
Cocaine Induces Cytoskeletal Changes in Cardiac Myocytes: Implications for Cardiac Morphology

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
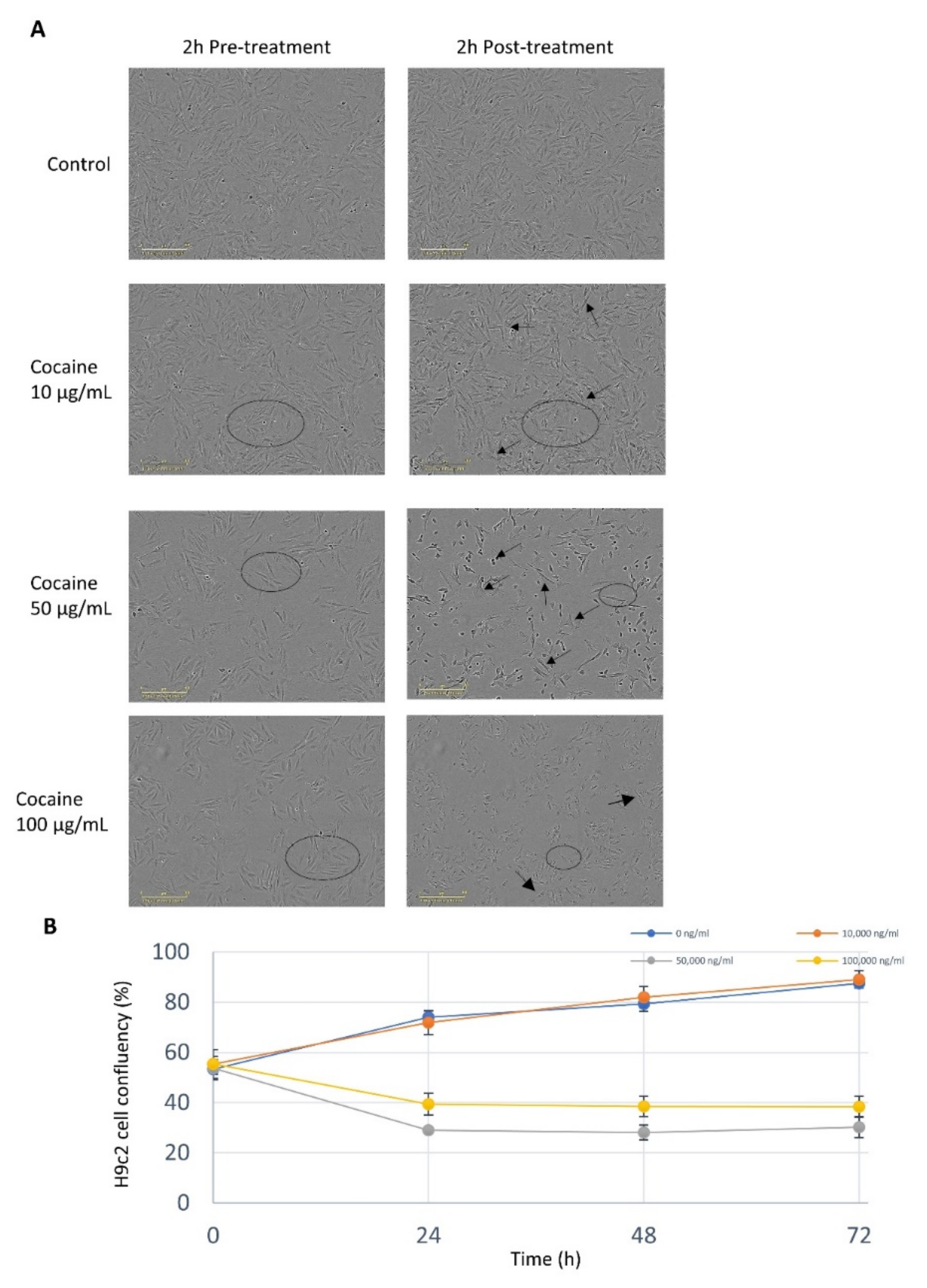
2.1. Cocaine Elicits a Dose Dependent Response on Cell Viability and Morphology
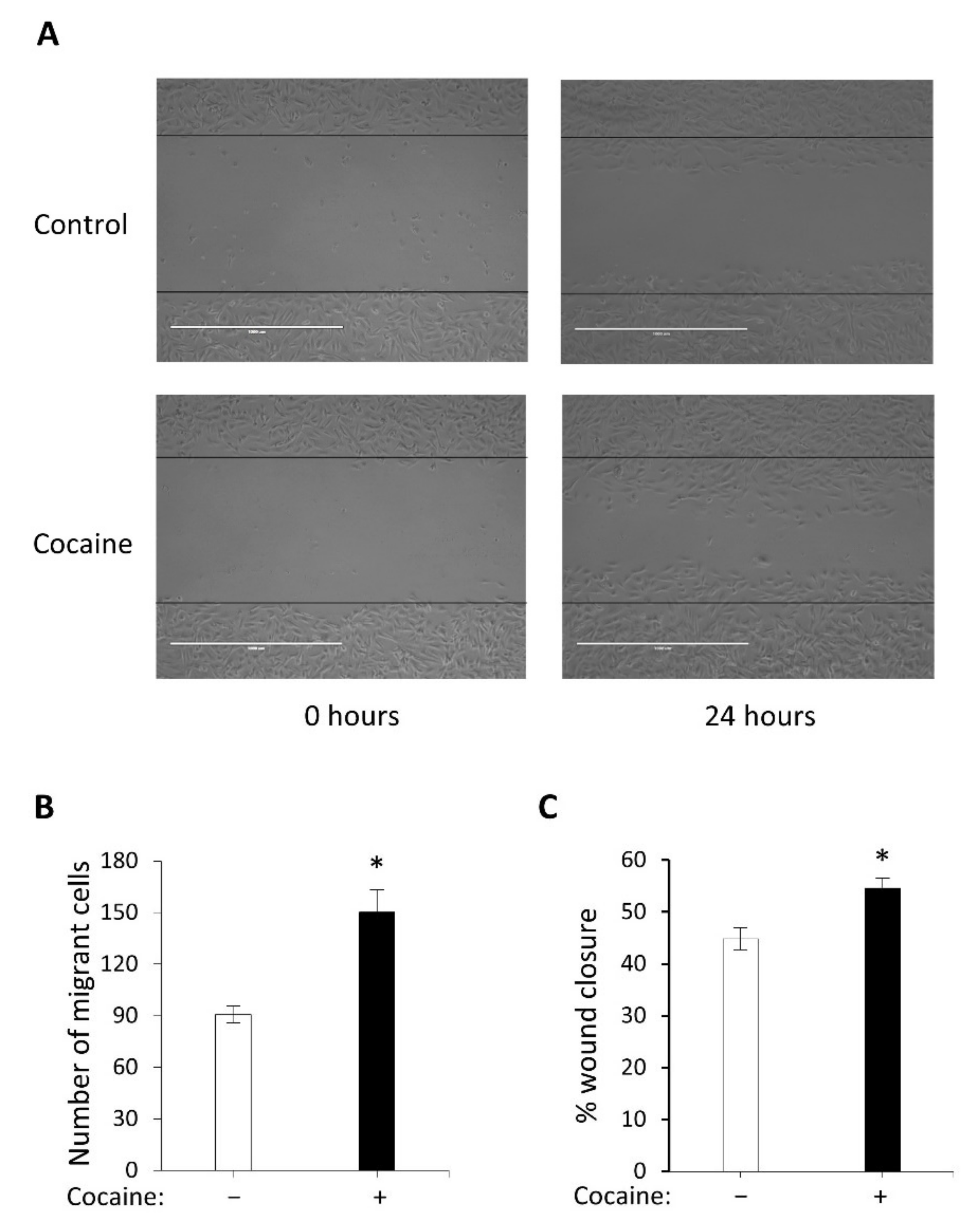
2.2. Cocaine Mediates Increased Cell Migration and Wound Closure
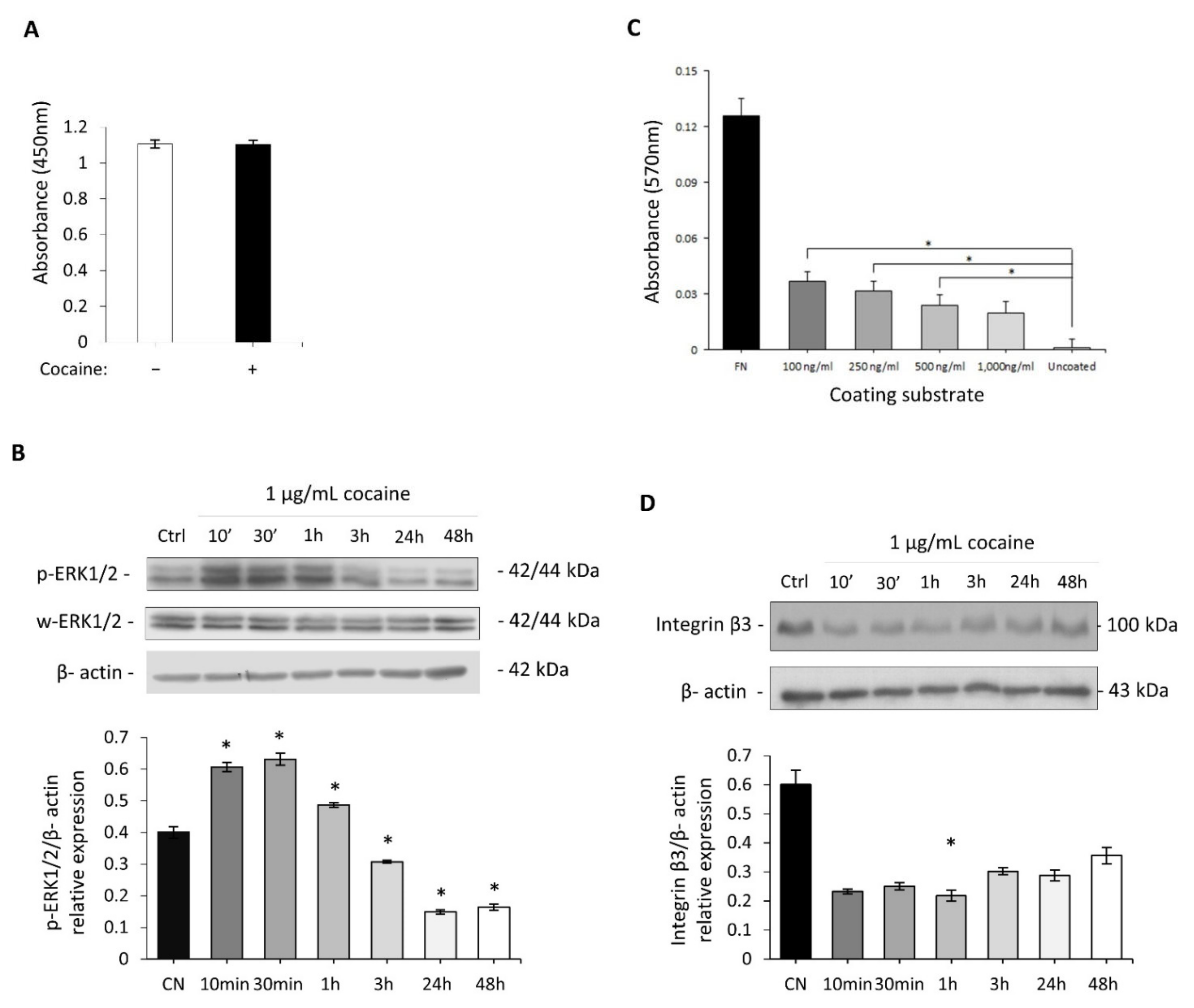
2.3. Investigation of Cocaine’s Effect on Cell Adhesion and Proliferation
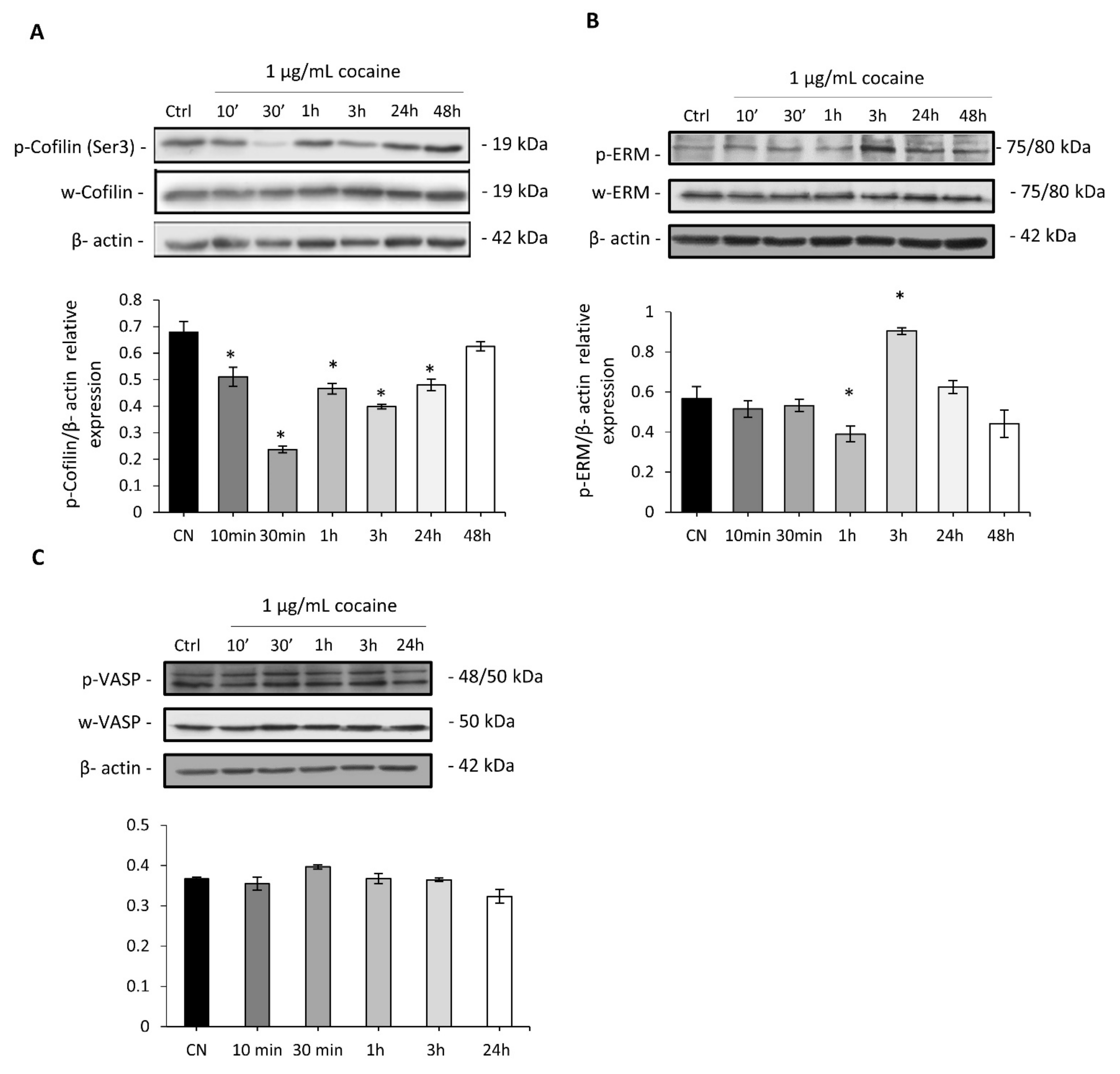
2.4. Cocaine Mediates Changes in Cardiomyocytic Actin Binding Protein Expression
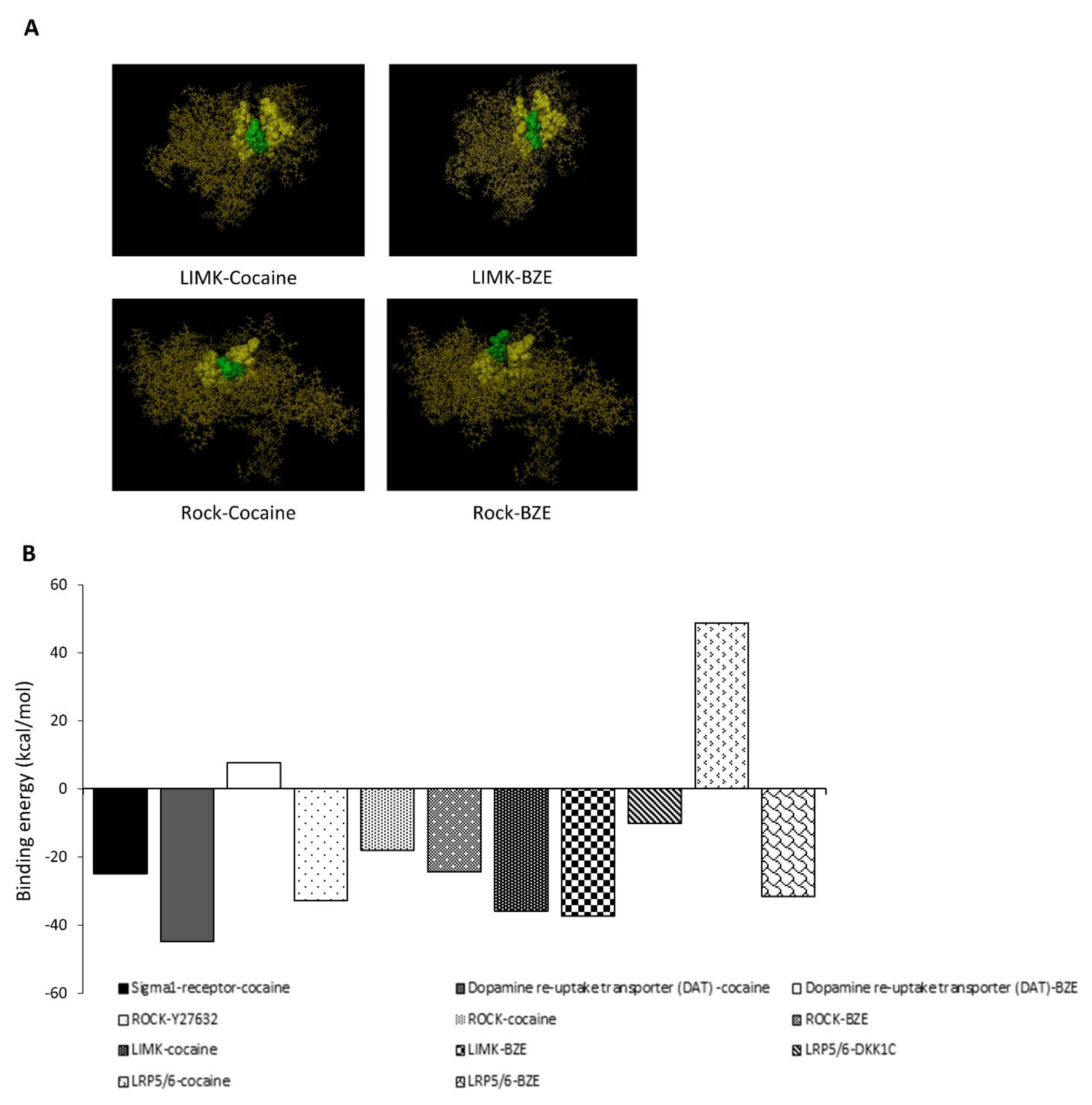
2.5. In-Silico Bioinformatics Docking Studies
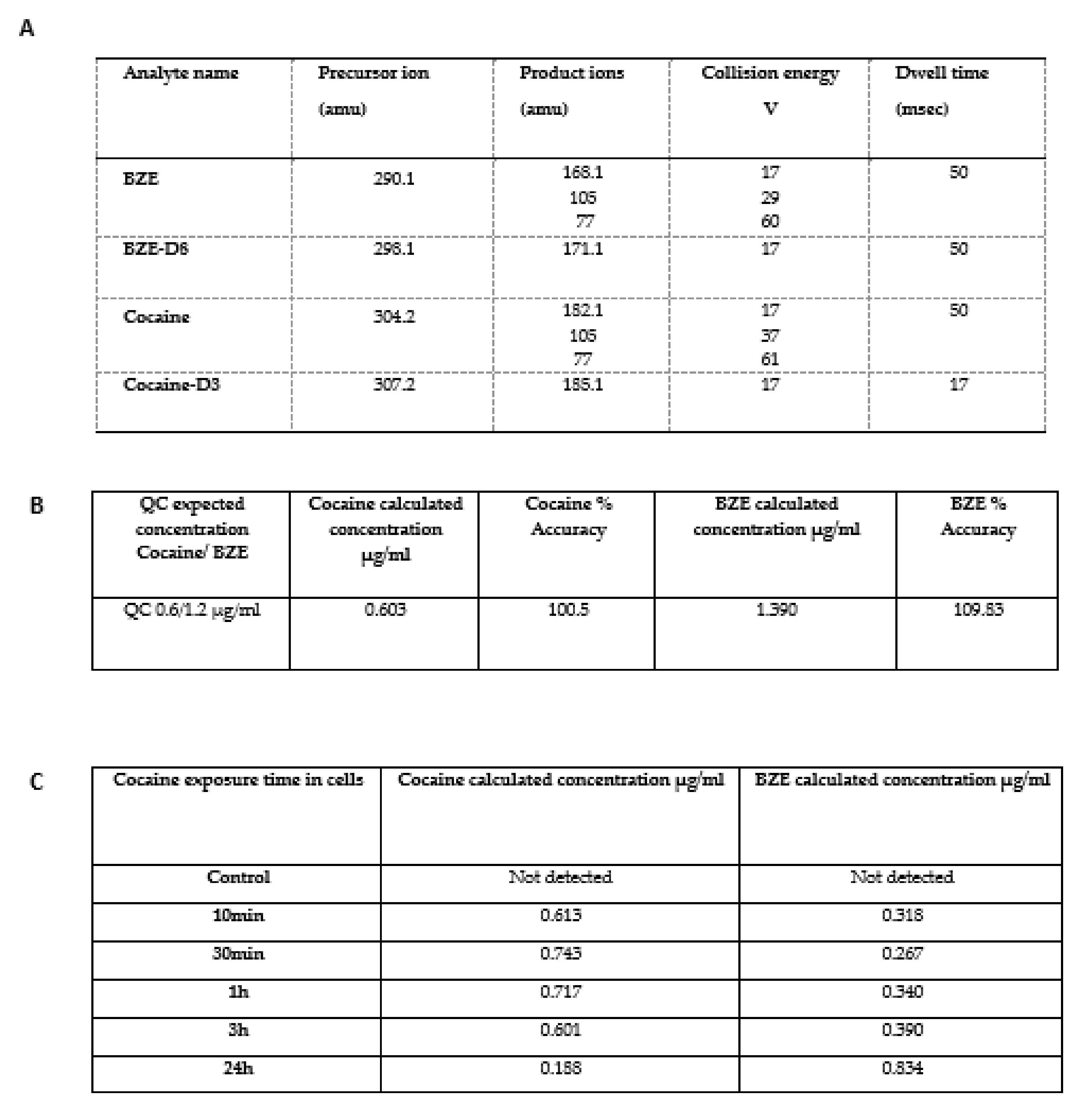
2.6. In Vitro Analysis of Cocaine Metabolism in Cardiomyocytes
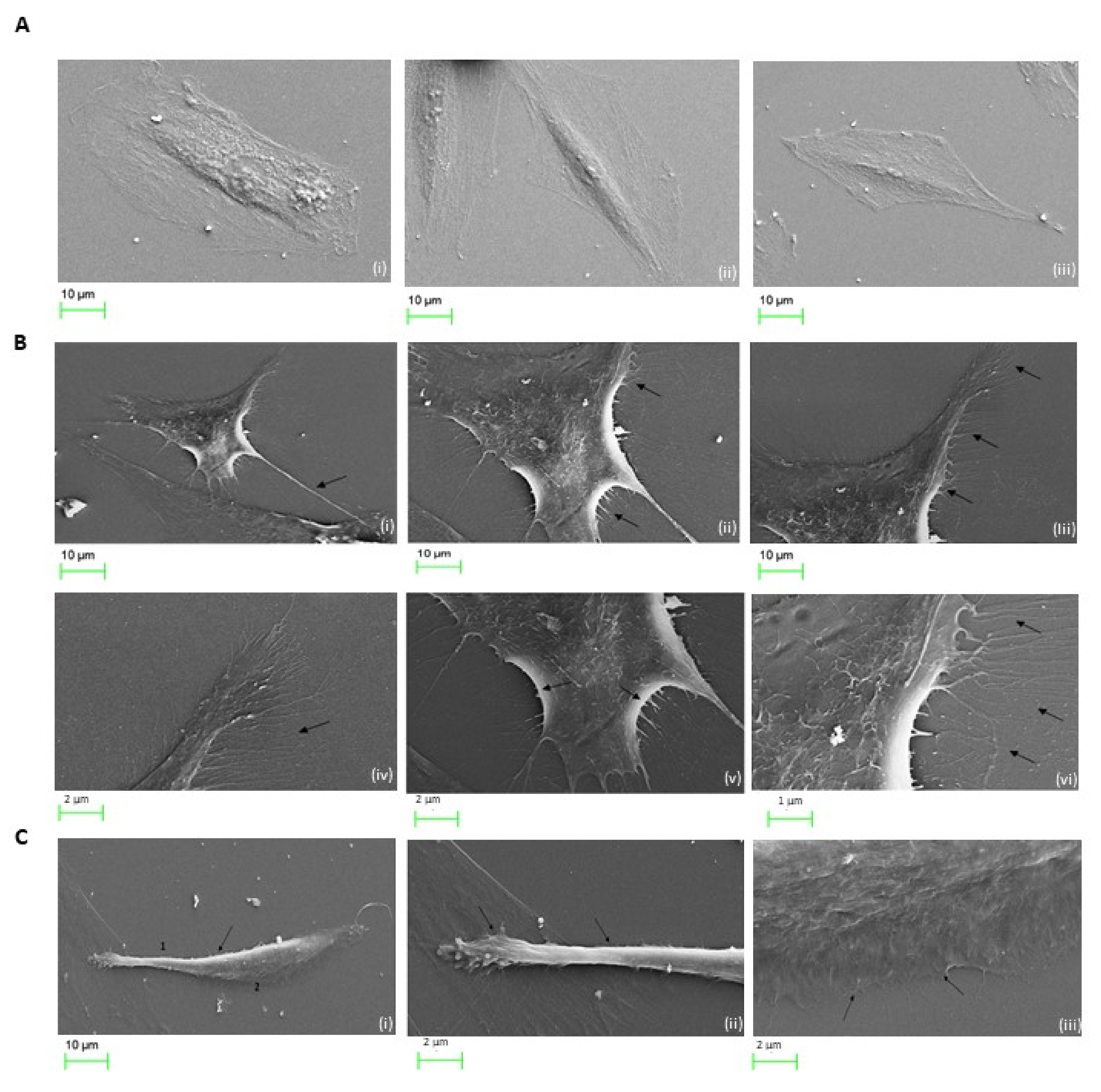
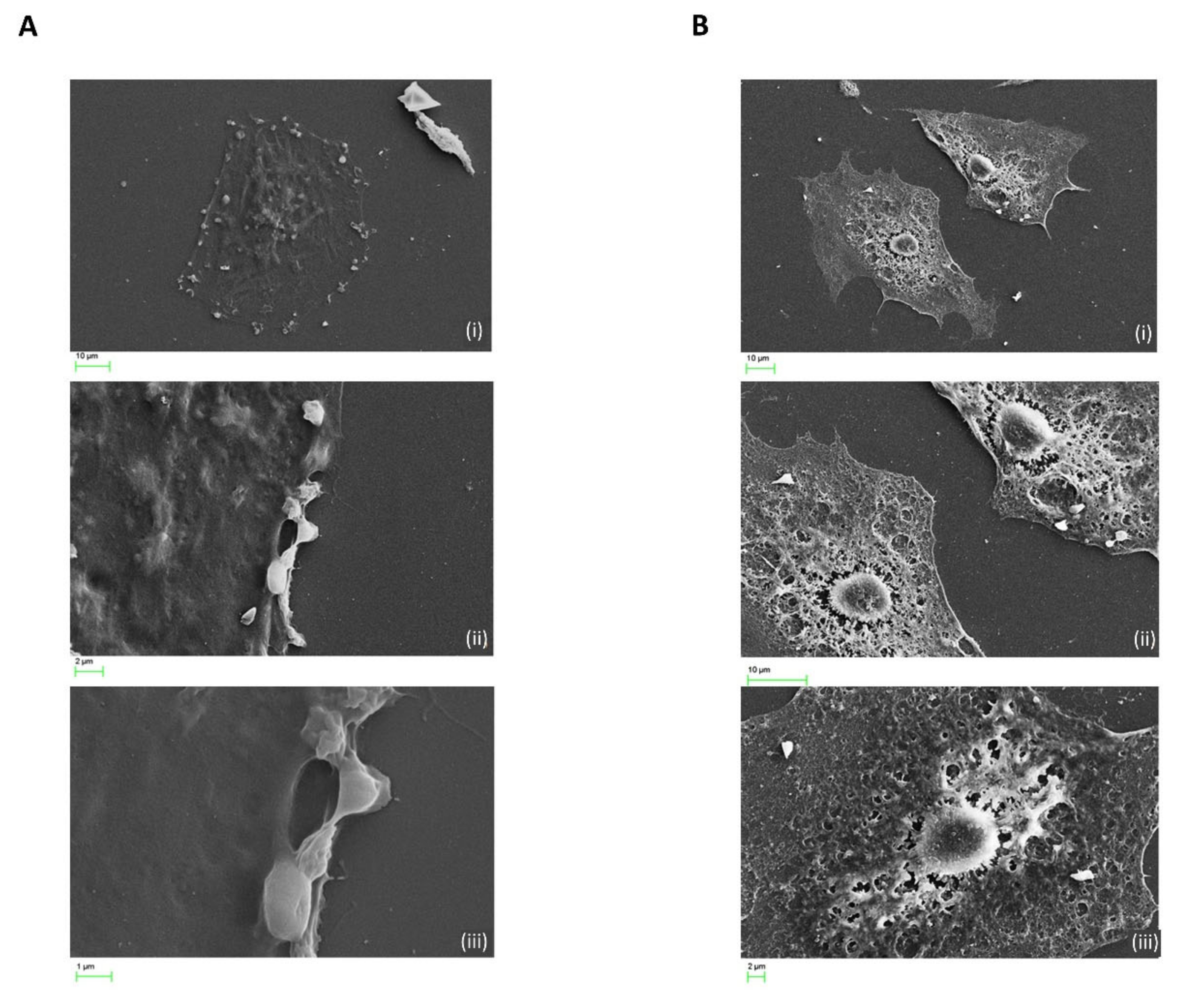
2.7. Cocaine Associated Cellular Microstructural Changes
3. Discussion
4. Materials and Methods
4.1. H9c2 Embryonic Cardiomyocyte Cell Lines
4.2. Chemicals and Reagents
4.3. Cell Viability and Toxicity Assay
4.4. Scratch Wound Assay
4.5. Immunoblotting
4.6. Cell Proliferation Assay
4.7. In-Vitro Cell-Substrate Adhesion Assay
4.8. LC-MS Analysis
4.9. Scanning Electron Microscopy
4.10. In-Silico Bioinformatic Docking Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABP | Actin binding protein |
| BrdU | Bromo-2′-deoxy-uridine |
| BZE | Benzoylecgonine |
| DPBS | Dulbecco’s Phosphate Buffered-Saline |
| DMEM | Dulbecco’s modified eagle medium |
| ECM | Extracellular matrix |
| ERK | Extracellular regulated kinase |
| ERM | Exrin Radixin Moesin |
| FASTA | Format DNA and Protein Sequence Alignment |
| FBS | Fetal bovine serum |
| FN | Fibronectin |
| LC-MS | Liquid Chromatography Mass Spectrometry |
| LIMK | LIM-kinase |
| MAPK | Mitogen-Activated Protein Kinase |
| NAc | Nucleus accumbens |
| RIPA | Radioimmunoprecipitation assay |
| ROCK | Rho- associated kinase |
| SEM | Scanning Electron Microscopy |
| VASP | Vasodilator stimulated phosphoprotein |
References
- Knuth, M.; Temme, O.; Daldrup, T.; Pawlik, E. Analysis of cocaine adulterants in human brain in cases of drug-related death. Forensic Sci. Int. 2018, 285, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Mittleman, M.A.; Mintzer, D.; Maclure, M.; Tofler, G.H.; Sherwood, J.B.; Muller, J.E. Triggering of Myocardial Infarction by Cocaine. Circulation 1999, 99, 