
DAMPs and RAGE Pathophysiology at the Acute Phase of Brain Injury: An Overview

 , ,
, ,
Abstract
:1. Introduction
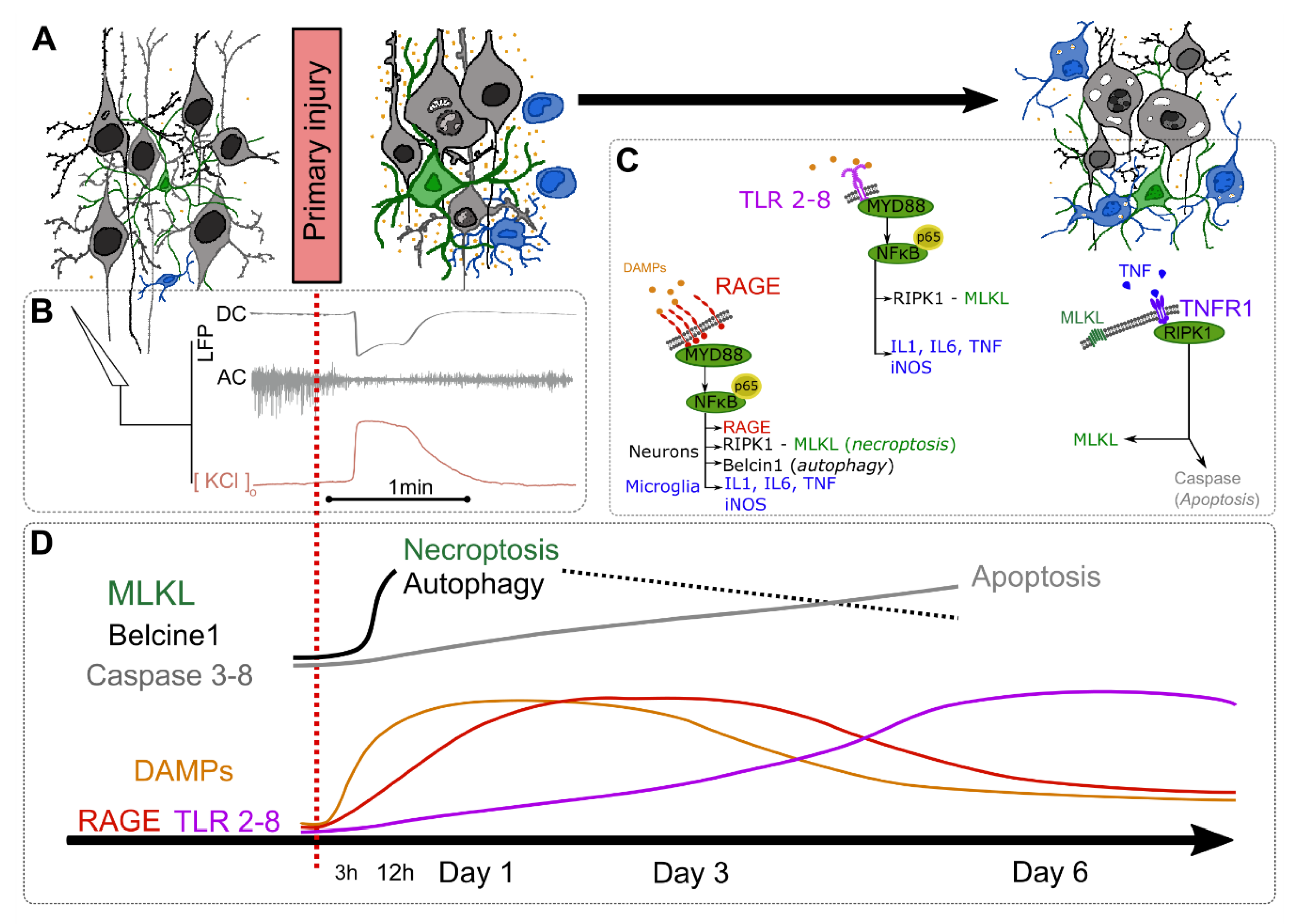
2. Acute Lesion Progression Pathophysiology
2.1. Kinetics of DAMPs and Consequences after the Primary Insult
2.2. DAMPs Clearence
3. A Biomarker Approach
4. Therapeutic Perspectives
5. Materials and Methods
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | Peptide amyloid β |
| ATP | Adenosine triphosphate |
| BBB | Blood–brain barrier |
| CSF | Cerebrospinal fluid |
| CXCL | Chemokine (C-X-C motif) ligand |
| DAMPs | Damage or danger-associated molecular patterns |
| EVD | External ventricular drain |
| GFAP | Glial fibrillary acidic protein |
| HMGB1 | High mobility group protein 1 |
| IL | Interleukin |
| IS | Ischemic stroke |
| MLKL | Mixed-lineage kinase domain-like pseudokinase |
| MSR1 | Macrophage scavenger receptor 1 |
| NF-κB | Nuclear factor-kappa B |
| NO | Nitric oxide |
| NOD | Nucleotide oligomerization domain |
| NSE | Neuron-specific enolase |
| PRRs | Pattern-recognition receptors |
| RAGE | Receptor for advanced glycation end-products |
| sRAGE | Soluble form of the receptor for advanced glycation end-products |
| RIPK | Receptor-interacting protein kinase |
| SAH | Subarachnoid hemorrhage |
| SD | Spreading depolarization |
| TBI | Traumatic brain injury |
| TNF | Tumor necrosis factor |
| TNFR1 | Tumor necrosis factor receptor 1 |
| TLRs | Toll-like receptors |
References
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef]
- Gao, T.-L.; Yuan, X.-T.; Yang, D.; Dai, H.-L.; Wang, W.-J.; Peng, X.; Shao, H.-J.; Jin, Z.-F.; Fu, Z.-J. Expression of HMGB1 and RAGE in rat and human brains after traumatic brain injury. J. Trauma Acute Care Surg. 2012, 72, 643–649. [Google Scholar] [CrossRef]
- Khan, M.A.; Schultz, S.; Othman, A.; Fleming, T.; Lebrón-Galán, R.; Rades, D.; Clemente, D.; Nawroth, P.P.; Schwaninger, M. Hyperglycemia in Stroke Impairs Polarization of Monocytes/Macrophages to a Protective Noninflammatory Cell Type. J. Neurosci. 2016, 36, 9313–9325. [Google Scholar] [CrossRef] [Green Version]
- Bsibsi, M.; Ravid, R.; Gveric, D.; Noort, J.M. van Broad Expression of Toll-Like Receptors in the Human Central Nervous System. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/Macrophage Polarization Dynamics Reveal Novel Mechanism of Injury Expansion After Focal Cerebral Ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Z.; Fan, L.; Zhao, L.; Xu, X.; Liu, Y.; Chao, H.; Liu, N.; You, Y.; Liu, Y.; Wang, X.; et al. Silencing of A20 Aggravates Neuronal Death and Inflammation After Traumatic Brain Injury: A Potential Trigger of Necroptosis. Front. Mol. Neurosci. 2019, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.; Hafez, A.; Jahromi, B.; Kinfe, T.; Lamprecht, A.; Niemelä, M.; Muhammad, S. Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2018, 19, 2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Zhang, X.-S.; Zhang, Z.-H.; Zhou, X.-M.; Gao, Y.-Y.; Liu, G.-J.; Wang, H.; Wu, L.-Y.; Li, W.; Hang, C.-H. Peroxiredoxin 2 activates microglia by interacting with Toll-like receptor 4 after subarachnoid hemorrhage. J. Neuroinflamm. 2018, 15, 87. [Google Scholar] [CrossRef]
- Tang, S.-C.; Wang, Y.-C.; Li, Y.-I.; Lin, H.-C.; Manzanero, S.; Hsieh, Y.-H.; Phipps, S.; Hu, C.-J.; Chiou, H.-Y.; Huang, Y.-S.; et al. Functional Role of Soluble Receptor for Advanced Glycation End Products in Stroke. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.; Tuttolomondo, A.; Raimondo, D.D.; Fernandez, P.; Licata, G. Risk factors profile and clinical outcome of ischemic stroke patients admitted in a Department of Internal Medicine and classified by TOAST classification. Int. Angiol. J. Int. Union Angiol. 2006, 25, 261–267. [Google Scholar]
- Shi, K.; Tian, D.-C.; Li, Z.-G.; Ducruet, A.F.; Lawton, M.T.; Shi, F.-D. Global brain inflammation in stroke. Lancet Neurol. 2019, 18, 1058–1066. [Google Scholar] [CrossRef]
- Mijajlović, M.D.; Pavlović, A.; Brainin, M.; Heiss, W.-D.; Quinn, T.J.; Ihle-Hansen, H.B.; Hermann, D.M.; Assayag, E.B.; Richard, E.; Thiel, A.; et al. Post-stroke dementia—A comprehensive review. BMC Med. 2017, 15, 11. [Google Scholar] [CrossRef] [Green Version]
- Kliper, E.; Bashat, D.B.; Bornstein, N.M.; Shenhar-Tsarfaty, S.; Hallevi, H.; Auriel, E.; Shopin, L.; Bloch, S.; Berliner, S.; Giladi, N.; et al. Cognitive Decline After Stroke. Stroke 2013, 44, 1433–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dénes, Á.; Ferenczi, S.; Kovács, K.J. Systemic inflammatory challenges compromise survival after experimental stroke via augmenting brain inflammation, blood- brain barrier damage and brain oedema independently of infarct size. J. Neuroinflamm. 2011, 8, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; Shuttleworth, C.W.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef] [PubMed]
- Silverman, W.R.; de Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The Pannexin 1 Channel Activates the Inflammasome in Neurons and Astrocytes. J. Biol. Chem. 2009, 284, 18143–18151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wu, Z.; Limnuson, K.; Cheyuo, C.; Wang, P.; Ahn, C.H.; Narayan, R.K.; Hartings, J.A. Development and application of a microfabricated multimodal neural catheter for neuroscience. Biomed. Microdevices 2016, 18, 8. [Google Scholar] [CrossRef]
