
Foam Cells as Therapeutic Targets in Atherosclerosis with a Focus on the Regulatory Roles of Non-Coding RNAs


 ,
,
Abstract
:1. Introduction
2. Pathophysiology of Atherosclerosis and the Key Role of Foam Cells
3. Foam Cells as Therapeutic Targets in Atherosclerosis
4. Non-Coding RNAs as Therapeutic Targets
5. Non-Coding RNAs That Stimulate Foam Cell Formation/Function
5.1. miR-33
5.2. miR-27a/b
5.3. miR-216a
5.4. miR-382-5p
6. Non-Coding RNAs That Attenuate Foam Cell Formation
6.1. miR-150
6.2. miR-155
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wakabayashi, I. Associations between alcohol drinking and multiple risk factors for atherosclerosis in smokers and nonsmokers. Angiology 2010, 61, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P.; Camici, P. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010, 9, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S. Modulation of nitric oxide synthases by oxidized LDLs: Role in vascular inflammation and atherosclerosis development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Liu, W.; Yin, Y.; Zhou, Z.; He, M.; Dai, Y. OxLDL-induced IL-1beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflammasome activation. Inflamm. Res. 2014, 63, 33–43. [Google Scholar] [CrossRef]
- Daub, K.; Seizer, P.; Stellos, K.; Krämer, B.F.; Bigalke, B.; Schaller, M.; Fateh-Moghadam, S.; Gawaz, M.; Lindemann, S. Oxidized LDL-activated platelets induce vascular inflammation. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2010; pp. 146–156. [Google Scholar]
- Chávez-Sánchez, L.; Espinosa-Luna, J.E.; Chávez-Rueda, K.; Legorreta-Haquet, M.V.; Montoya-Díaz, E.; Blanco-Favela, F. Innate immune system cells in atherosclerosis. Arch. Med. Res. 2014, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kiyan, Y.; Tkachuk, S.; Hilfiker-Kleiner, D.; Haller, H.; Fuhrman, B.; Dumler, I. oxLDL induces inflammatory responses in vascular smooth muscle cells via urokinase receptor association with CD36 and TLR4. J. Mol. Cell. Cardiol. 2014, 66, 72–82. [Google Scholar] [CrossRef]
- Seo, J.-W.; Yang, E.-J.; Yoo, K.-H.; Choi, I.-H. Macrophage differentiation from monocytes is influenced by the lipid oxidation degree of low density lipoprotein. Mediat. Inflamm. 2015, 2015, 235797. [Google Scholar] [CrossRef] [Green Version]
- Rios, F.J.; Koga, M.M.; Pecenin, M.; Ferracini, M.; Gidlund, M.; Jancar, S. Oxidized LDL induces alternative macrophage phenotype through activation of CD36 and PAFR. Mediat. Inflamm. 2013, 2013, 198193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfs, I.M.; Donners, M.M.; de Winther, M.P. Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb. Haemost. 2011, 106, 763–771. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Vainio, S.; Ikonen, E. Macrophage cholesterol transport: A critical player in foam cell formation. Ann. Med. 2003, 35, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Birck, M.M.; Saraste, A.; Hyttel, P.; Odermarsky, M.; Liuba, P.; Saukko, P.; Hansen, A.K.; Pesonen, E. Endothelial cell death and intimal foam cell accumulation in the coronary artery of infected hypercholesterolemic minipigs. J. Cardiovasc. Transl. Res. 2013, 6, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Khismatullin, D.B. Oxidized low-density lipoprotein contributes to atherogenesis via co-activation of macrophages and mast cells. PLoS ONE 2015, 10, e0123088. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef] [Green Version]
- Hultgårdh-Nilsson, A.; Durbeej, M. Role of the extracellular matrix and its receptors in smooth muscle cell function: Implications in vascular development and disease. Curr. Opin. Lipidol. 2007, 18, 540–545. [Google Scholar] [CrossRef]
- Wezel, A.; Lagraauw, H.M.; van der Velden, D.; de Jager, S.C.; Quax, P.H.; Kuiper, J.; Bot, I. Mast cells mediate neutrophil recruitment during atherosclerotic plaque progression. Atherosclerosis 2015, 241, 289–296. [Google Scholar] [CrossRef]
- Van der Donckt, C.; Van Herck, J.L.; Schrijvers, D.M.; Vanhoutte, G.; Verhoye, M.; Blockx, I.; Van Der Linden, A.; Bauters, D.; Lijnen, H.R.; Sluimer, J.C. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur. Heart J. 2015, 36, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, R.P.; Lee, J.M.; Greaves, D.R. Mechanisms of disease: Macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 309–315. [Google Scholar] [CrossRef]
- Peters, W.; Charo, I.F. Involvement of chemokine receptor 2 and its ligand, monocyte chemoattractant protein-1, in the development of atherosclerosis: Lessons from knockout mice. Curr. Opin. Lipidol. 2001, 12, 175–180. [Google Scholar] [CrossRef]
- Greaves, D.R.; Gordon, S. Thematic review series: The immune system and atherogenesis. Recent insights into the biology of macrophage scavenger receptors. J. Lipid Res. 2005, 46, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combadière, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.D.; Trogan, E.; Ginsberg, M.; Grigaux, C.; Tian, J.; Miyata, M. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc. Natl. Acad. Sci. USA 1995, 92, 8264–8268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipriani, S.; Francisci, D.; Mencarelli, A.; Renga, B.; Schiaroli, E.; D’Amore, C.; Baldelli, F.; Fiorucci, S. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation 2013, 127, 2114–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fotis, L.; Agrogiannis, G.; Vlachos, I.S.; Pantopoulou, A.; Margoni, A.; Kostaki, M.; Verikokos, C.; Tzivras, D.; Mikhailidis, D.P.; Perrea, D. Intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 at the early stages of atherosclerosis in a rat model. In Vivo 2012, 26, 243–250. [Google Scholar]
- Ley, K.; Huo, Y. VCAM-1 is critical in atherosclerosis. J. Clin. Investig. 2001, 107, 1209–1210. [Google Scholar] [CrossRef]
- Serebruany, V.; Malinin, A.; Scott, R. The in vitro effects of a novel vascular protectant, AGI-1067, on platelet aggregation and major receptor expression in subjects with multiple risk factors for vascular disease. J. Cardiovasc. Pharmacol. Ther. 2006, 11, 191–196. [Google Scholar] [CrossRef]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Coller, S.P.; Paulnock, D.M. Signaling pathways initiated in macrophages after engagement of type A scavenger receptors. J. Leukoc. Biol. 2001, 70, 142–148. [Google Scholar]