2737–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egred, M.; Viswanathan, G.; Davis, G.K. Myocardial infarction in young adults. Postgrad. Med. J. 2005, 81, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Maceira, A.M.; Ripoll, C.; Cosin-Sales, J.; Igual, B.; Gavilan, M.; Salazar, J.; Belloch, V.; Pennell, D.J. Long term effects of cocaine on the heart assessed by cardiovascular magnetic resonance at 3T. J. Cardiovasc. Magn. Reson. 2014, 16, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-J.; Martin, J.A.; Gancarz, A.M.; Adank, D.N.; Sim, F.J.; Dietz, D.M. Activin A is increased in the nucleus accumbens following a cocaine binge. Sci. Rep. 2017, 7, 43658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.-M.; Lin, S.-Z.; Chang, N.-C. Membrane ERα attenuates myocardial fibrosis via RhoA/ROCK-mediated actin remodeling in ovariectomized female infarcted rats. J. Mol. Med. 2013, 92, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.; Saez, C.G.; Pallavicini, J.; Pereira-Flores, K.; Mendoza, C.; Hernández, R.; Novoa, U.; Ocaranza, M.P.; Massardo, T.; Mezzano, D. Cocaine-Induced Endothelial Dysfunction: Role of RhoA/Rho Kinase Pathway Activation. Blood 2012, 120, 2177. [Google Scholar] [CrossRef]
- Pradhan, L.; Mondal, D.; Chandra, S.; Ali, M.; Agrawal, K.C. Molecular Analysis of Cocaine-Induced Endothelial Dysfunction: Role of Endothelin-1 and Nitric Oxide. Cardiovasc. Toxicol. 2008, 8, 161–171. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, G.; Zhao, H.; Wang, Z.; Li, F.; Zhang, P.; Zhang, J.; Wang, X.; Wang, W. Targeted inhibition of Focal Adhesion Kinase Attenuates Cardiac Fibrosis and Preserves Heart Function in Adverse Cardiac Remodeling. Sci. Rep. 2017, 7, srep43146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, M.J.; Bravo, R.R.; Enea, M.; Carmo, H.; Carvalho, F.; Bastos, M.D.L.; Dinis-Oliveira, R.J.; Da Silva, D.D. Ethanol addictively enhances the in vitro cardiotoxicity of cocaine through oxidative damage, energetic deregulation, and apoptosis. Arch. Toxicol. 2018, 92, 2311–2325. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, W.; Ma, L.; Wang, Y.; Ma, J.; Jiang, M.; Deng, X.; Huang, F.; Yang, T.; Chen, M. Profilin-1 contributes to cardiac injury induced by advanced glycation end-products in rats. Mol. Med. Rep. 2017, 16, 6634–6641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziani, M.; Sarti, P.; Arese, M.; Magnifico, M.C.; Badiani, A.; Saso, L. Cardiovascular Mitochondrial Dysfunction Induced by Cocaine: Biomarkers and Possible Beneficial Effects of Modulators of Oxidative Stress. Oxidative Med. Cell. Longev. