, TNF-a, VEGF) | Target activation and (patho)physiological response | Patients | [115] | |
| Hepatocyte growth factor | 0/1 (0%) | - | - | - | Patients | [116] | ||
| Small molecule | EAAT2 | 0/1 (0%) | - | - | - | Patients | [117] | |
| Putative mitochondrial modulation | 0/1 (0%) | - | - | - | HVs | [118] | ||
| Inflammatory macrophages and monocytes regulation | 1/1 (100%) | Blood monocyte immune activation markers CD16, HLA-DR | - | Target activation | Patients | [119] | ||
| SOD1 | 2/2 (100%) | Erythrocyte SOD1 enzymatic activity; Leukocyte actin-normalized SOD1 | CSF SOD1 protein and enzymic activity | Target activation | Patients | [120,121] | ||
| Supplement | Lysosomal Cathepsins B and L | 0/1 (0%) | - | - | - | Patients | [122] | |
| Stabilize the mitochondrial transition pore, buffer intracellular energy stores, stimulate synaptic glutamate uptake, and scavenge reactive oxygen species | 1/1 (100%) | - | MRS brain glutamate and glutamine (Glx) | Physiological response | Patients | [123] | ||
| Overall use of mechanistic biomarkers in early phase ALS trials | 14/27 (52%) * | |||||||
| ATTR amyloidosis | Antisense oligonucleotide | Transthyretin (TTR) | 1/1 (100%) | Plasma TTR | - | Target activation | HVs | [124] |
| RNA interference | Transthyretin amyloid | 1/1 (100%) | Serum transthyretin, retinol-binding protein and vitamin A | - | Target occupancy and activation | HVs and patients | [125] | |
| Overall use of mechanistic biomarkers in early phase ATTR trials | 2/2 (100%) | |||||||
| FRDA | Small molecule | FXN gene expression | 1/1 (100%) | Whole blood FXN mRNA, frataxin protein; PBMC chromatin modification via H3 lysine 9 acetylation | - | Target occupancy and activation | Patients | [126] |
| Supplement | FXN gene expression | 1/1 (100%) | PBMC FXN mRNA and frataxin protein); Blood heterochromatin modifications at the FXN locus | - | Target occupancy and activation | Patients | [127] | |
| Polyunsaturated fatty acid | Lipid peroxidation | 1/1 (100%) | RBC compartment D2-LA | - | Target occupancy | Patients | [128] | |
| Overall use of mechanistic biomarkers in early phase FRDA trials | 3/3 (100%) | |||||||
| FTD | Small molecule | Progranulin protein (PGRN) | 1/1 (100%) | Plasma PGRN, PGRN-related inflammatory markers (CRP, ESR), blood cytokines (IL-10, IL-2, IL-6, IL-8, TNFa) | CSF PGRN, NfL, Aβ42, tau, cytokines (IL-10, IL-2, IL-6, IL-8, TNFa); MRI volumetric assessment | Target activation and (patho)physiological response | Patients | [129] |
| Overall use of mechanistic biomarkers in early phase FTD trials | 1/1 (100%) | |||||||
| GM2 gangliosidosis | Small molecule | β-hexosaminidase (Hex) | 1/1 (100%) | Leucocyte and plasma Hex A, β-galactosidase and glucocerebrosidase activity, β-glucuronidase and acid phosphatase | - | Target activation | Patients | [130] |
| Overall use of mechanistic biomarkers in early phase GM2 gangliosidosis trials | 1/1 (100%) | |||||||
| HD | Antisense oligonucleotide | HTT mRNA | 1/1 (100%) | - | CSF mutant HTT, NfL; MRI ventricular volume | Target activation and pathophysiological response | Patients | [131] |
| Peptide | Cardiolipin | 1/1 (100%) | MRI skeletal muscle dynamic 31P-MRS; PBMC mitochondrial membrane potential (∆Ψm) | MRI brain 31P-MRS; CNS functional domain test battery (NeuroCart®) | Target activation and (patho)physiological response | Patients | [132] | |
| Overall use of mechanistic biomarkers in early phase HD trials | 2/2 (100%) | |||||||
| Leber Hereditary Optic Neuropathy | Gene therapy | Mitochondrial gene encoding NADH:ubiquinone oxidoreductase subunit 4 (ND4) | 1/2 (50%) | - | OCT average retinal nerve fiber layer (RNFL) thickness; Pattern electroretinogram amplitudes | Physiological response | Patients | [133,134] |