- Sikorski, K.; Czerwoniec, A.; Bujnicki, J.M.; Wesoly, J.; Bluyssen, H.A. STAT1 as a novel therapeutical target in pro-atherogenic signal integration of IFNγ, TLR4 and IL-6 in vascular disease. Cytokine Growth Factor Rev. 2011, 22, 211–219. [Google Scholar] [CrossRef]
- De Vos, J.; Mathijs, I.; Xavier, C.; Massa, S.; Wernery, U.; Bouwens, L.; Lahoutte, T.; Muyldermans, S.; Devoogdt, N. Specific targeting of atherosclerotic plaques in ApoE−/− mice using a new camelid sdAb binding the vulnerable plaque marker LOX-1. Mol. Imaging Biol. 2014, 16, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ogura, S.; Chen, J.; Little, P.J.; Moss, J.; Liu, P. LOX-1 in atherosclerosis: Biological functions and pharmacological modifiers. Cell. Mol. Life Sci. 2013, 70, 2859–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkinen, P.I.; Lappalainen, J.P.; Heinonen, S.E.; Leppänen, P.; Lähteenvuo, M.T.; Aarnio, J.V.; Heikkilä, J.; Turunen, M.P.; Ylä-Herttuala, S. Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc. Res. 2010, 88, 530–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.-Y.; Cai, Y.; Mao, D.-D.; Qi, Y.-F.; Tang, C.; Xu, Q.; Zhu, Y.; Xu, M.-J.; Wang, X. Increased stability of phosphatase and tensin homolog by intermedin leading to scavenger receptor A inhibition of macrophages reduces atherosclerosis in apolipoprotein E-deficient mice. J. Mol. Cell. Cardiol. 2012, 53, 509–520. [Google Scholar] [CrossRef]
- Sakashita, N.; Miyazaki, A.; Takeya, M.; Horiuchi, S.; Chang, C.C.; Chang, T.-Y.; Takahashi, K. Localization of human acyl-coenzyme A: Cholesterol acyltransferase-1 (ACAT-1) in macrophages and in various tissues. Am. J. Pathol. 2000, 156, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Sekiya, M.; Osuga, J.-I.; Igarashi, M.; Okazaki, H.; Ishibashi, S. The role of neutral cholesterol ester hydrolysis in macrophage foam cells. J. Atheroscler. Thromb. 2011, 18, 359–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, J.E.; Michael, D.R.; Ashlin, T.G.; Ramji, D.P. Cytokines, macrophage lipid metabolism and foam cells: Implications for cardiovascular disease therapy. Prog. Lipid Res. 2011, 50, 331–347. [Google Scholar] [CrossRef]
- Fazio, S.; Linton, M. Mouse models of hyperlipidemia and atherosclerosis. Front. Biosci 2001, 6, D515–D525. [Google Scholar] [CrossRef]
- Ikenoya, M.; Yoshinaka, Y.; Kobayashi, H.; Kawamine, K.; Shibuya, K.; Sato, F.; Sawanobori, K.; Watanabe, T.; Miyazaki, A. A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis 2007, 191, 290–297. [Google Scholar] [CrossRef]
- Perrey, S.; Legendre, C.; Matsuura, A.; Guffroy, C.; Binet, J.; Ohbayashi, S.; Tanaka, T.; Ortuno, J.C.; Matsukura, T.; Laugel, T. Preferential pharmacological inhibition of macrophage ACAT increases plaque formation in mouse and rabbit models of atherogenesis. Atherosclerosis 2001, 155, 359–370. [Google Scholar] [CrossRef]
- Sekiya, M.; Yamamuro, D.; Ohshiro, T.; Honda, A.; Takahashi, M.; Kumagai, M.; Sakai, K.; Nagashima, S.; Tomoda, H.; Igarashi, M. Absence of Nceh1 augments 25-hydroxycholesterol-induced ER stress and apoptosis in macrophages. J. Lipid Res. 2014, 55, 2082–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef]
- Silva, J.C.; César, F.A.; de Oliveira, E.M.; Turato, W.M.; Tripodi, G.L.; Castilho, G.; Machado-Lima, A.; de las Heras, B.; Boscá, L.; Rabello, M.M. New PPARγ partial agonist improves obesity-induced metabolic alterations and atherosclerosis in LDLr−/− mice. Pharmacol. Res. 2016, 104, 49–60. [Google Scholar] [CrossRef]
- Li, A.C.; Binder, C.J.; Gutierrez, A.; Brown, K.K.; Plotkin, C.R.; Pattison, J.W.; Valledor, A.F.; Davis, R.A.; Willson, T.M.; Witztum, J.L. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARα, β/δ, and γ. J. Clin. Investig. 2004, 114, 1564–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef] [Green Version]
- Joyce, C.W.; Wagner, E.M.; Basso, F.; Amar, M.J.; Freeman, L.A.; Shamburek, R.D.; Knapper, C.L.; Syed, J.; Wu, J.; Vaisman, B.L. ABCA1 overexpression in the liver of LDLr-KO mice leads to accumulation of pro-atherogenic lipoproteins and enhanced atherosclerosis. J. Biol. Chem. 2006, 281, 33053–33065. [Google Scholar] [CrossRef] [Green Version]
- Van Eck, M.; Bos, I.S.T.; Hildebrand, R.B.; Van Rij, B.T.; Van Berkel, T.J. Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am. J. Pathol. 2004, 165, 785–794. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, Y.; Chen, H.; Dan, J.; Cheng, J.; Guo, S.; Sun, X.; Wang, W.; Ai, Y.; Li, S. The predominant pathway of apoptosis in THP-1 macrophage-derived foam cells induced by 5-aminolevulinic acid-mediated sonodynamic therapy is the mitochondria-caspase pathway despite the participation of endoplasmic reticulum stress. Cell. Physiol. Biochem. 2014, 33, 1789–1801. [Google Scholar] [CrossRef]
- Tabas, I. Macrophage apoptosis in atherosclerosis: Consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid. Redox Signal. 2009, 11, 2333–2339. [Google Scholar] [CrossRef]
- Newby, A.C.; George, S.J.; Ismail, Y.; Johnson, J.L.; Sala-Newby, G.B.; Thomas, A.C. Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb. Haemost. 2009, 101, 1006–1011. [Google Scholar]
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Melnichenko, A.A.; Grechko, A.V.; Myasoedova, V.A.; Orekhov, A.N. Potential of anti-inflammatory agents for treatment of atherosclerosis. Exp. Mol. Pathol. 2018, 104, 114–124. [Google Scholar] [CrossRef]
- Tabas, I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: The importance of lesion stage and phagocytic efficiency. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2255–2264. [Google Scholar] [CrossRef]