- Yao, X.; Liu, S.; Ding, W.; Yue, P.; Jiang, Q.; Zhao, M.; Hu, F.; Zhang, H. TLR4 signal ablation attenuated neurological deficits by regulating microglial M1/M2 phenotype after traumatic brain injury in mice. J. Neuroimmunol. 2017, 310, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Kligman, D.; Marshak, D.R. Purification and characterization of a neurite extension factor from bovine brain. Proc. Natl. Acad. Sci. USA 1985, 82, 7136–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003, 60, 540–551. [Google Scholar] [CrossRef]
- Villarreal, A.; Seoane, R.; Torres, A.G.; Rosciszewski, G.; Angelo, M.F.; Rossi, A.; Barker, P.A.; Ramos, A.J. S100B protein activates a RAGE-dependent autocrine loop in astrocytes: Implications for its role in the propagation of reactive gliosis. J. Neurochem. 2014, 131, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wilson, G. A temporary local energy pool coupled to neuronal activity: Fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J. Neurochem. 1997, 69, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. HMGB1-Mediated Neuroinflammatory Responses in Brain Injuries: Potential Mechanisms and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 4609. [Google Scholar] [CrossRef]
- Manivannan, S.; Marei, O.; Elalfy, O.; Zaben, M. Neurogenesis after traumatic brain injury—The complex role of HMGB1 and neuroinflammation. Neuropharmacology 2021, 183, 108400. [Google Scholar] [CrossRef]
- Vincent, A.M.; Perrone, L.; Sullivan, K.A.; Backus, C.; Sastry, A.M.; Lastoskie, C.; Feldman, E.L. Receptor for Advanced Glycation End Products Activation Injures Primary Sensory Neurons via Oxidative Stress. Endocrinology 2007, 148, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Tang, D.; Lotze, M.T.; III, H.J.Z. RAGE regulates autophagy and apoptosis following oxidative injury. Autophagy 2011, 7, 442–444. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Aida, Y.; Kamide, T.; Ishii, H.; Kitao, Y.; Uchiyama, N.; Nakada, M.; Hori, O. Soluble receptor for advanced glycation end products as a biomarker of symptomatic vasospasm in subarachnoid hemorrhage. J. Neurosurg. 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cheng, S.; Hu, H.; Zhang, X.; Xu, J.; Wang, R.; Zhang, P. Progranulin protects against cerebral ischemia-reperfusion (I/R) injury by inhibiting necroptosis and oxidative stress. Biochem. Bioph. Res. Commun. 2019, 521, 569–576. [Google Scholar] [CrossRef]
- Croker, B.A.; Rickard, J.A.; Shlomovitz, I.; Al-Obeidi, A.; D’Cruz, A.A.; Gerlic, M. Apoptosis and Beyond; Wiley: Hoboken, NJ, USA, 2018; pp. 99–128. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Jitsuishi, T.; Hozumi, T.; Iwanami, J.; Kitajo, K.; Yamaguchi, H.; Mori, Y.; Mogi, M.; Sawai, S. Temporal expression profiling of DAMPs-related genes revealed the biphasic post-ischemic inflammation in the experimental stroke model. Mol. Brain 2020, 13, 57. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Sun, L.; Feng, D.; Sun, Q.; Dou, Y.; Liu, C.; Zhou, F.; Li, H.; Shen, H.; Wang, Z.; et al. HMGB1 promotes neurovascular remodeling via Rage in the late phase of subarachnoid hemorrhage. Brain Res. 2017, 1670, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, W.; Sun, Q.; Liu, M.; Li, W.; Zhang, X.; Zhou, M.; Hang, C. Expression and cell distribution of receptor for advanced glycation end-products in the rat cortex following experimental subarachnoid hemorrhage. Brain Res. 2014, 1543, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A.; et al. The HMGB1 Receptor RAGE Mediates Ischemic Brain Damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef] [Green Version]
- Pleines, U.E.; Morganti-Kossmann, M.C.; Rancan, M.; Joller, H.; Trentz, O.; Kossmann, T. S-100 reflects the extent of injury and outcome, whereas neuronal specific enolase is a better indicator of neuroinflammation in patients with severe traumatic brain injury. J. Neurotrauma 2001, 18, 491–498. [Google Scholar] [CrossRef]
- Okuma, Y.; Liu, K.; Wake, H.; Liu, R.; Nishimura, Y.; Hui, Z.; Teshigawara, K.; Haruma, J.; Yamamoto, Y.; Yamamoto, H.; et al. Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1–RAGE interaction. Neuropharmacology 2014, 85, 18–26. [Google Scholar] [CrossRef]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; Marchis, F.D.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4The HMGB1–CXCL12 heterocomplex acts via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, G.; Talcott, K.E.; Bhattacharya, S.K.; Crabb, J.W.; Sears, J.E. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp. Cell Res. 2006, 312, 3526–3538. [Google Scholar] [CrossRef] [PubMed]
- Urbonaviciute, V.; Meister, S.; Fürnrohr, B.G.; Frey, B.; Gückel, E.; Schett, G.; Herrmann, M.; Voll, R.E. Oxidation of the alarmin high-mobility group box 1 protein (HMGB1) during apoptosis. Autoimmunity 2009, 42, 305–307. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Cheh, C.-W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef] [Green Version]
- Hassid, B.G.; Nair, M.N.; Ducruet, A.F.; Otten, M.L.; Komotar, R.J.; Pinsky, D.J.; Schmidt, A.M.; Yan, S.F.; Connolly, E.S. Neuronal RAGE expression modulates severity of injury following transient focal cerebral ischemia. J. Clin. Neurosci. 2009, 16, 302–306. [Google Scholar] [CrossRef]