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Manninger, M.; Perl, S.; Brussee, H.; Toth, G.G. Sniff of coke breaks the heart: Cocaine-induced coronary vasospasm aggravated by therapeutic hypothermia and vasopressors after aborted sudden cardiac death: A case report. Eur. Hear. J. Case Rep. 2018, 2, yty041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calipari, E.S.; Godino, A.; Salery, M.; Damez-Werno, D.M.; Cahill, M.E.; Werner, C.T.; Gancarz, A.M.; Peck, E.G.; Jlayer, Z.; Rabkin, J.; et al. Synaptic Microtubule-Associated Protein EB3 and SRC Phosphorylation Mediate Structural and Behavioral Adaptations During Withdrawal From Cocaine Self-Administration. J. Neurosci. 2019, 39, 5634–5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.M.; Baldwin, G.T.; Compton, W.M. Recent Increases in Cocaine-Related Overdose Deaths and the Role of Opioids. Am. J. Public Heal. 2017, 107, 430–432. [Google Scholar] [CrossRef]
- Wu, S.-N.; Chang, H.-D.; Sung, R.J. Cocaine-Induced Inhibition of ATP-Sensitive K+ Channels in Rat Ventricular Myocytes and in Heart-Derived H9c2 Cells. Basic Clin. Pharmacol. Toxicol. 2006, 98, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sawbridge, D.; George, V.; Teng, L.; Bailey, A.; Kitchen, I.; Li, J.-M. Chronic Cocaine-Induced Cardiac Oxidative Stress and Mitogen-Activated Protein Kinase Activation: The Role of Nox2 Oxidase. J. Pharmacol. Exp. Ther. 2008, 328, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattanzio, F.A.; Tiangco, D.; Osgood, C.; Beebe, S.; Kerry, J.; Hargrave, B.Y. Cocaine Increases Intracellular Calcium and Reactive Oxygen Species, Depolarizes Mitochondria, and Activates Genes Associated With Heart Failure and Remodeling. Cardiovasc. Toxicol. 2005, 5, 377–390. [Google Scholar] [CrossRef]
- Havakuk, O.; Rezkalla, S.H.; Kloner, R.A. The Cardiovascular Effects of Cocaine. J. Am. Coll. Cardiol. 2017, 70, 101–113. [Google Scholar] [CrossRef]
- Moreira, F.P.; Medeiros, J.R.C.; Lhullier, A.C.; Souza, L.D.D.M.; Jansen, K.; Portela, L.V.; Lara, D.R.; Da Silva, R.A.; Wiener, C.D.; Oses, J.P. Cocaine abuse and effects in the serum levels of cytokines IL-6 and IL-10. Drug Alcohol Depend. 2016, 158, 181–185. [Google Scholar] [CrossRef] [Green Version]