- Liu, J.; Thewke, D.P.; Su, Y.R.; Linton, M.F.; Fazio, S.; Sinensky, M.S. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 174–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorp, E.; Li, Y.; Bao, L.; Yao, P.M.; Kuriakose, G.; Rong, J.; Fisher, E.A.; Tabas, I. Brief report: Increased apoptosis in advanced atherosclerotic lesions of Apoe−/− mice lacking macrophage Bcl-2. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Shelton, J.M.; Chen, M.; Bradley, M.N.; Castrillo, A.; Bookout, A.L.; Mak, P.A.; Edwards, P.A.; Mangelsdorf, D.J.; Tontonoz, P. A role for the apoptosis inhibitory factor AIM/Spα/Api6 in atherosclerosis development. Cell Metab. 2005, 1, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Grainger, D.J.; Reckless, J.; McKilligin, E. Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J. Immunol. 2004, 173, 6366–6375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, S. Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Kratzer, A.; Buchebner, M.; Pfeifer, T.; Becker, T.M.; Uray, G.; Miyazaki, M.; Miyazaki-Anzai, S.; Ebner, B.; Chandak, P.G.; Kadam, R.S. Synthetic LXR agonist attenuates plaque formation in apoE−/− mice without inducing liver steatosis and hypertriglyceridemia. J. Lipid Res. 2009, 50, 312–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahuczky, G.; Kristóf, E.; Majai, G.; Fésüs, L. Differentiation and glucocorticoid regulated apopto-phagocytic gene expression patterns in human macrophages. Role of Mertk in enhanced phagocytosis. PLoS ONE 2011, 6, e21349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majai, G.; Sarang, Z.; Csomós, K.; Zahuczky, G.; Fésüs, L. PPARγ-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur. J. Immunol. 2007, 37, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, M.; Volkova, N.; Aviram, M. Pomegranate phytosterol (β-sitosterol) and polyphenolic antioxidant (punicalagin) addition to statin, significantly protected against macrophage foam cells formation. Atherosclerosis 2013, 226, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Uitz, E.; Bahadori, B.; McCarty, M.F.; Moghadasian, M.H. Practical strategies for modulating foam cell formation and behavior. World J. Clin. Cases WJCC 2014, 2, 497. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Endres, M.; Custodis, F.; Gertz, K.; Nickenig, G.; Liao, J.K.; Böhm, M. Suppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of rho GTPase gene transcription. Circulation 2000, 102, 3104–3110. [Google Scholar] [CrossRef] [Green Version]
- Afshari, A.R.; Mollazadeh, H.; Henney, N.C.; Jamialahmad, T.; Sahebkar, A. Effects of statins on brain tumors: A review. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. The effects of statins on microglial cells to protect against neurodegenerative disorders: A mechanistic review. BioFactors 2020, 46, 309–325. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Kiaie, N.; Pirro, M.; Bianconi, V.; Jamialahmadi, T.; Sahebkar, A. Effects of statins on the biological features of mesenchymal stem cells and therapeutic implications. Heart Fail. Rev. 2020. [Google Scholar] [CrossRef]
- Kouhpeikar, H.; Delbari, Z.; Sathyapalan, T.; Simental-Mendía, L.E.; Jamialahmadi, T.; Sahebkar, A. The Effect of Statins through Mast Cells in the Pathophysiology of Atherosclerosis: A Review. Curr. Atheroscler. Rep. 2020, 22, 19. [Google Scholar] [CrossRef]
- Mollazadeh, H.; Tavana, E.; Fanni, G.; Bo, S.; Banach, M.; Pirro, M.; von Haehling, S.; Jamialahmadi, T.; Sahebkar, A. Effects of statins on mitochondrial pathways. J. Cachexia Sarcopenia Muscle 2021. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Serban, C.; Mikhailidis, D.P.; Undas, A.; Lip, G.Y.H.; Muntner, P.; Bittner, V.; Ray, K.K.; Watts, G.F.; Hovingh, G.K.; et al. Association between statin use and plasma d-dimer levels: A systematic review and meta-analysis of randomised controlled trials. Thromb. Haemost. 2015, 114, 546–557. [Google Scholar] [CrossRef]
- Sahebkar, A.; Serban, C.; Ursoniu, S.; Mikhailidis, D.P.; Undas, A.; Lip, G.Y.H.; Bittner, V.; Ray, K.K.; Watts, G.F.; Kees Hovingh, G.; et al. The impact of statin therapy on plasma levels of von Willebrand factor antigen: Systematic review and meta-analysis of Randomised placebo-controlled trials. Thromb. Haemost. 2016, 115, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Serban, C.; Sahebkar, A.; Ursoniu, S.; Mikhailidis, D.P.; Rizzo, M.; Lip, G.Y.H.; Kees Hovingh, G.; Kastelein, J.J.P.; Kalinowski, L.; Rysz, J.; et al. A systematic review and meta-analysis of the effect of statins on plasma asymmetric dimethylarginine concentrations. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.C.; Eijgelaar, W.J.; Daemen, M.J.; Newby, A.C. Foam cell formation in vivo converts macrophages to a pro-fibrotic phenotype. PLoS ONE 2015, 10, e0128163. [Google Scholar] [CrossRef] [Green Version]
- Peeters, W.; Moll, F.L.; Vink, A.; van der Spek, P.J.; de Kleijn, D.P.; de Vries, J.-P.P.; Verheijen, J.H.; Newby, A.C.; Pasterkamp, G. Collagenase matrix metalloproteinase-8 expressed in atherosclerotic carotid plaques is associated with systemic cardiovascular outcome. Eur. Heart J. 2011, 32, 2314–2325. [Google Scholar] [CrossRef] [PubMed]
- Laxton, R.C.; Hu, Y.; Duchene, J.; Zhang, F.; Zhang, Z.; Leung, K.-Y.; Xiao, Q.; Scotland, R.S.; Hodgkinson, C.P.; Smith, K. A role of matrix metalloproteinase-8 in atherosclerosis. Circ. Res. 2009, 105, 921–929. [Google Scholar] [CrossRef]
- Luttun, A.; Lutgens, E.; Manderveld, A.; Maris, K.; Collen, D.; Carmeliet, P.; Moons, L. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation 2004, 109, 1408–1414. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; George, S.J.; Newby, A.C.; Jackson, C.L. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc. Natl. Acad. Sci. USA 2005, 102, 15575–15580. [Google Scholar] [CrossRef] [Green Version]
- Daub, K.; Langer, H.; Seizer, P.; Stellos, K.; May, A.E.; Goyal, P.; Bigalke, B.; Schönberger, T.; Geisler, T.; Siegel-Axel, D. Platelets induce differentiation of human CD34+ progenitor cells into foam cells and endothelial cells. FASEB J. 2006, 20, 2559–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Schouteden, S.; Geenens, R.; Van Duppen, V.; Herijgers, P.; Holvoet, P.; Van Veldhoven, P.P.; Verfaillie, C.M. Hematopoietic stem/progenitor cell proliferation and differentiation is differentially regulated by high-density and low-density lipoproteins in mice. PLoS ONE 2012, 7, e47286. [Google Scholar] [CrossRef] [Green Version]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Yu, B.; Wong, M.M.; Potter, C.M.; Simpson, R.M.; Karamariti, E.; Zhang, Z.; Zeng, L.; Warren, D.; Hu, Y.; Wang, W. Vascular stem/progenitor cell migration induced by smooth muscle cell-derived chemokine (C-C Motif) ligand 2 and chemokine (C-X-C motif) ligand 1 contributes to neointima formation. Stem Cells 2016, 34, 2368–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivan, L.; Antohe, F. Hyperlipidemia induces endothelial-derived foam cells in culture. J. Recept. Signal. Transduct. 2010, 30, 106–114. [Google Scholar] [CrossRef]