- Lei, C.; Zhang, S.; Cao, T.; Tao, W.; Liu, M.; Wu, B. HMGB1 may act via RAGE to promote angiogenesis in the later phase after intracerebral hemorrhage. Neuroscience 2015, 295, 39–47. [Google Scholar] [CrossRef]
- Iliff, J.; Wang, M.; Liao, Y.; Plogg, B.; Peng, W.; Gundersen, G.; Benveniste, H.; Vates, G.; Deane, R.; Goldman, S.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid B. Sci. Transl. Med. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.; Wang, M.; Zeppenfeld, D.; Venkataraman, A.; Plog, B.; Liao, Y.; Deane, R.; Nedergaard, M. Cerebral Arterial Pulsation Drives Paravascular CSF-Interstitial Fluid Exchange in the Murine Brain. J. Neurosci. 2019, 33. [Google Scholar] [CrossRef] [Green Version]
- Plog, B.A.; Dashnaw, M.L.; Hitomi, E.; Peng, W.; Liao, Y.; Lou, N.; Deane, R.; Nedergaard, M. Biomarkers of Traumatic Injury Are Transported from Brain to Blood via the Glymphatic System. J. Neurosci. 2015, 35, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Risher, W.; Ard, D.; Yuan, J. Recurrent spontaneous spreading depolarizations facilitate acute dendritic injury in the ischemic penumbra. J. Neurosci. 2010, 29, 9859–9868. [Google Scholar] [CrossRef]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 2333. [Google Scholar] [CrossRef]
- Mestre, H.; Du, T.; Sweeney, A.M.; Liu, G.; Samson, A.J.; Peng, W.; Mortensen, K.N.; Stæger, F.F.; Bork, P.A.R.; Bashford, L.; et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 2020, 367, eaax7171. [Google Scholar] [CrossRef]
- Iliff, J.; Chen, M.; Plog, B.; Zeppenfeld, D.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34. [Google Scholar] [CrossRef] [Green Version]
- Gaberel, T.; Gakuba, C.; Goulay, R.; Lizarrondo, S.; Hanouz, J.-L.; Emery, E.; Touze, E.; Vivien, D.; Gauberti, M. Impaired Glymphatic Perfusion After Strokes Revealed by Contrast-Enhanced MRI. Stroke 2014, 45, 3092–3096. [Google Scholar] [CrossRef] [Green Version]
- Goulay, R.; Flament, J.; Gauberti, M.; Naveau, M.; Pasquet, N.; Gakuba, C.; Emery, E.; Hantraye, P.; Vivien, D.; Aron-Badin, R.; et al. Subarachnoid Hemorrhage Severely Impairs Brain Parenchymal Cerebrospinal Fluid Circulation in Nonhuman Primate. Stroke 2017, 48, 2301–2305. [Google Scholar] [CrossRef]
- Lindblad, C.; Nelson, D.W.; Zeiler, F.A.; Ercole, A.; Ghatan, P.H.; von Horn, H.; Risling, M.; Svensson, M.; Agoston, D.V.; Bellander, B.-M.; et al. Influence of Blood–Brain Barrier Integrity on Brain Protein Biomarker Clearance in Severe Traumatic Brain Injury: A Longitudinal Prospective Study. J. Neurotraum. 2020, 37, 1381–1391. [Google Scholar] [CrossRef] [Green Version]
- Shichita, T.; Ito, M.; Morita, R.; Komai, K.; Noguchi, Y.; Ooboshi, H.; Koshida, R.; Takahashi, S.; Kodama, T.; Yoshimura, A. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat. Med. 2017, 23, 723–732. [Google Scholar] [CrossRef]
- Lasič, E.; Galland, F.; Vardjan, N.; Šribar, J.; Križaj, I.; Leite, M.C.; Zorec, R.; Stenovec, M. Time-dependent uptake and trafficking of vesicles capturing extracellular S100B in cultured rat astrocytes. J. Neurochem. 2016, 139, 309–323. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Pasvogel, A.E.; Miketova, P.; Moore, I.M. (Ki) Cerebrospinal Fluid Phospholipid Changes Following Traumatic Brain Injury. Biol. Res. Nurs. 2008, 10, 113–120. [Google Scholar] [CrossRef]
- Pasvogel, A.E.; Miketova, P.; Moore, I.M. Differences in CSF Phospholipid Concentration by Traumatic Brain Injury Outcome. Biol. Res. Nurs. 2010, 11, 325–331. [Google Scholar] [CrossRef]
- Deane, R.; Yan, S.D.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hasu, M.; Popov, D.; Zhang, J.H.; Chen, J.; Yan, S.D.; Brett, J.; Cao, R.; Kuwabara, K.; Costache, G. Receptor for advanced glycation end products (AGEs) has a central role in vessel wall interactions and gene activation in response to circulating AGE proteins. Proc. Natl. Acad. Sci. 1994, 91, 8807–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.H.; Yeon, S.I.; Youn, J.H.; Choi, J.E.; Sasaki, N.; Choi, I.-H.; Shin, J.-S. Expression of a novel secreted splice variant of the receptor for advanced glycation end products (RAGE) in human brain astrocytes and peripheral blood mononuclear cells. Mol. Immunol. 2004, 40, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). Faseb J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef]
- Wang, W.; Lu, R.; Feng, D.; Zhang, H. Sevoflurane Inhibits Glutamate-Aspartate Transporter and Glial Fibrillary Acidic Protein Expression in Hippocampal Astrocytes of Neonatal Rats Through the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) Pathway. Anest. Analg. 2016, 123, 93. [Google Scholar] [CrossRef]
- Tang, S.-C.; Yeh, S.-J.; Tsai, L.-K.; Hu, C.-J.; Lien, L.-M.; Peng, G.-S.; Yang, W.-S.; Chiou, H.-Y.; Jeng, J.-S. Cleaved but not endogenous secretory RAGE is associated with outcome in acute ischemic stroke. Neurology 2016, 86, 270–276. [Google Scholar] [CrossRef]
- Yang, D.-B.; Dong, X.-Q.; Du, Q.; Yu, W.-H.; Zheng, Y.-K.; Hu, W.; Wang, K.-Y.; Chen, F.-H.; Xu, Y.-S.; Wang, Y.; et al. Clinical relevance of cleaved RAGE plasma levels as a biomarker of disease severity and functional outcome in aneurysmal subarachnoid hemorrhage. Clin. Chim. Acta 2018, 486, 335–340. [Google Scholar] [CrossRef]