| Overall use of mechanistic biomarkers in early phase Leber Hereditary Optic Neuropathy trials | 1/2 (50%) | |||||||
| MS | Antibody | Semaphorin 4D | 1/1 (100%) | T-cell cSEMA4D expression and saturation; Serum sSEMA4D | - | Target occupancy and activation | Patients | [135] |
| Cell therapy | Neurotrophic and immunomodulatory effects, neurogenesis | 2/2 (100%) | Lymphocyte subsets (CD4+, CD25+ and CD40+ lymphocytes and CD83+, CD86+, and HLA-DR+ myeloid dendritic cells); PBMC cytokine production | MRI labeled cell localization and volumetric assessment; OCT average retinal nerve fiber layer (RNFL); Vision (HCVA, LCLA) | Target occupancy and (patho)physiological response | Patients | [136,137] | |
| Small molecule | Anti-inflammatory | 1/1 (100%) | PBMC monocyte and 6-sulpho LacNAc + dendritic cell (slanDC) frequency, properties, and activation status | - | Target activation and physiological response | Patients | [138] | |
| Mitochondrial ATP production (coenzyme Q10) | 1/1 (100%) | - | CSF mitochondrial dysfunction markers (GDF15, lactate), NfL, sCD14; BBB leakage (albumin quotient); OCT retinal nerve fiber layer thinning; MRI brain ventricular volume | (patho)physiological response | Patients | [139] | ||
| Overall use of mechanistic biomarkers in early phase MS trials | 5/5 (100%) | |||||||
| MSA | Cell therapy | Neurotrophic factors secretion | 1/1 (100%) | - | CSF neurotrophic factors (NGF, GDNF, BDNF) | Physiological response | Patients | [140] |
| Immunotherapy | α-Synuclein | 1/1 (100%) | Serum immunopeptide titers, α-synuclein native epitope titers | - | Target activation | Patients | [141] | |
| Overall use of mechanistic biomarkers in early phase MSA trials | 2/2 (100%) | |||||||
| NCLs | Cell therapy | Palmitoyl-protein thioesterase 1 (PPT-1) and tripeptidyl peptidase 1 (TPP1) enzymes production | 0/1 (0%) | - | - | - | Patients | [142] |
| CLN2 disease | Enzyme replacement | Lysosomal enzyme TPP1 | 0/1 (0%) | - | - | - | Patients | [143] |
| Overall use of mechanistic biomarkers in early phase NCLs trials | 0/2 (0%) | |||||||
| NPC1 | Cyclodextrin | Neuronal cholesterol homoeostasis | 1/1 (100%) | Serum a24(S)-hydroxycholesterol (24[S]-HC) | CSF a24(S)-hydroxycholesterol (24[S]-HC), fatty acid binding protein 3 (FABP3) and calbindin D19 | Target activation and (patho)physiological response | Patients | [144] |
| Overall use of mechanistic biomarkers in early phase NPC1 trials | 1/1 (100%) | |||||||
| PD | Antibody | α-synuclein | 3/3 (100%) | Plasma antibody/α-syn complexes; Serum total and free α-synuclein | CSF total and free α-synuclein, total Aβ, Aβ42, DJ-1, DAT scan | Target occupancy, activation, and pathophysiological response | HVs and patients | [145,146,147] |
| Cell therapy | Neurotrophic factors to restore dopaminergic cell function | 0/1 (0%) | - | - | - | Patients | [148] | |
| Gene therapy | Aromatic L-amino acid decarboxylase (AADC) | 3/3 (100%) | - | PET FMT brain AADC expression and activity | Target occupancy and activation | Patients | [149,150,151] | |
| Tyrosine hydroxylase, AADC, cyclohydrolase 1 | 1/1 (100%) | - | PET cortical excitability and reflex recordings | Physiological response | Patients | [152] | ||
| Growth factor | Granulocyte colony-stimulating factor (G-CSF) | 0/1 (0%) | - | PET 18 F-DOPA for disease progression | Pathophysiological response | Patients | [153] | |
| Granulocyte-macrophage colony-stimulating factor (GM-CSF) | 1/1 (100%) | Expression of Treg phenotype and function (CD4+ Teffs (CD4+CD127hiCD25hi), CD4+ Tregs (CD4+CD127loCD25hi), FOXP3+CD4+ Tregs, iCTLA4+CD4+ Tregs, CD39+CD4+ Tregs, and f FAS+CD4+ Tregs), T cell proliferation mRNA (GATA4, IL2, HOXA10, and KIF2C), anti-inflammatory gene expression (PPARG, LRRC32, FOSL1, IL1R2, IL13RA1, NR4A3, GFI1), tryptophan pathway targeted metabolomics | - | Target activation and physiological response | Patients | [154] | ||