- Xu, S.; Kamato, D.; Little, P.J.; Nakagawa, S.; Pelisek, J.; Jin, Z.G. Targeting epigenetics and non-coding RNAs in atherosclerosis: From mechanisms to therapeutics. Pharmacol. Ther. 2019, 196, 15–43. [Google Scholar] [CrossRef]
- Uszczynska-Ratajczak, B.; Lagarde, J.; Frankish, A.; Guigó, R.; Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 2018, 19, 535–548. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Mehler, M.F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 2012, 13, 528–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.M.; Hutvagner, G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene 2006, 25, 6163–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beermann, J.; Piccoli, M.-T.; Viereck, J.; Thum, T. Non-coding RNAs in development and disease: Background, mechanisms, and therapeutic approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [Green Version]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution—Trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Magny, E.G.; Pueyo, J.I.; Pearl, F.M.; Cespedes, M.A.; Niven, J.E.; Bishop, S.A.; Couso, J.P. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science 2013, 341, 1116–1120. [Google Scholar] [CrossRef]
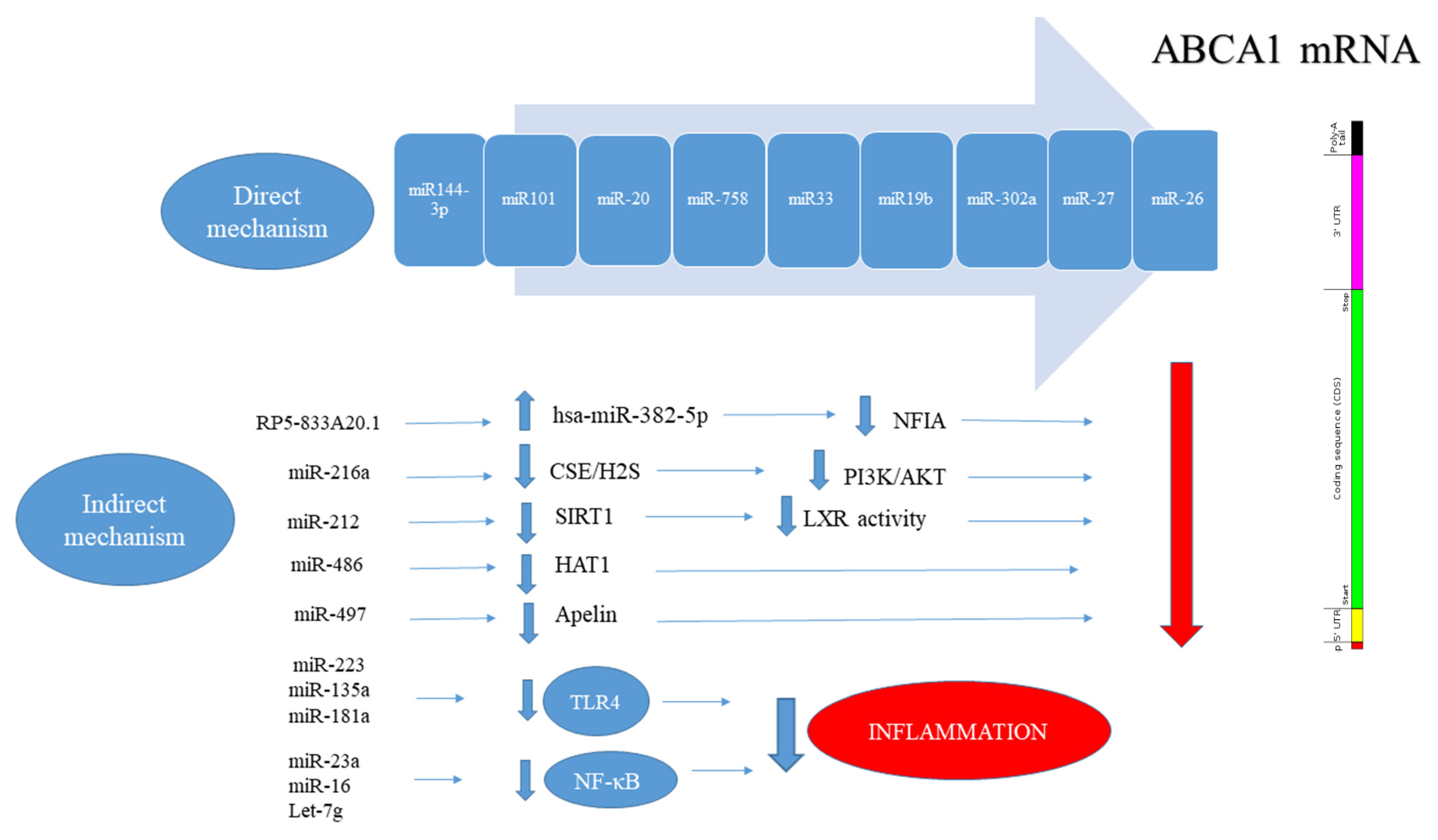
- Hu, Y.W.; Hu, Y.R.; Zhao, J.Y.; Li, S.F.; Ma, X.; Wu, S.G.; Lu, J.B.; Qiu, Y.R.; Sha, Y.H.; Wang, Y.C.; et al. An agomir of miR-144-3p accelerates plaque formation through impairing reverse cholesterol transport and promoting pro-inflammatory cytokine production. PLoS ONE 2014, 9, e94997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquart, T.J.; Wu, J.; Lusis, A.J.; Baldán, Á. Anti-miR-33 therapy does not alter the progression of atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 455–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotllan, N.; Ramírez, C.M.; Aryal, B.; Esau, C.C.; Fernández-Hernando, C. Therapeutic silencing of microRNA-33 inhibits the progression of atherosclerosis in Ldlr−/− mice—Brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1973–1977. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.-C.; Tang, Y.-Y.; Peng, J.; Zhao, G.-J.; Yang, J.; Yao, F.; Ouyang, X.-P.; He, P.-P.; Xie, W.; Tan, Y.-L. MicroRNA-19b promotes macrophage cholesterol accumulation and aortic atherosclerosis by targeting ATP-binding cassette transporter A1. Atherosclerosis 2014, 236, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Lei, J.; Lei, H.; Ruan, X.; Liu, Q.; Chen, Y.; Huang, W. MicroRNA-101 overexpression by IL-6 and TNF-α inhibits cholesterol efflux by suppressing ATP-binding cassette transporter A1 expression. Exp. Cell Res. 2015, 336, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yan, X.; Xia, M.; Yang, Y.; Li, D.; Li, X.; Song, F.; Ling, W. Coenzyme Q10 promotes macrophage cholesterol efflux by regulation of the activator protein-1/miR-378/ATP-binding cassette transporter G1–signaling pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1860–1870. [Google Scholar] [CrossRef] [Green Version]
- Meiler, S.; Baumer, Y.; Toulmin, E.; Seng, K.; Boisvert, W.A. MicroRNA 302a is a novel modulator of cholesterol homeostasis and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wu, J.-F.; Chen, W.-J.; Tang, S.-L.; Mo, Z.-C.; Tang, Y.-Y.; Li, Y.; Wang, J.-L.; Liu, X.-Y.; Peng, J. MicroRNA-27a/b regulates cellular cholesterol efflux, influx and esterification/hydrolysis in THP-1 macrophages. Atherosclerosis 2014, 234, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhang, J.; Xie, J.; Wei, W.; Chen, M.; Zhao, X. MiR-26 controls LXR-dependent cholesterol efflux by targeting ABCA1 and ARL7. FEBS Lett. 2012, 586, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Lan, G.; Xie, W.; Li, L.; Zhang, M.; Liu, D.; Tan, Y.-L.; Cheng, H.-P.; Gong, D.; Huang, C.; Zheng, X.-L. MicroRNA-134 actives lipoprotein lipase-mediated lipid accumulation and inflammatory response by targeting angiopoietin-like 4 in THP-1 macrophages. Biochem. Biophys. Res. Commun. 2016, 472, 410–417. [Google Scholar] [CrossRef]
- Tian, F.-J.; An, L.-N.; Wang, G.-K.; Zhu, J.-Q.; Li, Q.; Zhang, Y.-Y.; Zeng, A.; Zou, J.; Zhu, R.-F.; Han, X.-S. Elevated microRNA-155 promotes foam cell formation by targeting HBP1 in atherogenesis. Cardiovasc. Res. 2014, 103, 100–110. [Google Scholar] [CrossRef]
- Gong, D.; Cheng, H.-P.; Xie, W.; Zhang, M.; Liu, D.; Lan, G.; Huang, C.; Zhao, Z.-W.; Chen, L.-Y.; Yao, F. Cystathionine γ-lyase (CSE)/hydrogen sulfide system is regulated by miR-216a and influences cholesterol efflux in macrophages via the PI3K/AKT/ABCA1 pathway. Biochem. Biophys. Res. Commun. 2016, 470, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, M.; Xie, W.; Lan, G.; Cheng, H.-P.; Gong, D.; Huang, C.; Lv, Y.-C.; Yao, F.; Tan, Y.-L. MiR-486 regulates cholesterol efflux by targeting HAT1. Biochem. Biophys. Res. Commun. 2016, 472, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Zeng, H.; Gong, H. microRNA-212 promotes lipid accumulation and attenuates cholesterol efflux in THP-1 human macrophages by targeting SIRT1. Gene 2018, 643, 55–60. [Google Scholar] [CrossRef]
- Chen, H.; Li, X.; Liu, S.; Gu, L.; Zhou, X. MircroRNA-19a promotes vascular inflammation and foam cell formation by targeting HBP-1 in atherogenesis. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Ren, Z.; Zou, W.; Jiang, Y. miR-497 accelerates oxidized low-density lipoprotein-induced lipid accumulation in macrophages by repressing the expression of apelin. Cell Biol. Int. 2017, 41, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Wang, X.; Song, X.; Bai, R.; Yang, H.; Yang, Z.; Xiao, C.; Bian, Y. MicroRNA-20a/b regulates cholesterol efflux through post-transcriptional repression of ATP-binding cassette transporter A1. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 929–938. [Google Scholar] [CrossRef]