- Prasad, K. Low Levels of Serum Soluble Receptors for Advanced Glycation End Products, Biomarkers for Disease State: Myth or Reality. Int. J. Angiol. 2014, 23, 011–016. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Ming, J.; Liu, S.; Lai, B.; Fang, F.; Cang, J. Sevoflurane enhanced the clearance of Aβ1-40 in hippocampus under surgery via up-regulating AQP-4 expression in astrocyte. Life Sci. 2019, 221, 143–151. [Google Scholar] [CrossRef]
- Group, B.D.W. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Christ-Crain, M.; Opal, S.M. Clinical review: The role of biomarkers in the diagnosis and management of community-acquired pneumonia. Crit. Care 2010, 14, 203. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, A.D.; Niogi, S.N.; Whitlow, C.J.; Tsiouris, A.J. Traumatic Brain Injury: Imaging Patterns and Complications. Radiographics 2019, 39, 1571–1595. [Google Scholar] [CrossRef]
- Clement, M.O. Imaging of Brain Trauma. Radiol. Clin. N. Am. 2019, 57, 733–744. [Google Scholar] [CrossRef]
- Jadhav, A.P.; Desai, S.M.; Liebeskind, D.S.; Wechsler, L.R. Neuroimaging of Acute Stroke. Neurol. Clin. 2019, 38, 185–199. [Google Scholar] [CrossRef]
- Araki, T.; Yokota, H.; Morita, A. Pediatric Traumatic Brain Injury: Characteristic Features, Diagnosis, and Management. Neurol. Med. Chir. 2016, 57, ra.2016-0191. [Google Scholar] [CrossRef] [Green Version]
- Madsen, T.E.; Khoury, J.; Cadena, R.; Adeoye, O.; Alwell, K.A.; Moomaw, C.J.; McDonough, E.; Flaherty, M.L.; Ferioli, S.; Woo, D.; et al. Potentially Missed Diagnosis of Ischemic Stroke in the Emergency Department in the Greater Cincinnati/Northern Kentucky Stroke Study. Acad. Emerg. Med. 2016, 23, 1128–1135. [Google Scholar] [CrossRef] [Green Version]
- Liberman, A.L.; Prabhakaran, S. Stroke Chameleons and Stroke Mimics in the Emergency Department. Curr. Neurol. Neurosci. 2017, 17, 15. [Google Scholar] [CrossRef]
- Balança, B.; Ritzenthaler, T.; Gobert, F.; Richet, C.; Bodonian, C.; Carrillon, R.; Terrier, A.; Desmurs, L.; Perret-Liaudet, A.; Dailler, F. Significance and Diagnostic Accuracy of Early S100B Serum Concentration after Aneurysmal Subarachnoid Hemorrhage. J. Clin. Med. 2020, 9, 1746. [Google Scholar] [CrossRef]
- Ren, C.; Kobeissy, F.; Alawieh, A.; Li, N.; Li, N.; Zibara, K.; Zoltewicz, S.; Guingab-Cagmat, J.; Larner, S.F.; Ding, Y.; et al. Assessment of Serum UCH-L1 and GFAP in Acute Stroke Patients. Sci. Rep. 2016, 6, 24588. [Google Scholar] [CrossRef] [Green Version]
- Quintard, H.; Leduc, S.; Ferrari, P.; Petit, I.; Ichai, C. Early and persistent high level of PS 100β is associated with increased poor neurological outcome in patients with SAH: Is there a PS 100β threshold for SAH prognosis? Crit. Care 2016, 20, 33. [Google Scholar] [CrossRef] [Green Version]
- Kawata, K.; Liu, C.Y.; Merkel, S.F.; Ramirez, S.H.; Tierney, R.T.; Langford, D. Blood biomarkers for brain injury: What are we measuring? Neurosci. Biobehav. Rev. 2016, 68, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, S.; Jæger, H.S.; Dixon, J.; Bohmann, F.O.; Schaefer, J.; Richieri, S.P.; Larsen, K.; Hov, M.R.; Bache, K.G.; Foerch, C.; et al. Diagnostic Accuracy of Glial Fibrillary Acidic Protein and Ubiquitin Carboxy-Terminal Hydrolase-L1 Serum Concentrations for Differentiating Acute Intracerebral Hemorrhage from Ischemic Stroke. Neurocrit. Care 2020, 33, 39–48. [Google Scholar] [CrossRef]
- Katsanos, A.H.; Makris, K.; Stefani, D.; Koniari, K.; Gialouri, E.; Lelekis, M.; Chondrogianni, M.; Zompola, C.; Dardiotis, E.; Rizos, I.; et al. Plasma Glial Fibrillary Acidic Protein in the Differential Diagnosis of Intracerebral Hemorrhage. Stroke 2018, 48, 2586–2588. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Bao, J.; Wang, Y.; Pan, S. S100β as a biomarker for differential diagnosis of intracerebral hemorrhage and ischemic stroke. Neurol. Res. 2016, 38, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Branco, J.P.; Oliveira, S.; Sargento-Freitas, J.; Galego, O.; Cordeiro, G.; Cunha, L.; Gonçalves, A.F.; Pinheiro, J. Neuroimaging, serum biomarkers, and patient characteristics as predictors of upper limb functioning 12 weeks after acute stroke: An observational, prospective study. Top. Stroke Rehabil. 2018, 25, 1–7. [Google Scholar] [CrossRef]
- Kedziora, J.; Burzynska, M.; Gozdzik, W.; Kübler, A.; Kobylinska, K.; Adamik, B. Biomarkers of Neurological Outcome After Aneurysmal Subarachnoid Hemorrhage as Early Predictors at Discharge from an Intensive Care Unit. Neurocrit. Care 2020, 1–11. [Google Scholar] [CrossRef]
- Kiiski, H.; Långsjö, J.; Tenhunen, J.; Ala-Peijari, M.; Huhtala, H.; Hämäläinen, M.; Moilanen, E.; Peltola, J. S100B, NSE and MMP-9 fail to predict neurologic outcome while elevated S100B associates with milder initial clinical presentation after aneurysmal subarachnoid hemorrhage. J. Neurol. Sci. 2018, 390, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Abboud, T.; Mende, K.C.; Jung, R.; Czorlich, P.; Vettorazzi, E.; Priefler, M.; Kluge, S.; Westphal, M.; Regelsberger, J. Prognostic Value of Early S100 Calcium Binding Protein B and Neuron-Specific Enolase in Patients with Poor-Grade Aneurysmal Subarachnoid Hemorrhage: A Pilot Study. World Neurosurg. 2017, 108, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Mahan, M.Y.; Thorpe, M.; Ahmadi, A.; Abdallah, T.; Casey, H.; Sturtevant, D.; Judge-Yoakam, S.; Hoover, C.; Rafter, D.; Miner, M.J.; et al. GFAP Outperforms S100B and UCH-L1 as Predictor for Positive Head CT in Trauma Subjects. World Neurosurg. 2019, 128, e434–e444. [Google Scholar] [CrossRef]
