
Deficiency of Lipin2 Results in Enhanced NF-κB Signaling and Osteoclast Formation in RAW-D Murine Macrophages

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
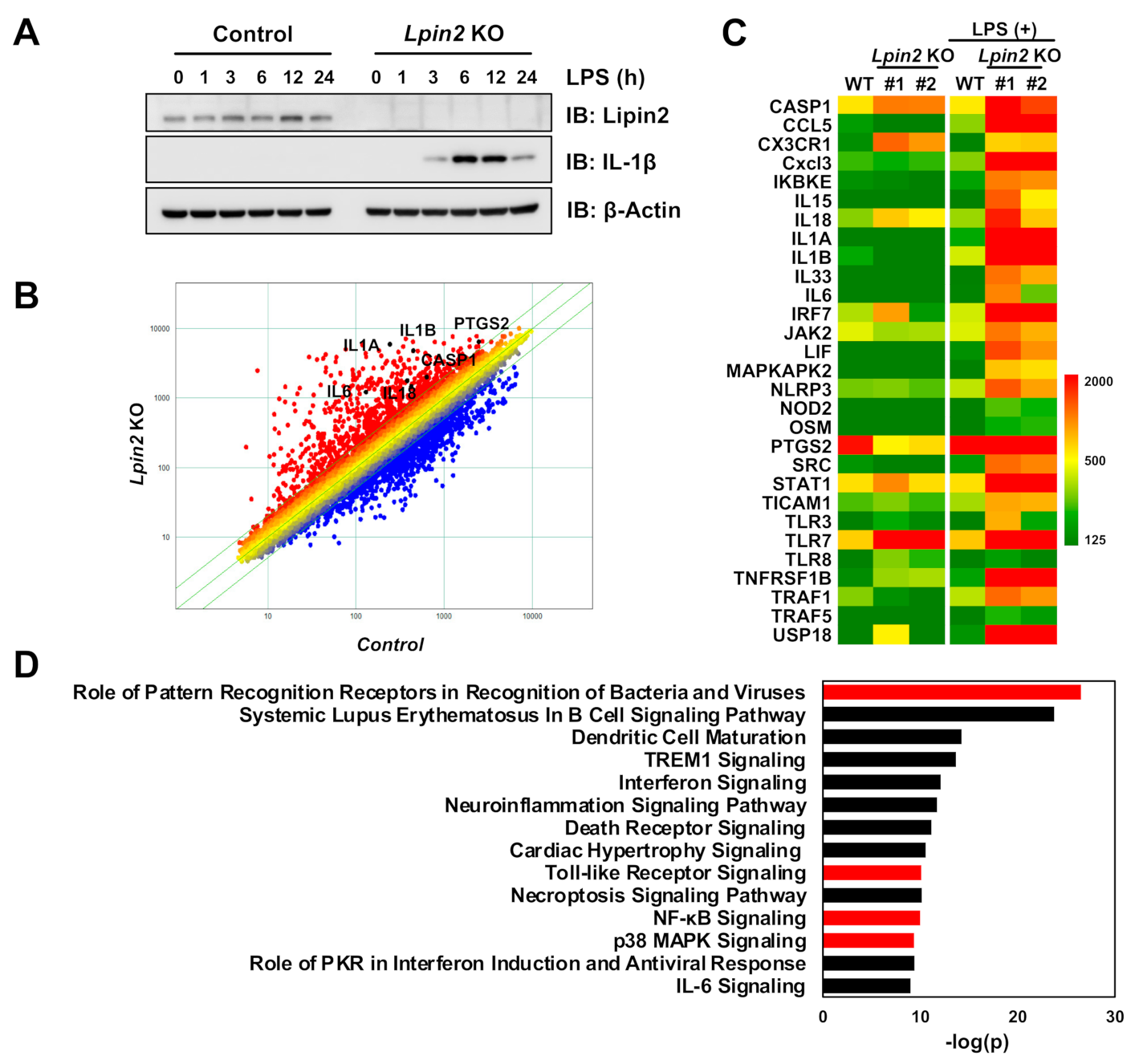
2.1. Lipin2 Deficiency Enhances Proinflammatory Responses in Macrophages by Modulating Inflammatory Transcription
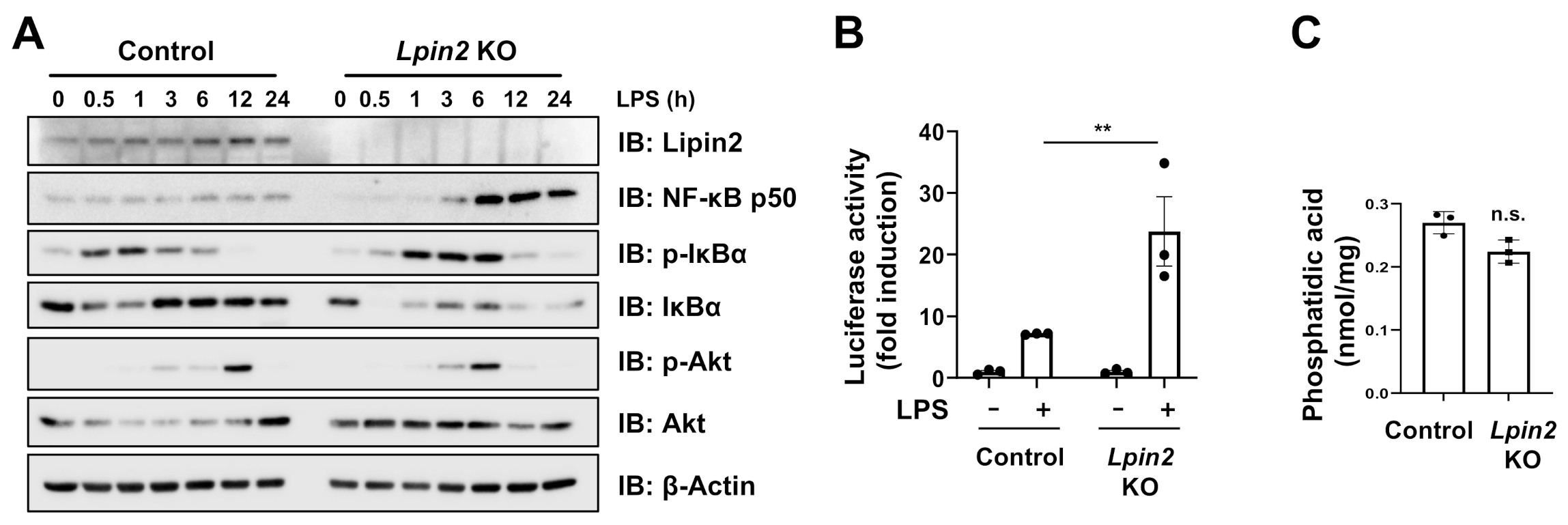