- Badisa, R.B.; Wi, S.; Jones, Z.; Mazzio, E.; Zhou, Y.; Rosenberg, J.T.; Latinwo, L.M.; Grant, S.C.; Goodman, C.B. Cellular and molecular responses to acute cocaine treatment in neuronal-like N2a cells: Potential mechanism for its resistance in cell death. Cell Death Discov. 2018, 4, 13. [Google Scholar] [CrossRef]
- Chandra, R.; Engeln, M.; Schiefer, C.; Patton, M.H.; Martin, J.A.; Werner, C.T.; Riggs, L.M.; Francis, T.C.; McGlincy, M.; Evans, B.; et al. Drp1 Mitochondrial Fission in D1 Neurons Mediates Behavioral and Cellular Plasticity during Early Cocaine Abstinence. Neuron 2017, 96, 1327–1341. [Google Scholar] [CrossRef] [Green Version]
- Werner, C.T.; Mitra, S.; Auerbach, B.D.; Wang, Z.-J.; Martin, J.A.; Stewart, A.F.; Gobira, P.H.; Iida, M.; An, C.; Cobb, M.M.; et al. Neuroadaptations in the Dorsal Hippocampus Underlie Cocaine Seeking During Prolonged Abstinence; National Academy of Sciences: Washington, DC, USA, 2020; Volume 117, pp. 26460–26469. [Google Scholar]
- Badisa, R.B.; Kumar, S.S.; Mazzio, E.; Haughbrook, R.D.; Allen, J.R.; Davidson, M.W.; Fitch-Pye, C.A.; Goodman, C.B. N-Acetyl Cysteine Mitigates the Acute Effects of Cocaine-Induced Toxicity in Astroglia-Like Cells. PLoS ONE 2015, 10, e0114285. [Google Scholar] [CrossRef]
- Mardal, M.; Kinyua, J.; Ramin, P.; Miserez, B.; Van Nuijs, A.L.N.; Covaci, A.; Meyer, M.R. Screening for illicit drugs in pooled human urine and urinated soil samples and studies on the stability of urinary excretion products of cocaine, MDMA, and MDEA in wastewater by hyphenated mass spectrometry techniques. Drug Test. Anal. 2016, 9, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.W.; Holmgren, A. Concentrations of Cocaine and Benzoylecgonine in Femoral Blood from Cocaine-Related Deaths Compared with Venous Blood from Impaired Drivers. J. Anal. Toxicol. 2013, 38, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandhare, J.; Addai, A.B.; Mantri, C.K.; Hager, C.C.; Smith, R.M.; Barnett, L.; Villalta, F.; Kalams, S.A.; Dash, C. Cocaine Enhances HIV-1–Induced CD4+ T-Cell Apoptosis. Am. J. Pathol. 2014, 184, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Peretti, F.J.; Isenschmid, D.S.; Levine, B.; Caplan, Y.H.; Smialek, J.E. Cocaine fatality: An unexplained blood concentration in a fatal overdose. Forensic Sci. Int. 1990, 48, 135–138. [Google Scholar] [CrossRef]