- Meier, T.B.; Nelson, L.D.; Huber, D.L.; Bazarian, J.J.; Hayes, R.L.; McCrea, M.A. Prospective Assessment of Acute Blood Markers of Brain Injury in Sport-Related Concussion. J. Neurotraum. 2017, 34, 3134–3142. [Google Scholar] [CrossRef]
- Çevik, S.; Özgenç, M.M.; Güneyk, A.; Evran, Ş.; Akkaya, E.; Çalış, F.; Katar, S.; Soyalp, C.; Hanımoğlu, H.; Kaynar, M.Y. NRGN, S100B and GFAP levels are significantly increased in patients with structural lesions resulting from mild traumatic brain injuries. Clin. Neurol. Neurosurg. 2019, 183, 105380. [Google Scholar] [CrossRef]
- Kellermann, I.; Kleindienst, A.; Hore, N.; Buchfelder, M.; Brandner, S. Early CSF and Serum S100B Concentrations for Outcome Prediction in Traumatic Brain Injury and Subarachnoid Hemorrhage. Clin. Neurol. Neurosurg. 2016, 145, 79–83. [Google Scholar] [CrossRef]
- Park, D.-W.; Park, S.-H.; Hwang, S.-K. Serial measurement of S100B and NSE in pediatric traumatic brain injury. Child. Nerv. Syst. 2019, 35, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Hwang, S.-K. Prognostic Value of Serum Levels of S100 Calcium-Binding Protein B, Neuron-Specific Enolase, and Interleukin-6 in Pediatric Patients with Traumatic Brain Injury. World Neurosurg. 2018, 118, e534–e542. [Google Scholar] [CrossRef] [PubMed]
- Thelin, E.P.; Jeppsson, E.; Frostell, A.; Svensson, M.; Mondello, S.; Bellander, B.-M.; Nelson, D.W. Utility of neuron-specific enolase in traumatic brain injury; relations to S100B levels, outcome, and extracranial injury severity. Crit. Care 2016, 20, 285. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jiang, Y.; Zeng, D.; Zhou, W.; Hong, X. Prognostic value of plasma HMGB1 in ischemic stroke patients with cerebral ischemia-reperfusion injury after intravenous thrombolysis. J. Stroke Cerebrovasc. Dis. 2020, 29, 105055. [Google Scholar] [CrossRef]
- Lai, P.M.; Du, R. Association between S100B Levels and Long-Term Outcome after Aneurysmal Subarachnoid Hemorrhage: Systematic Review and Pooled Analysis. PLoS ONE 2016, 11, e0151853. [Google Scholar] [CrossRef] [Green Version]
- Vourc’h, M.; Roquilly, A.; Asehnoune, K. Trauma-Induced Damage-Associated Molecular Patterns-Mediated Remote Organ Injury and Immunosuppression in the Acutely Ill Patient. Front. Immunol. 2018, 9, 1330. [Google Scholar] [CrossRef] [Green Version]
- Thelin, E.; Nimer, F.A.; Frostell, A.; Zetterberg, H.; Blennow, K.; Nyström, H.; Svensson, M.; Bellander, B.-M.; Piehl, F.; Nelson, D.W. A Serum Protein Biomarker Panel Improves Outcome Prediction in Human Traumatic Brain Injury. J. Neurotraum. 2019, 36, 2850–2862. [Google Scholar] [CrossRef] [Green Version]
- Battista, A.P.D.; Buonora, J.E.; Rhind, S.G.; Hutchison, M.G.; Baker, A.J.; Rizoli, S.B.; Diaz-Arrastia, R.; Mueller, G.P. Blood Biomarkers in Moderate-To-Severe Traumatic Brain Injury: Potential Utility of a Multi-Marker Approach in Characterizing Outcome. Front. Neurol. 2015, 6, 110. [Google Scholar] [CrossRef]
- Posti, J.P.; Takala, R.S.K.; Raj, R.; Luoto, T.M.; Azurmendi, L.; Lagerstedt, L.; Mohammadian, M.; Hossain, I.; Gill, J.; Frantzén, J.; et al. Admission Levels of Interleukin 10 and Amyloid β 1–40 Improve the Outcome Prediction Performance of the Helsinki Computed Tomography Score in Traumatic Brain Injury. Front. Neurol. 2020, 11, 549527. [Google Scholar] [CrossRef]
- Posti, J.P.; Takala, R.S.K.; Lagerstedt, L.; Dickens, A.M.; Hossain, I.; Mohammadian, M.; Ala-Seppälä, H.; Frantzén, J.; van Gils, M.; Hutchinson, P.J.; et al. Correlation of Blood Biomarkers and Biomarker Panels with Traumatic Findings on Computed Tomography after Traumatic Brain Injury. J. Neurotraum. 2019, 36, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Soosaipillai, A.; Fraser, D.D.; Diamandis, E.P. Discovery of novel plasma biomarker ratios to discriminate traumatic brain injury. F1000Research 2019, 8, 1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, L.H.M.; Korse, C.M.; Bonfrer, J.M.G. Comparison of four different assays for determination of serum S-100B. Int. J. Biol. Mark. 2005, 20, 34–42. [Google Scholar] [CrossRef]
- Einav, S.; Itshayek, E.; Kark, J.D.; Ovadia, H.; Weiniger, C.F.; Shoshan, Y. Serum S100B levels after meningioma surgery: A comparison of two laboratory assays. BMC Clin. Pathol. 2008, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudeux, J.L.; Léger, P.; Dequen, L.; Gandjbakhch, I.; Coriat, P.; Foglietti, M.J. Influence of hemolysis on the measurement of S-100beta protein and neuron-specific enolase plasma concentrations during coronary artery bypass grafting. Clin. Chem. 2000, 46, 989–990. [Google Scholar] [CrossRef] [Green Version]
- Raabe, A.; Kopetsch, O.; Groß, U.; Zimmermann, M.; Gebhart, P. Measurements of Serum S-100B Protein: Effects of Storage Time and Temperature on Pre-Analytical Stability. Clin. Chem. Lab. Med. 2003, 41, 700–703. [Google Scholar] [CrossRef]
- Kawata, K.; Steinfeldt, J.A.; Huibregtse, M.E.; Nowak, M.K.; Macy, J.T.; Kercher, K.; Rettke, D.J.; Shin, A.; Chen, Z.; Ejima, K.; et al. Association Between Proteomic Blood Biomarkers and DTI/NODDI Metrics in Adolescent Football Players: A Pilot Study. Front. Neurol. 2020, 11, 581781. [Google Scholar] [CrossRef]