- Ramirez, C.; Dávalos, A.; Goedeke, L.; Salerno, A.; Warrier, N.; Cirera-Salinas, D.; Suárez, Y.; Fernández-Hernando, C. miR-758 regulates cholesterol efflux through post-transcriptional repression of ABCA1. Arter. Thromb Vasc. Biol. 2011, 31, 2707–2714. [Google Scholar] [CrossRef] [Green Version]
- Huangfu, N.; Xu, Z.; Zheng, W.; Wang, Y.; Cheng, J.; Chen, X. LncRNA MALAT1 regulates oxLDL-induced CD36 expression via activating β-catenin. Biochem. Biophys. Res. Commun. 2018, 495, 2111–2117. [Google Scholar] [CrossRef]
- Cai, C.; Zhu, H.; Ning, X.; Li, L.; Yang, B.; Chen, S.; Wang, L.; Lu, X.; Gu, D. LncRNA ENST00000602558. 1 regulates ABCG1 expression and cholesterol efflux from vascular smooth muscle cells through a p65-dependent pathway. Atherosclerosis 2019, 285, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ma, R.; Fu, W.; Zhang, C.; Du, X. LncRNA UCA1 sponges miR-206 to exacerbate oxidative stress and apoptosis induced by ox-LDL in human macrophages. J. Cell. Physiol. 2019, 234, 14154–14160. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, W.; Fu, X.; Huang, Y.; Wen, Y.; Xu, Q.; He, X.; Wang, K.; Huang, S.; Lv, Z. Blood miR-1275 is associated with risk of ischemic stroke and inhibits macrophage foam cell formation by targeting ApoC2 gene. Gene 2020, 731, 144364. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, F.; Xie, Y.; Xu, W.; Xiong, G.; Xu, Y.; Huang, S.; Wu, Y.; Jiang, X. MiR-202-3p Inhibits Foam Cell Formation and is Associated with Coronary Heart Disease Risk in a Chinese Population. Int. Heart J. 2020, 61, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Du, X.J.; Lu, J.M.; Sha, Y. MiR-181a inhibits vascular inflammation induced by ox-LDL via targeting TLR4 in human macrophages. J. Cell. Physiol. 2018, 233, 6996–7003. [Google Scholar] [CrossRef]
- Du, X.J.; Lu, J.M. MiR-135a represses oxidative stress and vascular inflammatory events via targeting toll-like receptor 4 in atherogenesis. J. Cell. Biochem. 2018, 119, 6154–6161. [Google Scholar] [CrossRef]
- Xu, J.; Hu, G.; Lu, M.; Xiong, Y.; Li, Q.; Chang, C.C.; Song, B.; Chang, T.; Li, B. MiR-9 reduces human acyl-coenzyme A: Cholesterol acyltransferase-1 to decrease THP-1 macrophage-derived foam cell formation. Acta Biochim. Biophys. Sin. 2013, 45, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Canfrán-Duque, A.; Rotllan, N.; Zhang, X.; Fernández-Fuertes, M.; Ramírez-Hidalgo, C.; Araldi, E.; Daimiel, L.; Busto, R.; Fernández-Hernando, C.; Suárez, Y. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol. Med. 2017, 9, 1244–1262. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S. microRNA-150 inhibits the formation of macrophage foam cells through targeting adiponectin receptor 2. Biochem. Biophys. Res. Commun. 2016, 476, 218–224. [Google Scholar] [CrossRef]
- Peng, X.-P.; Huang, L.; Liu, Z.-H. miRNA-133a attenuates lipid accumulation via TR4-CD36 pathway in macrophages. Biochimie 2016, 127, 79–85. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, J.; Sun, D.; Wei, N. MiR-155 inhibits transformation of macrophages into foam cells via regulating CEH expression. Biomed. Pharmacother. 2018, 104, 645–651. [Google Scholar] [CrossRef]
- Li, X.; Kong, D.; Chen, H.; Liu, S.; Hu, H.; Wu, T.; Wang, J.; Chen, W.; Ning, Y.; Li, Y. miR-155 acts as an anti-inflammatory factor in atherosclerosis-associated foam cell formation by repressing calcium-regulated heat stable protein 1. Sci. Rep. 2016, 6, 21789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Wu, X.; Dai, D.; Li, J.; Mehta, J.L. MicroRNA-98 regulates foam cell formation and lipid accumulation through repression of LOX-1. Redox Biol. 2018, 16, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bai, X.; Song, Q.; Fan, F.; Hu, Z.; Cheng, G.; Zhang, Y. miR-223 inhibits lipid deposition and inflammation by suppressing toll-like receptor 4 signaling in macrophages. Int. J. Mol. Sci. 2015, 16, 24965–24982. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Wang, C.; Kou, J.; Han, D.; Huo, D.; Li, F.; Zhou, X.; Meng, D.; Xu, J.; Murtaza, G. MicroRNA-23a suppresses the apoptosis of inflammatory macrophages and foam cells in atherogenesis by targeting HSP90. Gene 2020, 729, 144319. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; An, Y. Blockade of 146b-5p promotes inflammation in atherosclerosis-associated foam cell formation by targeting TRAF6. Exp. Ther. Med. 2017, 14, 5087–5092. [Google Scholar] [CrossRef]
- Zhuang, X.; Li, R.; Maimaitijiang, A.; Liu, R.; Yan, F.; Hu, H.; Gao, X.; Shi, H. miR-221-3p inhibits oxidized low-density lipoprotein induced oxidative stress and apoptosis via targeting a disintegrin and metalloprotease-22. J. Cell. Biochem. 2019, 120, 6304–6314. [Google Scholar] [CrossRef]
- Liang, X.; Xu, Z.; Yuan, M.; Zhang, Y.; Zhao, B.; Wang, J.; Zhang, A.; Li, G. MicroRNA-16 suppresses the activation of inflammatory macrophages in atherosclerosis by targeting PDCD4. Int. J. Mol. Med. 2016, 37, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Li, S.; Li, N.; Yang, X.; Ma, S.; Yang, A.; Zhang, H.; Yang, S.; Mao, C.; Xu, L. miR-34a targets HDAC1-regulated H3K9 acetylation on lipid accumulation induced by homocysteine in foam cells. J. Cell. Biochem. 2017, 118, 4617–4627. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Hsi, E.; Cheng, H.-Y.; Hsu, S.-H.; Liao, Y.-C.; Juo, S.-H.H. Let-7g suppresses both canonical and non-canonical NF-κB pathways in macrophages leading to anti-atherosclerosis. Oncotarget 2017, 8, 101026. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Han, S.; Sun, Q.; Yao, Y.; Li, S.; Yuan, C.; Zhang, B.; Jing, B.; Wu, J.; Song, Y. Long non-coding RNA CDKN2B-AS1 reduces inflammatory response and promotes cholesterol efflux in atherosclerosis by inhibiting ADAM10 expression. Aging (Albany N. Y.) 2019, 11, 1695. [Google Scholar] [CrossRef]