- Del Olmo-Turrubiarte, A.; Calzada-Torres, A.; Díaz-Rosas, G.; Palma-Lara, I.; Sanchezurbina, R.; Balderrábano-Saucedo, N.; Gonzalezmarquez, H.; Garcia-Alonso, P.; Contrerasramos, A. Mouse models for the study of postnatal cardiac hypertrophy. IJC Hear. Vasc. 2015, 7, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.L.-H. From channels to systems: Ca2+ -sensitive K+ currents, alternans and cardiac arrhythmia. J. Physiol. 2017, 595, 2299–2300. [Google Scholar] [CrossRef]
- Welder, A.; Smith, M.; Ramos, K.; Acosta, D. Cocaine-induced cardiotoxicity in vitro. Toxicol. Vitr. 1988, 2, 205–213. [Google Scholar] [CrossRef]
- Chen, C.W.R.; Makkiya, M.; Aronow, W.; Spevack, D.M. Heightened risk of cardiac events following percutane-ous coronary intervention for cocaine-associated myocardial infarction. Arch. Med. Sci. AMS 2020, 16, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Bachi, K.; Mani, V.; Jeyachandran, D.; Fayad, Z.A.; Goldstein, R.Z.; Alia-Klein, N. Vascular disease in cocaine addiction. Atherosclerosis 2017, 262, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; He, J.; Gilbert, R.D.; Zhang, L. Cocaine induces apoptosis in fetal myocardial cells through a mitochon-dria-dependent pathway. J. Pharmacol. Exp. Ther. 2000, 292, 8–14. [Google Scholar]
- Sinha-Hikim, I.; Shen, R.; Nzenwa, I.; Gelfand, R.; Mahata, S.K.; Sinha-Hikim, A.P. Minocycline suppresses oxidative stress and attenuates fetal cardiac myocyte apoptosis triggered by in utero cocaine exposure. Apoptosis 2011, 16, 563–573. [Google Scholar] [CrossRef]
- Alvaro-Bartolome, M.; La Harpe, R.; Callado, L.F.; Meana, J.J.; Garcia-Sevilla, J. Molecular adaptations of apoptotic pathways and signaling partners in the cerebral cortex of human cocaine addicts and cocaine-treated rats. Neuroscience 2011, 196, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, J.E.N.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An introduction to the wound healing assay using live-cell microscopy. Cell Adhes. Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Itou, J.; Oishi, I.; Kawakami, H.; Glass, T.J.; Richter, J.; Johnson, A.; Lund, T.C.; Kawakami, Y. Migration of cardiomyocytes is essential for heart regeneration in zebrafish. Development 2012, 139, 4133–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo, X.; Curós, A.; Muga, R.; Serra, J.; Sanvisens, A.; Bayes-Genis, A. Acute coronary syndrome and cocaine use: 8-year prevalence and inhospital outcomes. Eur. Hear. J. 2011, 32, 1244–1250. [Google Scholar] [CrossRef] [Green Version]