- Tsukagawa, T.; Katsumata, R.; Fujita, M.; Yasui, K.; Akhoon, C.; Ono, K.; Dohi, K.; Aruga, T. Elevated Serum High-Mobility Group Box-1 Protein Level Is Associated with Poor Functional Outcome in Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2017, 26, 2404–2411. [Google Scholar] [CrossRef]
- Kiiski, H.; Långsjö, J.; Tenhunen, J.; Ala-Peijari, M.; Huhtala, H.; Hämäläinen, M.; Moilanen, E.; Öhman, J.; Peltola, J. Time-courses of plasma IL-6 and HMGB-1 reflect initial severity of clinical presentation but do not predict poor neurologic outcome following subarachnoid hemorrhage. Eneurologicalsci 2017, 6, 55–62. [Google Scholar] [CrossRef]
- Osier, N.D.; Conley, Y.P.; Okonkwo, D.O.; Puccio, A.M. Variation in Candidate Traumatic Brain Injury Biomarker Genes Are Associated with Gross Neurological Outcomes after Severe Traumatic Brain Injury. J. Neurotrauma 2018, 35, 2684–2690. [Google Scholar] [CrossRef]
- Chen, X.; Wu, S.; Chen, C.; Xie, B.; Fang, Z.; Hu, W.; Chen, J.; Fu, H.; He, H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-κB pathway following experimental traumatic brain injury. J. Neuroinflamm. 2017, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Genrikhs, E.E.; Stelmashook, E.V.; Voronkov, D.N.; Novikova, S.V.; Alexandrova, O.P.; Fedorov, A.V.; Isaev, N.K. The single intravenous administration of methylene blue after traumatic brain injury diminishes neurological deficit, blood-brain barrier disruption and decrease in the expression of S100 protein in rats. Brain Res. 2020, 1740, 146854. [Google Scholar] [CrossRef]
- Gu, X.-J.; Xu, J.; Ma, B.-Y.; Chen, G.; Gu, P.-Y.; Wei, D.; Hu, W.-X. Effect of glycyrrhizin on traumatic brain injury in rats and its mechanism. Chin. J. Traumatol. 2014, 17, 1–7. [Google Scholar]
- Li, Y.; Sun, F.; Jing, Z.; Wang, X.; Hua, X.; Wan, L. Glycyrrhizic acid exerts anti-inflammatory effect to improve cerebral vasospasm secondary to subarachnoid hemorrhage in a rat model. Neurol. Res. 2017, 39, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Ieong, C.; Sun, H.; Wang, Q.; Ma, J. Glycyrrhizin suppresses the expressions of HMGB1 and ameliorates inflammative effect after acute subarachnoid hemorrhage in rat model. J. Clin. Neurosci. 2018, 47, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Güiza, F.; Depreitere, B.; Piper, I.; Citerio, G.; Chambers, I.; Jones, P.A.; Lo, T.-Y.; Enblad, P.; Nillson, P.; Feyen, B.; et al. Visualizing the pressure and time burden of intracranial hypertension in adult and paediatric traumatic brain injury. Intensive Care Med. 2015, 41, 1067–1076. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- He, Y.; Wan, S.; Hua, Y.; Keep, R.F.; Xi, G. Autophagy after Experimental Intracerebral Hemorrhage. J. Cereb. Blood Flow Metab. 2007, 28, 897–905. [Google Scholar] [CrossRef]
- Zille, M.; Karuppagounder, S.S.; Chen, Y.; Gough, P.J.; Bertin, J.; Finger, J.; Milner, T.A.; Jonas, E.A.; Ratan, R.R. Neuronal Death After Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 2018, 48, 1033–1043. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Hou, L.; Liu, N.; Ji, J. Melatonin attenuates traumatic brain injury-induced inflammation: A possible role for mitophagy. J. Pineal Res. 2016, 61, 177–186. [Google Scholar] [CrossRef]
- Lu, J.; Sun, Z.; Fang, Y.; Zheng, J.; Xu, S.; Xu, W.; Shi, L.; Mei, S.; Wu, H.; Liang, F.; et al. Melatonin Suppresses Microglial Necroptosis by Regulating Deubiquitinating Enzyme A20 After Intracerebral Hemorrhage. Front. Immunol. 2019, 10, 1360. [Google Scholar] [CrossRef] [PubMed]
- Thangudu, S.; Cheng, F.-Y.; Su, C.-H. Advancements in the Blood–Brain Barrier Penetrating Nanoplatforms for Brain Related Disease Diagnostics and Therapeutic Applications. Polymers 2020, 12, 3055. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Soler, Y.; Karuppan, M.K.M.; Zhao, Y.; Batrakova, E.V.; El-Hage, N. Targeting Beclin1 as an Adjunctive Therapy against HIV Using Mannosylated Polyethylenimine Nanoparticles. Pharmaceutics 2021, 13, 223. [Google Scholar] [CrossRef]
- Tóth, O.M.; Menyhárt, Á.; Frank, R.; Hantosi, D.; Farkas, E.; Bari, F. Tissue Acidosis Associated with Ischemic Stroke to Guide Neuroprotective Drug Delivery. Biology 2020, 9, 460. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.C.; Amorim, D.; Laranjeira, I.; Almeida, A.; Reis, R.L.; Ferreira, H.; Pinto-Ribeiro, F.; Neves, N.M. Modulating inflammation through the neutralization of Interleukin-6 and tumor necrosis factor-α by biofunctionalized nanoparticles. J. Control. Release 2021. [Google Scholar] [CrossRef]
- Nalamolu, K.R.; Challa, S.R.; Fornal, C.A.; Grudzien, N.A.; Jorgenson, L.C.; Choudry, M.M.; Smith, N.J.; Palmer, C.J.; Pinson, D.M.; Klopfenstein, J.D.; et al. Attenuation of the Induction of TLRs 2 and 4 Mitigates Inflammation and Promotes Neurological Recovery After Focal Cerebral Ischemia. Transl. Stroke Res. 2021, 1–14. [Google Scholar] [CrossRef]