| rhPDGF-BB (proliferation of SOX-2/Olig-1– positive periventricular progenitor cells) | 1/1 (100%) | - | [11C]PE2I DAT binding | Pathophysiological response | Patients | [155] | ||
| Immunotherapy | α-Synuclein | 1/1 (100%) | Serum antibody titres | CSF antibody titres, total α-synuclein, Aβ1–42, p-tau | Target activation and pathophysiological response | Patients | [156] | |
| Deep brain stimulation | Unknown | 0/1 (0%) | - | - | N/A | Patients | [157] | |
| Small molecule | Glucosylceramide synthase (GCS) | 1/1 (100%) | Plasma glucosylceramide (GL-1), globostriaosylceramide (GL-3), and GM3 ganglioside (GM3) | - | Target activation | HVs | [158] | |
| Myeloperoxidase | 1/1 (100%) | - | PET distribution volume of 11C-PBR28 binding to microglia marker TSPO | Target occupancy | Patients | [159] | ||
| Flavonoid (regulating dopaminergic system function, anti-oxidative damage and anti-inflammatory effects) | 0/1 (0%) | - | - | N/A | HVs | [160] | ||
| Supplement | Antioxidant | 0/1 (0%) | - | - | N/A | Patients | [161] | |
| Overall use of mechanistic biomarkers in early phase PD trials | 12/17 (71%) | |||||||
| PSP | Antibody | Tau protein | 0/2 (0%) | - | - | N/A | Patients | [31,162] |
| Cell therapy | Trophic, anti-apoptotic and regenerative effects | 0/1 (0%) | - | MRI, SPECT and PET with tropanic tracers (FP-CIT and Beta-CIT) longitudinal neuroimaging | Pathophysiological response | Patients | [163] | |
| Small molecule/Blood product | Acetylation of tau/unknown | 1/1 (100%) | Plasma NfL concentrations | CSF amyloid beta Aβ, t-tau, p-tau181; MRI brain volumetric assessment | (patho)physiological response | Patients | [164] | |
| Overall use of mechanistic biomarkers in early phase PSP trials | 1/4 (25%) | |||||||
| SCA | Cell therapy | Trophic factor secretion, immunomodulation | 1/1 (100%) | - | PET brain glucose metabolism | Physiological response | Patients | [165] |
| Growth factor | Antiapoptotic, antioxidative, anti-inflammatory, neurotrophic and angio- genic properties | 0/1 (0%) | - | - | N/A | HVs | [166] | |
| Overall use of mechanistic biomarkers in early phase SCA trials | 1/2 (50%) | |||||||
| SMA | Antisense oligonucleotide | SMN2 mRNA splicing | 1/1 (100%) | - | CSF SMN protein | Target activation | Patients | [167] |
| Small molecule | SMN2 splicing | 2/2 (100%) | Blood mRNA (full-length SMN2, SMN1, SMNΔ7), SMN protein | - | Target activation | HVs and patients | [168,169] | |
| Gene therapy | SMN | 0/1 (0%) | - | - | N/A | Patients | [170] | |
| Overall use of mechanistic biomarkers in early phase SMA trials | 3/4 (25%) | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vissers, M.F.J.M.; Heuberger, J.A.A.C.; Groeneveld, G.J. Targeting for Success: Demonstrating Proof-of-Concept with Mechanistic Early Phase Clinical Pharmacology Studies for Disease-Modification in Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 1615. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041615
Vissers MFJM, Heuberger JAAC, Groeneveld GJ. Targeting for Success: Demonstrating Proof-of-Concept with Mechanistic Early Phase Clinical Pharmacology Studies for Disease-Modification in Neurodegenerative Disorders. International Journal of Molecular Sciences. 2021; 22(4):1615. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041615
Chicago/Turabian StyleVissers, Maurits F. J. M., Jules A. A. C. Heuberger, and Geert Jan Groeneveld. 2021. "Targeting for Success: Demonstrating Proof-of-Concept with Mechanistic Early Phase Clinical Pharmacology Studies for Disease-Modification in Neurodegenerative Disorders" International Journal of Molecular Sciences 22, no. 4: 1615. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041615