- Song, S.H.; Park, K.; Kim, S.W.; Paick, J.; Cho, M.C. Involvement of Rho-Kinase/LIM Kinase/Cofilin Signaling Pathway in Corporal Fibrosis after Cavernous Nerve Injury in Male Rats. J. Sex. Med. 2015, 12, 1522–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, K.; Luan, Y.; Wang, T.; Zhuan, L.; Rao, K.; Wang, S.-G.; Ye, Z.-Q.; Liu, J.-H.; Wang, D.-W. Reduced corporal fibrosis to protect erectile function by inhibiting the Rho-kinase/LIM-kinase/cofilin pathway in the aged transgenic rat harboring human tissue kallikrein 1. Asian J. Androl. 2016, 19, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Kligys, K.; Claiborne, J.N.; DeBiase, P.J.; Hopkinson, S.B.; Wu, Y.; Mizuno, K.; Jones, J.C.R. The Slingshot Family of Phosphatases Mediates Rac1 Regulation of Cofilin Phosphorylation, Laminin-332 Organization, and Motility Behavior of Keratinocytes. J. Biol. Chem. 2007, 282, 32520–32528. [Google Scholar] [CrossRef] [Green Version]
- Toda, S.; Shen, H.-W.; Peters, J.; Cagle, S.; Kalivas, P.W. Cocaine Increases Actin Cycling: Effects in the Reinstatement Model of Drug Seeking. J. Neurosci. 2006, 26, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Kim, K.; Duan, M.; Hayashi, T.; Guo, M.; Morgello, S.; Prat, A.; Wang, J.; Su, T.-P.; Buch, S. Cocaine Hijacks 1 Receptor to Initiate Induction of Activated Leukocyte Cell Adhesion Molecule: Implication for Increased Monocyte Adhesion and Migration in the CNS. J. Neurosci. 2011, 31, 5942–5955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Heyob, K.; Tipple, T.E.; Pryhuber, G.S.; Rogers, L.K. Alterations in VASP phosphorylation and profilin1 and cofilin1 expression in hyperoxic lung injury and BPD. Respir. Res. 2018, 19, 229. [Google Scholar] [CrossRef]
- Okayama, T.; Kikuchi, S.; Ochiai, T.; Ikoma, H.; Kubota, T.; Ichikawa, D.; Fujiwara, H.; Okamoto, K.; Sakakura, C.; Sonoyama, T.; et al. Attenuated response to liver injury in moesin-deficient mice: Impaired stellate cell migration and decreased fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2008, 1782, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpin, M.; Chirivino, D.; Naba, A.; Zwaenepoel, I. Emerging role for ERM proteins in cell adhesion and migration. Cell Adhes. Migr. 2011, 5, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Eldik, W.; Adel, B.D.; Monshouwer-Kloots, J.; Salvatori, D.; Maas, S.; Van Der Made, I.; Creemers, E.E.; Frank, D.; Frey, N.; Boontje, N.; et al. Z-disc protein CHAPb induces cardiomyopathy and contractile dysfunction in the postnatal heart. PLoS ONE 2017, 12, e0189139. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Shin, S.; Kim, S.; Jeon, S.; Kim, J.-H. Cocaine regulates ezrin–radixin–moesin proteins and RhoA signaling in the nucleus accumbens. Neuroscience 2009, 163, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Sartoretto, J.L.; Jin, B.Y.; Bauer, M.; Gertler, F.B.; Liao, R.; Michel, T. Regulation of VASP phosphorylation in cardiac myocytes: Differential regulation by cyclic nucleotides and modulation of protein expression in diabetic and hypertrophic heart. Am. J. Physiol. Circ. Physiol. 2009, 297, H1697–H1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, N.; Waschke, J. VASP is involved in cAMP-mediated Rac 1 activation in microvascular endothelial cells. Am. J. Physiol. Physiol. 2009, 296, C453–C462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmbhatt, A.A.; Klemke, R.L. ERK and RhoA Differentially Regulate Pseudopodia Growth and Retraction during Chemotaxis. J. Biol. Chem. 2003, 278, 13016–13025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.G.H.; Pierotti, V.; Storez, H.; Lindberg, E.; Thuret, A.; Muntaner, O.; Labbé-Jullié, C.; Pitcher, J.A.; Marullo, S. Cooperative Regulation of Extracellular Signal-Regulated Kinase Activation and Cell Shape Change by Filamin A and β-Arrestins. Mol. Cell. Biol. 2006, 26, 3432–3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, M.C.; Er, E.E.; Zhang, W.; Ballif, B.A.; Elliott, H.L.; Danuser, G.; Blenis, J. ERK-MAPK Drives Lamellipodia Protrusion by Activating the WAVE2 Regulatory Complex. Mol. Cell 2011, 41, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Lai, N.C.; Kawaraguchi, Y.; Liao, P.; Copps, J.; Sugano, Y.; Okada-Maeda, S.; Banerjee, I.; Schilling, J.M.; Gingras, A.R.; et al. Integrins protect cardiomyocytes from ischemia/reperfusion injury. J. Clin. Investig. 2013, 123, 4294–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shewchuk, L.J.; Bryan, S.; Ulanova, M.; Khaper, N. Integrin β3 prevents apoptosis of HL-1 cardiomyocytes under conditions of oxidative stressThis article is one of a selection of papers published in a Special Issue on Oxidative Stress in Health and Disease. Can. J. Physiol. Pharmacol. 2010, 88, 324–330. [Google Scholar] [CrossRef]
- Cerretani, D.; Fineschi, V.; Bello, S.; Riezzo, I.; Turillazzi, E.; Neri, M. Role of oxidative stress in cocaine-induced cardiotoxicity and cocaine-related death. Curr. Med. Chem. 2012, 19, 5619–5623. [Google Scholar] [CrossRef]