- Szanto, A.; Narkar, V.; Shen, Q.; Uray, I.P.; Davies, P.J.A.; Nagy, L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004, 11, S126–S143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, M.; Nakamura, M.; Tran, M.T.N.; Moriguchi, T.; Hong, C.; Ohsumi, T.; Dinh, T.T.H.; Kusakabe, M.; Hattori, M.; Katsumata, T.; et al. MafB promotes atherosclerosis by inhibiting foam-cell apoptosis. Nat. Commun. 2014, 5, 3147. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, A.; Asou, N.; Atsuta, Y.; Sakura, T.; Ueda, Y.; Sawa, M.; Dobashi, N.; Taniguchi, Y.; Suzuki, R.; Nakagawa, M.; et al. Tamibarotene maintenance improved relapse-free survival of acute promyelocytic leukemia: A final result of prospective, randomized, JALSG-APL204 study. Leukemia 2019, 33, 358–370. [Google Scholar] [CrossRef]
- Oddo, M.; Crippa, I.; Mehta, S.; Menon, D.; Payen, J.-F.; Taccone, F.; Citerio, G. Optimizing sedation in patients with acute brain injury. Crit. Care 2016, 20, 128. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Wang, X.; Wang, L.; Meng, Q.; Guo, D.; Chen, L.; Dai, M.; Wang, G.; Cooney, R.; Luo, J. A nanotrap improves survival in severe sepsis by attenuating hyperinflammation. Nat. Commun. 2020, 11, 3384. [Google Scholar] [CrossRef] [PubMed]
- Gruda, M.C.; Ruggeberg, K.-G.; O’Sullivan, P.; Guliashvili, T.; Scheirer, A.R.; Golobish, T.D.; Capponi, V.J.; Chan, P.P. Broad adsorption of sepsis-related PAMP and DAMP molecules, mycotoxins, and cytokines from whole blood using CytoSorb® sorbent porous polymer beads. PLoS ONE 2018, 13, e0191676. [Google Scholar] [CrossRef]
- McKinley, T.O.; Lei, Z.; Kalbas, Y.; White, F.A.; Shi, Z.; Wu, F.; Xu, Z.C.; Rodgers, R.B. Blood purification by nonselective hemoadsorption prevents death after traumatic brain injury and hemorrhagic shock in rats. J. Trauma Acute Care 2018, 85, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thelin, E.P.; Nelson, D.W.; Ghatan, P.H.; Bellander, B.-M. Microdialysis Monitoring of CSF Parameters in Severe Traumatic Brain Injury Patients: A Novel Approach. Front. Neurol. 2014, 5, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Author (Year) | Marker | Study Design | Applications | Outcome | Results |
|---|---|---|---|---|---|
| Ren et al. [79] (2016) | GFAP | Case-control (132 IS; 57 controls) | Diagnosis | Stroke subtype | GFAP discriminated IS from ICH within 4.5 h of symptoms onset (Se = 61%, Sp = 96%, AUC = 0.86). |
| Luger et al. [82] (2020) | GFAP | Prospective observational (251 IS) | Diagnosis | Stroke subtype | ICH patients had higher serum level of GFAP than IS patients and mimics. |
| Clinical severity | CT lesion volume | GFAP was correlated with ICH volume (r = 0.296). | |||
| Katsanos et al. [83] (2017) | GFAP | Case-control (191 IS; 79 controls) | Diagnosis | Stroke subtype | GFAP discriminated IS from ICH (Se = 91%, Sp = 97%, AUC = 0.97). |
| Clinical severity | NIHSS | No correlation has been found between serum levels of GFAP and stroke severity on admission in IS of different subtype. | |||
| Zhou et al. [84] (2016) | S100B | Prospective observational (46 ICH; 71 IS) | Diagnosis | Stroke subtype | ICH patients had higher plasma level of S100B than IS patients. |
| Clinical severity | NIHSS | Positive correlation between S100B and infarct size (r = 0.820). | |||
| Prognosis | 90-day mRS | S100B predicted a poor prognosis (Se = 100%, Sp = 76%, AUC = 0.92). | |||
| Balança et al. [78] (2020) | S100B | Prospective observational (81 SAH) | Clinical severity | 3-day GOS | Severe EBI was associated with higher S100B concentration at admission or day 1 (Cliff’s delta = 0.73, 95% CI [0.46; 0.88]), which predicted early recovery (AUC = 0.87). |
| Branco et al. [85] (2018) | S100B | Prospective observational (131 IS) | Prognosis | 12-week upper limb functioning | S100B predicted hand functioning (Se = 69%, Sp = 90%, AUC = 0.84). |
| Kedziora et al. [86] (2020) | S100B | Prospective observational (55 SAH) | Prognosis | GOS at ICU discharge | S100B predicted ICU outcome (Se = 91%, Sp = 63%, AUC = 0.81). |
| Kellermann et al. [92] (2016) | S100B | Prospective observational (45 SAH) | Prognosis | 6-month GOS | S100N at day 1 predicted poor outcome (OR = 4.38, 95% CI, [1.08; 17]). A negative correlation was found between serum level of S100B and 6 months GOS (r = 0.434). |
| Kiiski et al. [87] (2018) | S100B; NSE | Prospective observational (47 SAH) | Prognosis | 6-month mRS | No correlation has been found between biomarker concentrations and the neurological outcome. |
| Abboud et al. [88] (2018) | S100B; NSE | Prospective observational (52 SAH) | Prognosis | 6-month GOS | S100B and NSE at day 1 predicted good outcome with 100% specificity. |
| Quintard et al. [80] (2015) | S100B; NSE | Prospective observational (48 SAH) | Prognosis | 6-month GOS | Poor neurological outcome was predicted by S100B levels at day 5 (AUC = 0.91) and NSE level at day 7 (AUC = 0.83). |
| Aida et al. [31] (2019) | sRAGE | Prospective observational (627 SAH) | Complication | symptomatic vasospasm | sRAGE level was lower in symptomatic vasospasm group on day 7, and predicted poor outcome (Se = 70%, Sp = 86%, AUC = 0.77). |
| Yang et al. [67] (2018) | sRAGE | Case-control (108 SAH and 108 controls) | Prognosis | 6-month GOS score | sRAGE within 24 h after SAH was associated with clinical severity and poor 6-month outcomes (Se = 83%, Sp = 75%, AUC = 0.82). |
| Tang et al. [66] (2015) | sRAGE | Case-control (106 IS and 150 controls) | Prognosis | 3-month mRS | sRAGE level was higher in the IS group and predicted poor neurological score (OR = 2.44, 95% CI [1.16; 5.16]). |