- Li, Y.; Sun, T.; Shen, S.; Wang, L.; Yan, J. LncRNA DYNLRB2-2 inhibits THP-1 macrophage foam cell formation by enhancing autophagy. Biol. Chem. 2019, 400, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F.; Vaughan, A.M. ABCA1-mediated transport of cellular cholesterol and phospholipids to HDL apolipoproteins. Curr. Opin. Lipidol. 2000, 11, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Attie, A.D. ABCA1: At the nexus of cholesterol, HDL and atherosclerosis. Trends Biochem. Sci. 2007, 32, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.R.; Dove, D.E.; Major, A.S.; Hasty, A.H.; Boone, B.; Linton, M.F.; Fazio, S. Reduced ABCA1-mediated cholesterol efflux and accelerated atherosclerosis in Apolipoprotein E–deficient mice lacking macrophage-derived ACAT1. Circulation 2005, 111, 2373–2381. [Google Scholar] [CrossRef] [Green Version]
- Ragozin, S.; Niemeier, A.; Laatsch, A.; Loeffler, B.; Merkel, M.; Beisiegel, U.; Heeren, J. Knockdown of hepatic ABCA1 by RNA interference decreases plasma HDL cholesterol levels and influences postprandial lipemia in mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1433–1438. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tang, C. Regulation of ABCA1 functions by signaling pathways. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2012, 1821, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Yin, K.; Liao, D.-F.; Tang, C.-K. ATP-binding membrane cassette transporter A1 (ABCA1): A possible link between inflammation and reverse cholesterol transport. Mol. Med. 2010, 16, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Ranalletta, M.; Wang, N.; Han, S.; Terasaka, N.; Li, R.; Welch, C.; Tall, A.R. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Investig. 2007, 117, 3900–3908. [Google Scholar] [CrossRef] [Green Version]
- Goedeke, L.; Vales-Lara, F.M.; Fenstermaker, M.; Cirera-Salinas, D.; Chamorro-Jorganes, A.; Ramírez, C.M.; Mattison, J.A.; de Cabo, R.; Suárez, Y.; Fernández-Hernando, C. A regulatory role for microRNA 33* in controlling lipid metabolism gene expression. Mol. Cell. Biol. 2013, 33, 2339–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, O. MicroRNA-33 Deficiency Reduces the Progression of Atherosclerotic Plaque in ApoE−/− Mice. Ph.D. Thesis, Kyoto University, Kyoto, Japan, 2014. [Google Scholar]
- Zadelaar, S.; Kleemann, R.; Verschuren, L.; de Vries-Van der Weij, J.; van der Hoorn, J.; Princen, H.M.; Kooistra, T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arter. Thromb. Vasc. Biol. 2007, 27, 1706–1721. [Google Scholar] [CrossRef] [Green Version]
- Price, N.L.; Rotllan, N.; Canfrán-Duque, A.; Zhang, X.; Pati, P.; Arias, N.; Moen, J.; Mayr, M.; Ford, D.A.; Baldán, Á.; et al. Genetic dissection of the impact of miR-33a and miR-33b during the progression of atherosclerosis. Cell Rep. 2017, 21, 1317–1330. [Google Scholar] [CrossRef] [Green Version]
- Price, N.L.; Singh, A.K.; Rotllan, N.; Goedeke, L.; Wing, A.; Canfran-Duque, A.; Diaz-Ruiz, A.; Araldi, E.; Baldan, A.; Camporez, J.P.; et al. Genetic Ablation of miR-33 Increases Food Intake, Enhances Adipose Tissue Expansion, and Promotes Obesity and Insulin Resistance. Cell Rep. 2018, 22, 2133–2145. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-J.; Yin, K.; Zhao, G.-J.; Fu, Y.-C.; Tang, C.-K. The magic and mystery of microRNA-27 in atherosclerosis. Atherosclerosis 2012, 222, 314–323. [Google Scholar] [CrossRef]
- Vickers, K.C.; Shoucri, B.M.; Levin, M.G.; Wu, H.; Pearson, D.S.; Osei-Hwedieh, D.; Collins, F.S.; Remaley, A.T.; Sethupathy, P. MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology 2013, 57, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirasaki, T.; Honda, M.; Shimakami, T.; Horii, R.; Yamashita, T.; Sakai, Y.; Sakai, A.; Okada, H.; Watanabe, R.; Murakami, S. MicroRNA-27a regulates lipid metabolism and inhibits hepatitis C virus replication in human hepatoma cells. J. Virol. 2013, 87, 5270–5286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, S.H.; Kwok, W.K.; To, K.F.; Lau, J.Y.W. Anti-atherogenic effect of hydrogen sulfide by over-expression of cystathionine gamma-lyase (CSE) gene. PLoS ONE 2014, 9, e113038. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.; Fasanaro, P.; Castelvecchio, S.; D’Alessandra, Y.; Arcelli, D.; Di Donato, M.; Malavazos, A.; Capogrossi, M.C.; Menicanti, L.; Martelli, F. MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes 2012, 61, 1633–1641. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.; Li, H.; Untereiner, A.; Wu, L.; Yang, G.; Austin, R.C.; Dickhout, J.G.; Lhoták, Š.; Meng, Q.H.; Wang, R. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 2013, 127, 2523–2534. [Google Scholar] [CrossRef] [Green Version]
- Peh, M.T.; Anwar, A.B.; Ng, D.S.; Atan, M.S.B.M.; Kumar, S.D.; Moore, P.K. Effect of feeding a high fat diet on hydrogen sulfide (H2S) metabolism in the mouse. Nitric Oxide 2014, 41, 138–145. [Google Scholar] [CrossRef]
- Guo, Z.; Li, C.S.; Wang, C.M.; Xie, Y.J.; Wang, A.L. CSE/H2S system protects mesenchymal stem cells from hypoxia and serum deprivation-induced apoptosis via mitochondrial injury, endoplasmic reticulum stress and PI3K/Akt activation pathways. Mol. Med. Rep. 2015, 12, 2128–2134. [Google Scholar] [CrossRef] [PubMed]
- Waki, H.; Nakamura, M.; Yamauchi, T.; Wakabayashi, K.-I.; Yu, J.; Hirose-Yotsuya, L.; Take, K.; Sun, W.; Iwabu, M.; Okada-Iwabu, M. Global mapping of cell type-specific open chromatin by FAIRE-seq reveals the regulatory role of the NFI family in adipocyte differentiation. PLoS Genet. 2011, 7, e1002311. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-J.; Lim, S.H.; Yeh, Y.-T.; Lien, S.-C.; Chiu, J.-J. Roles of microRNAs in atherosclerosis and restenosis. J. Biomed. Sci. 2012, 19, 79. [Google Scholar] [CrossRef] [Green Version]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; Van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celermajer, D.S.; Chow, C.K.; Marijon, E.; Anstey, N.M.; Woo, K.S. Cardiovascular disease in the developing world: Prevalences, patterns, and the potential of early disease detection. J. Am. Coll. Cardiol. 2012, 60, 1207–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
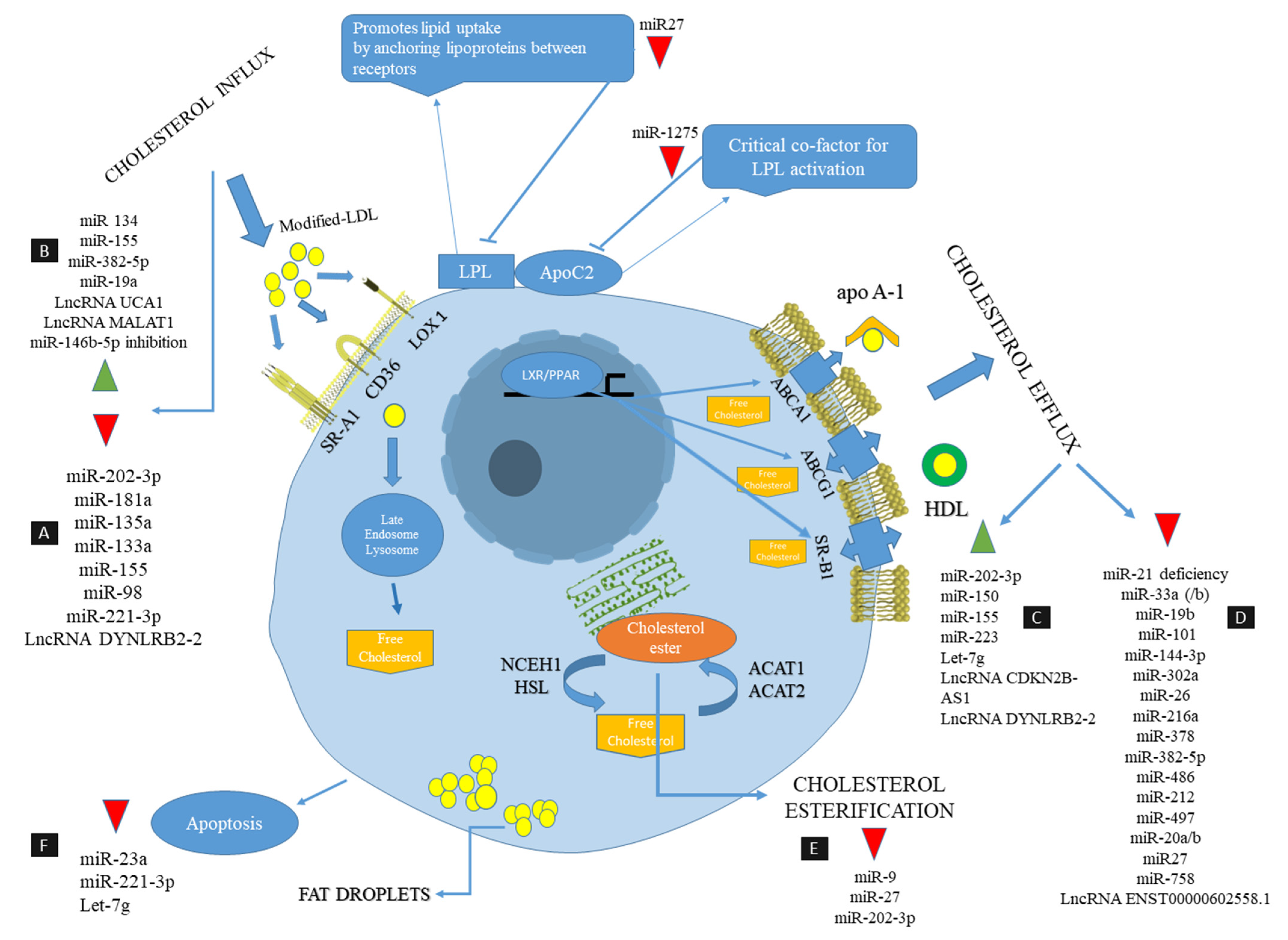
 = decrease,
= decrease,  = increase.