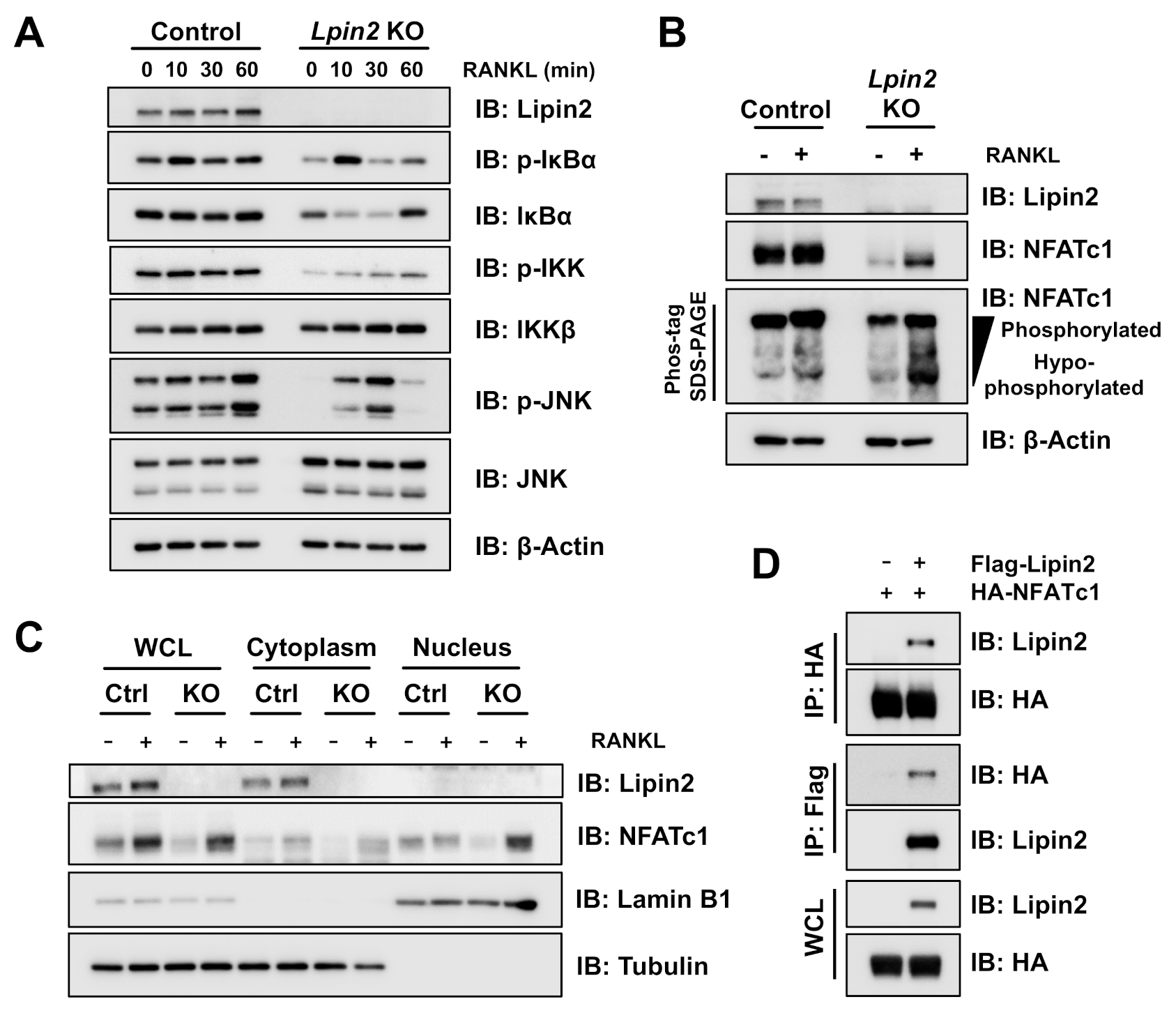
2.2. Lpin2 Knockout Enhances the NF-κB Signaling Pathway in RAW-D Cells
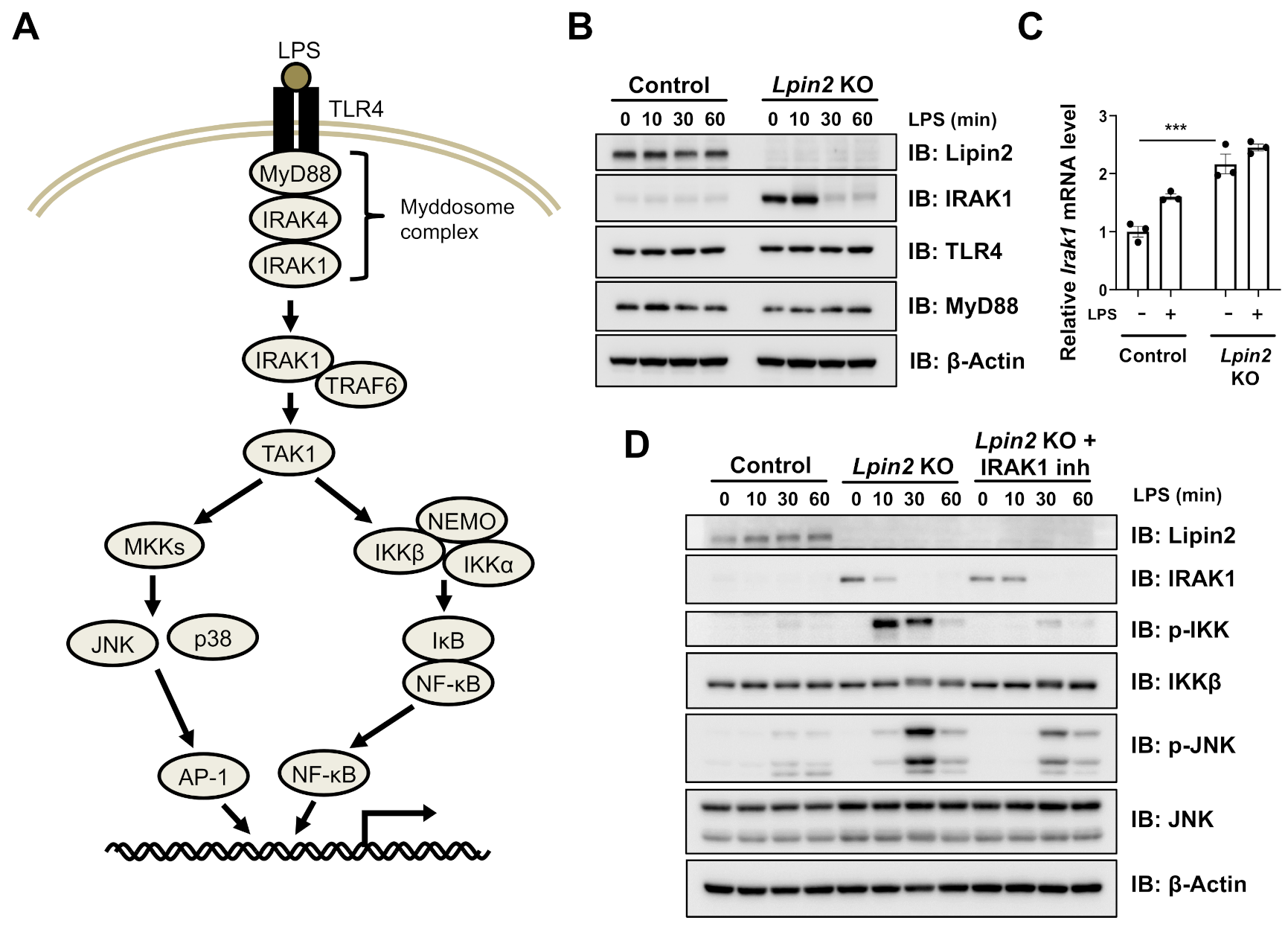
2.3. Elevated IRAK Signaling