| Tsukagawa et al. [109] (2017) | HMGB1 | Case-control (183 IS and 16 controls) | Prognosis | 1-year mRS | HMGB1 level on admission was a significant independent predictor of poor outcome (OR = 2.34, 95% CI [1.02; 5.38]). |
| Wang et al. [96] (2020) | HMGB1 | Prospective observational (132 IS) | Prognosis | 3-month mRS | High concentration of HMGB1 at 6 h after thrombolytic therapy was associated with poor outcome (Se = 87%, Sp = 74%, AUC = 0.87). |
| Kiiski et al. [110] (2017) | HMGB1 | Prospective observational (47 SAH) | Prognosis | 6-month mRS | No correlation has been found between biomarker concentrations and the neurological outcome. |
| Author (Year) | Marker | Study Design | Applications | Outcome | Results |
|---|---|---|---|---|---|
| Osier et al. [111] (2018) | S100B SNP in genes | Prospective study (305 severe TBI) | Risk factor | 3, 6, 12, 24-months GOS | The variant allele of one S100B SNP (rs1051169) was identified as a protective factor at 3 months (OR = 0.39), 6 months (OR = 0.34), 12 months (OR = 0.32) and 24 months (OR = 0.30). |
| Mahan et al. [89] (2019) | GFAP; S100B | Case-control (118 TBI and 37 controls) | Diagnosis | CT lesion (CT+) | These biomarkers were significantly higher in patients CT+ than patients CT−. GFAP had the greatest prognostic capacity (0–8 h: AUC = 0.89; and 12–32 h: AUC = 0.94). |
| Meier et al. [90] (2017) | GFAP; S100B | Case-control (32 TBI and 29 controls) | Diagnosis | Sport-related concussion | S100B could predict concussion in athletes (AUC = 0.72) but not for GFAP. |
| Çevik et al. [91] (2019) | GFAP; S100B | Cross-sectional study (48 mild TBI) | Diagnosis | CT lesion (CT+) | S100B and GFPA were significantly higher in mild TBI patients with CT+. |
| Kellermann et al. [92] (2016) | S100B | Prospective observational (57 TBI) | Prognosis | 6-month GOS | S100B at day 1 predicted a poor outcome (OR = 7.6, 95% CI [2.24; 25.80]). A negative correlation was found between serum level of S100B and 6 months GOS (r = 0.494). |
| Park et al. [94] (2018) | S100B; NSE; IL-6 | Prospective observational (15 pediatric patients with TBI) | Prognosis | 6-month GOS | S100B and NSE correlated with the severity of brain injury and predicted poor neurological outcome, but not IL6. |
| Park et al. [93] (2019) | S100B; NSE | Prospective observational (10 pediatric patients with TBI) | Prognosis | 6-month GOS | High concentration of S100B at 1 week after admission was associated with poor outcome. No statistical difference was found for NSE. |
| Thelin et al. [95] (2016) | S100B; NSE | Prospective observational (417 TBI) | Prognosis | 3-month GOS | CSF level of S100B at day 1 predicted poor outcome (OR = 4.15, 95% CI [1.34; 12.84]). |
| 6-month GOS | S100B had a betted diagnostic accuracy than NSE. |
| Author (Year) | Marker | Study Design | Applications | Outcome | Results |
|---|---|---|---|---|---|
| Posti et al. [101] (2020) | Aβ40; Aβ42; GFAP; H-FABP; IL-10; NF-L; S100β; Tau | Prospective observational (136 TBI patients) | Diagnosis | CT lesion (CT+) | The combination of two biomarkers (Aβ40 and IL-10) and clinical characteristics (HCTS) were better to discriminate CT+ and CT− patients (Se = 91%, Sp = 59%) than HCTS alone (Se = 97%, Sp = 22%). |
| Posti et al. [102] (2019) | Aβ40; Aβ42; GFAP; H-FABP; IL-10; NF-L; S100β; Tau | Prospective observational (160 TBI including 93 mild TBI) | Diagnosis | CT lesion (CT+) | H-FABP + S100B + Tau were the best association to diagnose CT+ in mild TBI (Se = 100%, Sp = 46%) whereas it was a question of the combination of GFAP + H-FAB + IL-10 in group with all severities TBI (Se = 100%, Sp = 39%). |
| Chen et al. [103] (2019) | S100β; NSE; hK6; PGDS | Case-control (10 severe TBI and 10 controls) | Diagnosis | Subtype severe TBI | S100B/hK6 and NSE/PGDS ratios (both AUC = 1.00) diagnosed severe TBI with greater accuracy than S100B and NSE alone (both AUC = 0.97). |
| Thelin et al. [99] (2019) | S100β; NSE; GFAP; Tau; NF-L; UCH-L1 | Prospective observational (172 TBI) | Prognosis | 12-month GOS | GFAP + NF-L was the best association to predict poor outcome. |
| Di Battista et al. [100] (2015) | S100β; GFAP; NSE; BDNF; MCP-1; ICAM-5; PRDX-6 | Prospective observational (85 TBI) | Prognosis | Death and 6-month GOS | S100B and GFAP levels were the most discriminating to predict poor outcome (AUC = 0.82 and AUC = 0.71, respectively) and mortality (AUC = 0.86 and AUC = 0.79, respectively). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balança, B.; Desmurs, L.; Grelier, J.; Perret-Liaudet, A.; Lukaszewicz, A.-C. DAMPs and RAGE Pathophysiology at the Acute Phase of Brain Injury: An Overview. Int. J. Mol. Sci. 2021, 22, 2439. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052439
Balança B, Desmurs L, Grelier J, Perret-Liaudet A, Lukaszewicz A-C. DAMPs and RAGE Pathophysiology at the Acute Phase of Brain Injury: An Overview. International Journal of Molecular Sciences. 2021; 22(5):2439. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052439
Chicago/Turabian StyleBalança, Baptiste, Laurent Desmurs, Jérémy Grelier, Armand Perret-Liaudet, and Anne-Claire Lukaszewicz. 2021. "DAMPs and RAGE Pathophysiology at the Acute Phase of Brain Injury: An Overview" International Journal of Molecular Sciences 22, no. 5: 2439. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052439