= decrease, = increase.
= increase.
= decrease, = increase.


{kind=link}
{kind=link}
| Non-Coding RNAs | Expression Type | Target Gene | Genetic Information (Human) | Experimental Model | Effect on Foam Cell Formation | Regulation in Lipid Metabolism | Reference | |
|---|---|---|---|---|---|---|---|---|
| In Vivo | In Vitro | |||||||
| miR-144-3p |  | ABCA1 | LXR and FXR control miR-144 expression, with both causing an upregulation in this miRNA | ApoE−/− mice | human THP-1 macrophage-derived foam cells | (+) Decrease cholesterol efflux to both apoAI and HDL | decreased HDL-C circulation and impaired RCT in vivo 144-3p mimics (agomir) increases the expression of inflammatory factors such as IL-1b, IL-6 and TNF-α in vivo and in vitro | [103] |
| miR-33(a/b) | | ABCA1, NPC1 ABCG1 | encoded within intron 16 of SREBF2, a gene that encodes a key transcriptional regulator of cholesterol uptake and synthesis | miR-33−/− ApoE−/− mice | THP-1 macrophage-derived foam cells | (+) Decrease cholesterol efflux | miR-33 coordinates cholesterol homeostasis | [104,105] |
| miR-19b | | ABCA1 | located on chromosome 13q31.3 By an unknown mechanism, expression was increased compared with the control group in advanced human atherosclerotic plaques obtained from patients during endarterectomy | ApoE−/− mice | human THP-1 macrophage-derived foam cells | (+) Decrease the efflux of cholesterol to apoAI | Decrease the levels of HDL (inhibitory role in RCT) | [106] |
| miR-101 | | ABCA1 | located on chromosome 1p31.3 | - | human THP-1-derived macrophages | (+) Decrease the efflux of cholesterol under inflammatory conditions | regulates the availability of free cholesterol for cellular efflux by inhibiting autophagy | [107] |
| miR-378 | | ABCG1 | The level of miR-378 in the aorta is elevated during the progression of atherosclerosis in apoE−/− mice | ApoE−/− mice | ox-LDL-treated human THP-1 macrophages | (+) Decrease cholesterol efflux | activator protein-1/miRNA-378/ABCG1 is a novel cascade for CoQ10 in facilitating macrophage cholesterol efflux in vitro and in vivo | [108] |
| miR-302a | | ABCA1 | located on chromosome 4q25 | Ldlr−/− mice | primary human and murine macrophages | (+) Decreases efflux of cholesterol | inhibiting miR-302a in vivo increases ABCA1 in aorta and liver of Ldlr−/− mice and increases circulating HDL | [109] |
| miR-27 | | LPL, ACAT1, ABCA1 CD36 | located on chromosome 9q22 | - | ox-LDL-treated THP-1 macrophages | (+) Reduced cholesterol efflux reduces cholesteryl ester formation blocks lipid uptake | regulates the ratio of cellular free cholesterol (FC) and cholesterol ester (CE) in THP-1 macrophages | [110] |
| miR-26 | | ARL7, ABCA1 | located on chromosome 3p22.2 | - | mouse and human LXR activated macrophages | (+) Downregulate LXR dependent cholesterol efflux | blocks the expression of two important LXR target genes (ABCA1 and ARL7) required for cholesterol efflux | [111] |
| miR-134 | | ANGPTL4 | located on chromosome 14q32.31 | - | THP-1 macrophages | (+) Increase LPL-mediated lipid accumulation | By suppressing the expression of ANGPTL4, which is a regulator of lipoprotein lipase (LPL) activity, it promotes oxLDL uptake and inflammatory responses in vitro | [112] |
| miR-155 | | HBP1 | located within a region known as the B-cell integration cluster (BIC) | ApoE−/− mice | ox-LDL-treated human THP-1 macrophages | (+) Enhanced lipid uptake and enhanced ROS production | Silencing of miR-155 in ApoE−/− mice by injecting antagomiR-155 decreased the lipid-laden macrophages and the formation of atherosclerotic plaques | [113] |
| miR-216a | | CSE | located on chromosome 2p16.1 | - | THP-1 macrophages-derived foam cells | (+) Decreased ABCA1 expression and cholesterol efflux | Downregulation of CSE/H2S leads to an increase in cholesterol accumulation in foam cells | [114] |
| miR-382-5p | | NFIA | located on chromosome 14q32.31 | - | THP-1 macrophage-derived foam cells | (+) Reduces cholesterol efflux Increases lipid uptake | RP5-833A20.1/miR-382-5p/NFIA pathway regulates cholesterol homeostasis | [107] |
| miR-486 | | HAT1 | located on chromosome 8p11.21 | - | THP-1 macrophage-derived foam cells | (+) Reduces cholesterol efflux | HAT1 is capable of acetylating H4K5 and H4K12 and increasing ABCA1 expression | [115] |
| miR-212 | | SirT1 | located on chromosome 17p13.3 | ApoE−/− mice | THP-1 human macrophages treated with oxLDL | (+) Suppresses ABCA1 dependent cholesterol efflux | SIRT1 has capable of inducing LXR activity to increase ABCA1 expression in human macrophages | [116] |
| miR-19a | | HBP-1 | miR-19a is an important member of the miR-17–92 polycistronic gene cluster MiR-19a is abundant in the blood and tissues of patients with atherosclerotic coronary artery disease | ApoE−/− mice | THP-1 derived macrophages RAW264.7 cells | (+) Increases lipid uptake of macrophages | HBP-1 participates in inhibiting the expression of the macrophage migration inhibitory factor (MIF) and lipid uptake by macrophages size of the atherosclerotic plaques in antagmiR-19a treated mice was reduced | [117] |
| miR-497 | | Apelin | Located on chromosome 17q13.1 | - | Human THP-1 macrophages treated with oxLDL | (+) Decrease cholesterol efflux to apoA-I. | Apelin is an adipokine that is involved in the pathophysiology of cardiovascular diseases | [118] |
| miR-20a/b | | ABCA1 | Mir 20a Located on chromosome 13q31.3 Mir 20b Located on chromosome Xq26.2 | ApoE−/− Mice | THP-1 and RAW 264.7 Macrophage-derived foam cells. | (+) Reducing cholesterol Efflux, impairs RCT in vivo | Both in in vitro studies and in ApoE−/− mice treated with miR-20a/b, the hepatic expression of ABCA1, as well as reverse cholesterol transport, are decreased | [119] |
| miR-758 | | ABCA1 | miR-758 was widely expressed in mouse tissues and particularly abundant in the brain, heart and aorta localized in an intergenic region within chromosome 14 | Ldlr−/− mouse | Mouse and human macrophage | (+) Cholesterol efflux to apoA1 | effect of miR-758 on cholesterol efflux to ApoA1 was significantly attenuated after ABCA1 silencing decrease in peritoneal macrophages obtained from hypercholesterolemia LDLR−/− mice | [120] |
| LncRNA MALAT1 | | CD36, b-catenin | Located on chromosome 11q13.1 | - | THP-1-derived macrophage | (+) Enhances lipid uptake | oxLDL induces MALAT1 transcription via the NF-kB pathway Knockdown of MALAT1 using siRNA transfection reduces CD36 expression and affects lipid uptake in macrophages | [121] |
| LncRNA ENST00000602558.1 | | ABCG1 | - | vascular smooth muscle cells (VSMCs) | (+) Reduce ABCG1-mediated cholesterol efflux to HDL | ENST00000602558.1 induces p65, which is a specific inhibitor of NF-kB and mediates a decrease in the expression of ABCG1 | [122] | |