- Chi, Z.; Melendez, A.J. Role of Cell Adhesion Molecules and Immune-Cell Migration in the Initiation, Onset and Development of Atherosclerosis. Cell Adhes. Migr. 2007, 1, 171–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrizi, R.; Pasceri, V.; Sciahbasi, A.; Summaria, F.; Rosano, G.M.; Lioy, E. Evidence of Cocaine-Related Coronary Atherosclerosis in Young Patients With Myocardial Infarction. J. Am. Coll. Cardiol. 2006, 47, 2120–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrose, J.A.; Singh, M. Pathophysiology of coronary artery disease leading to acute coronary syndromes. F1000Prime Rep. 2015, 7, 08. [Google Scholar] [CrossRef]
- Camera, M.; Rossetti, L.; Barbieri, S.S.; Zanotti, I.; Canciani, B.; Trabattoni, D.; Ruscica, M.; Tremoli, E.; Ferri, N. PCSK9 as a Positive Modulator of Platelet Activation. J. Am. Coll. Cardiol. 2018, 71, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Park, T. Acute and Chronic Effects of Cocaine on Cardiovascular Health. Int. J. Mol. Sci. 2019, 20, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witek, P.; Korga, A.; Burdan, F.; Ostrowska, M.; Nosowska, B.; Iwan, M.; Dudka, J. The effect of a number of H9C2 rat cardiomyocytes passage on repeatability of cytotoxicity study results. Cytotechnology 2016, 68, 2407–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verma, A.; Orme Merve, A.; Remeškevičius, V.; Sobiecka, P.; Taylor, L.; Lawton, S.; Jones, B.P.; Polycarpou, E.; Bennett, J.; Rooney, B. Cocaine Induces Cytoskeletal Changes in Cardiac Myocytes: Implications for Cardiac Morphology. Int. J. Mol. Sci. 2021, 22, 2263. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052263
Verma A, Orme Merve A, Remeškevičius V, Sobiecka P, Taylor L, Lawton S, Jones BP, Polycarpou E, Bennett J, Rooney B. Cocaine Induces Cytoskeletal Changes in Cardiac Myocytes: Implications for Cardiac Morphology. International Journal of Molecular Sciences. 2021; 22(5):2263. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052263
Chicago/Turabian StyleVerma, Avnish, Ayse Orme Merve, Vytautas Remeškevičius, Pola Sobiecka, Luke Taylor, Scott Lawton, Ben P Jones, Elena Polycarpou, Jason Bennett, and Brian Rooney. 2021. "Cocaine Induces Cytoskeletal Changes in Cardiac Myocytes: Implications for Cardiac Morphology" International Journal of Molecular Sciences 22, no. 5: 2263. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052263