| LncRNA UCA1 | | Mi R-206 | downregulation of UCA1 inhibits oxidative stress and induces apoptosis of THP-1 cells, Located on chromosome 19p13.12 | - | THP-1 cells | (+) Increase oxidative stress process In addition, CD36 levels | oxLDL greatly increases UCA1 expression, UCA1 ‘sponges’ miR-206 to exacerbate atherosclerosis | [123] |
| Non-Coding RNA | Expression Type | Target Gene | Genetic Information (Human) | Experimental Model | Effect on Foam Cell Formation | Regulation in Lipid Metabolism | Reference | |
|---|---|---|---|---|---|---|---|---|
| In Vivo | In Vitro | |||||||
| miR-1275 | | ApoC2 | located on chromosome 6p21.31 | - | THP-1 derived macrophages | (−) Inhibited the cellular uptake of Ox-LDL and lipid accumulation | ApoC2 most important cofactor for LPL lipolytic activity | [124] |
| miR-202-3p | | SCARB2, ABCG4, NCEH1I | located on chromosome 10q26.3 | - | Human THP-1 macrophage-derived foam cell | (−) Attenuates lipid uptake Enhances cholesterol efflux | Reduces the expression of scavenger receptor SCARB2 Increases the expression of NCEH1 and ABCG4 | [125] |
| miR-181a | | TLR4 | located on chromosome 1q32.1 | - | THP-1 | (−) Attenuates lipid uptake and apoptosis and inflammation | Inhibits protein levels of CD36 (scavenger receptor class B) | [126] |
| miR-135a | | TLR4 | located on chromosome 3p21.2 | - | RAW264.7 and MOVAS cells | (−) Attenuates lipid uptake and inhibit oxidative stress and vascular inflammation | Inhibits CD36 expression | [127] |
| miR-9 | | ACAT1 | located on chromosome 1q22 | - | Human THP-1 macrophage-derived foam cell | (−) Decrease the cholesterol ester formation | Reduces the levels of the ACAT1 protein (Acyl-coenzyme A:cholesterol acyltransferase ) | [128] |
| miR-21 |  | MKK3 MERTK | located on chromosome 17q23.1 | Ldlr−/− or miR21−/− mice | Peritoneal macrophages from adult miR21−/− mice | (−) Attenuated cholesterol efflux and promoting the lipid accumulation, Reduce apoptotic cell uptake | Knock down of miR-21 increases the expression of MKK3, promoting the induction of p38-CHOP and jNK signaling, which results in degradation of ABCG1, Decreases expression of MERTK; a key receptor that mediates the clearance of apoptotic cells | [129] |
| miR-150 | | AdipoR2 | located on chromosome 19q13.33 | - | THP-1 macrophages | (−) Attenuates lipid uptake Increases cholesterol efflux | Decreases CD36, upregulates ABCA1 and ABCG1 via the PPARγ- and LXRα-dependent pathways. | [130] |
| miR-133a | | TR4 (testicular orphan nuclear receptor 4) | located on chromosome 18q11.2 | - | RAW 264.7 macrophage cells | (−) Decreased oxLDL uptake In addition, lipid accumulation | Decreases expression of CD36 | [131] |
| miR-155 | | Tim-3, CEH (cholesterol ester hydrolase) | located on chromosome 21q21.3 | - | THP-1 | (−) Increases cholesterol efflux, Decreased lipid uptake | inhibits Tim-3 expression and increases the expression of CEH resulting in increased expression of ABCA1; inhibits the expression of SR-A, | [132] |
| miR-155 | | CARHSP1 | located on chromosome 21q21.3 | - | THP-1 | (−) Decrease inflammation and lipid uptake | suppresses TNF-α production by directly targeting CARHSP1, which is required for TNF-α mRNA stabilization | [133] |
| miR-98 | | LOX-1 (Lectin-like ox-LDL scavenger receptor-1) | located on chromosome Xp11.22 | ApoE−/− mice | Macrophages were collected from C57BL/6 mice | (−) Decreases the lipid uptake and lipid accumulation | decreases LOX-1 expression | [134] |
| miR-223 | | TLR4-NF-κB signaling pathway | located on chromosome Xq12 | ApoE−/− mouse, C57BL/6J wild-type mice(control) | Murine macrophage cell line RAW 264.7 | (−) Increase cholesterol efflux to apoA-I, Decrease inflammation | activates the PI3K/AKT signaling pathway | [135] |
| miR-23a | | HSP90 | located on chromosome 19p13.12 | - | THP-1 macrophages | (−) Inhibit lipid accumulation | Decreased apoptosis of foam cells; decreases inflammation factors by inhibiting the activation of NF-κB pathways | [136] |
| miR-146b-5p | | TRAF6 | located on chromosome 10q24.32 | - | THP-1 human monocytic and the HEK293T human embryonic kidney cell line | (−) Decrease lipid uptake | miR-146-5p inhibitor increases TRAF6-mediated activation of NF-κB (p65) and increases inflammatory response | [137] |
| miR-221-3p | | ADAM22 | located on chromosome Xp11.3 | - | RAW264.7 | (−) Decrease lipid uptake | Decreases CD36; decrease oxidative stress and apoptosis | [138] |
| miR-16 | | PDCD4 (Programmed cell death 4) | located on chromosome 13q14 | ApoE−/− mice | macrophage-derived foam cell (RAW264.7) | (−) | Suppresses the activation of inflammatory macrophages and decreases release of inflammatory cytokines via the MAPK and NF-κB pathways | [139] |
| miR-34a | | HDAC1 (histone deacetylase 1) | located on chromosome 1p36.22 | Male C57BL/6J and ApoE−/− mice | Human THP-1 macrophages | (−) Prevents the accumulation of triglyceride and total and free cholesterol | Hyperhomocysteinemia (HHcy) accelerates atherogenesis via decreased expression of miR-34a | [140] |
| Let-7g | | NF-κB complex MEKK1 | located on chromosome 3p21.2 | ApoE−/− mouse, IKKαf/f:MLysCre/apoE−/− mice | Human THP-1 macrophage | (−) increase cholesterol efflux, Decrease intracellular lipid accumulation | up-regulates SREBF2, which is a critical regulator of cholesterol/lipid homeostasis, up-regulates ABCA1 via suppression of miR-33a, decreases p53-dependent apoptosis | [141] |
| LncRNA CDKN2B-AS1 | | ADAM10 | located on chromosome 9p21.3 | ApoE−/− Mice, C57BL/6J Mice | THP-1 macrophage-derived foam cells | (−) Increases Cholesterol Efflux, Decrease lipid accumulation | Inhibits inflammatory responses by suppressing the transcription of ADAM10 | [142] |
| LncRNA DYNLRB2-2 | | miR-298 | located on chromosome 16q23.2 | - | THP-1 macrophage-derived foam cells | (−) Impairing oxLDL uptake, increases cholesterol efflux | Induces autophagy by activating the LKB1/AMPK/mTOR signaling pathway via the miR-298/SIRT3 axis; decreases the expression of TLR2 and enhances the expression of ABCA1 | [143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javadifar, A.; Rastgoo, S.; Banach, M.; Jamialahmadi, T.; Johnston, T.P.; Sahebkar, A. Foam Cells as Therapeutic Targets in Atherosclerosis with a Focus on the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 2529. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052529
Javadifar A, Rastgoo S, Banach M, Jamialahmadi T, Johnston TP, Sahebkar A. Foam Cells as Therapeutic Targets in Atherosclerosis with a Focus on the Regulatory Roles of Non-Coding RNAs. International Journal of Molecular Sciences. 2021; 22(5):2529. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052529
Chicago/Turabian StyleJavadifar, Amin, Sahar Rastgoo, Maciej Banach, Tannaz Jamialahmadi, Thomas P. Johnston, and Amirhossein Sahebkar. 2021. "Foam Cells as Therapeutic Targets in Atherosclerosis with a Focus on the Regulatory Roles of Non-Coding RNAs" International Journal of Molecular Sciences 22, no. 5: 2529. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052529