Induces Downstream Pathways in Lpin2 Knockout Macrophages
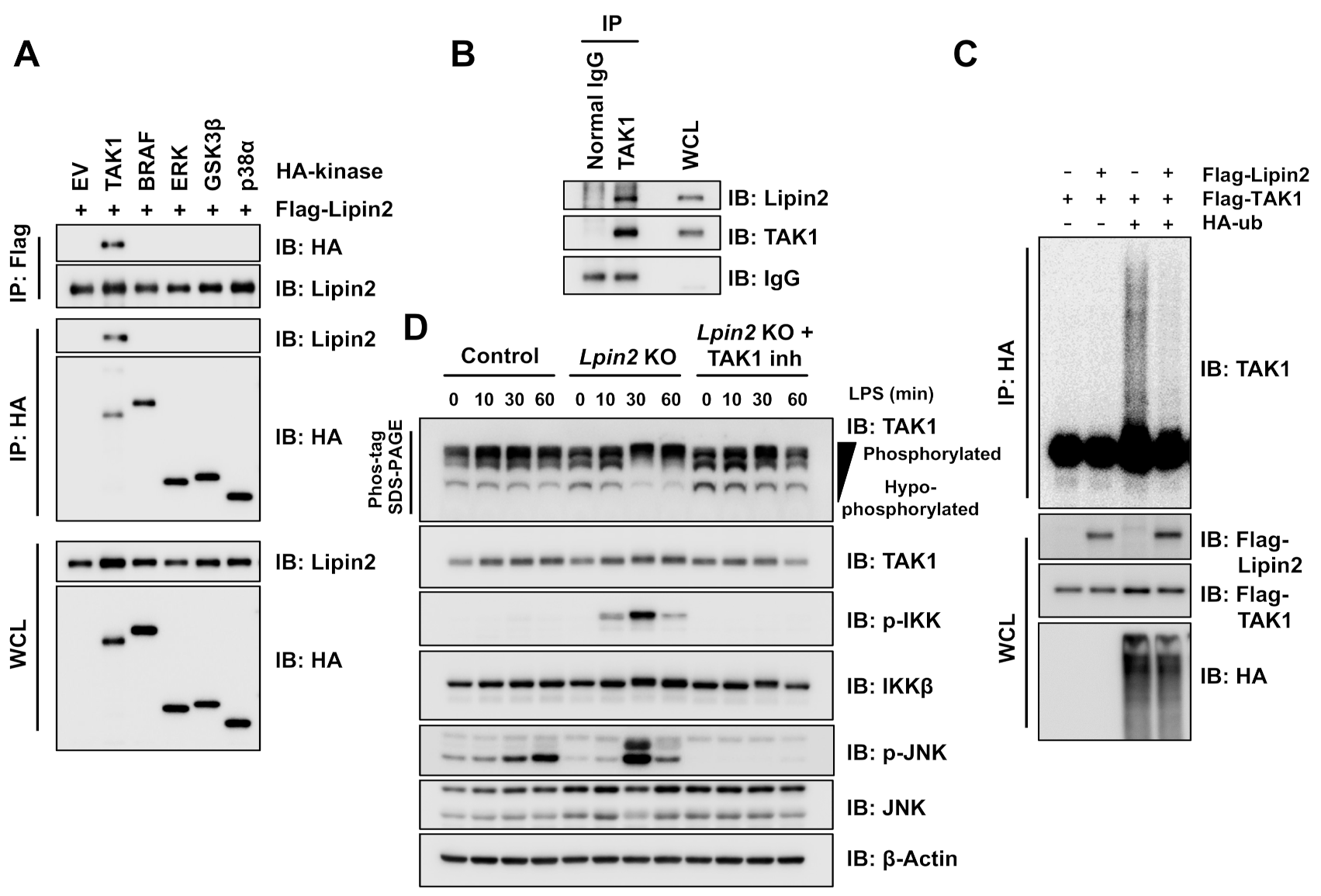
2.4. Elevated TAK1 Activity Induces Downstream Signaling in Lpin2 Knockout Macrophages
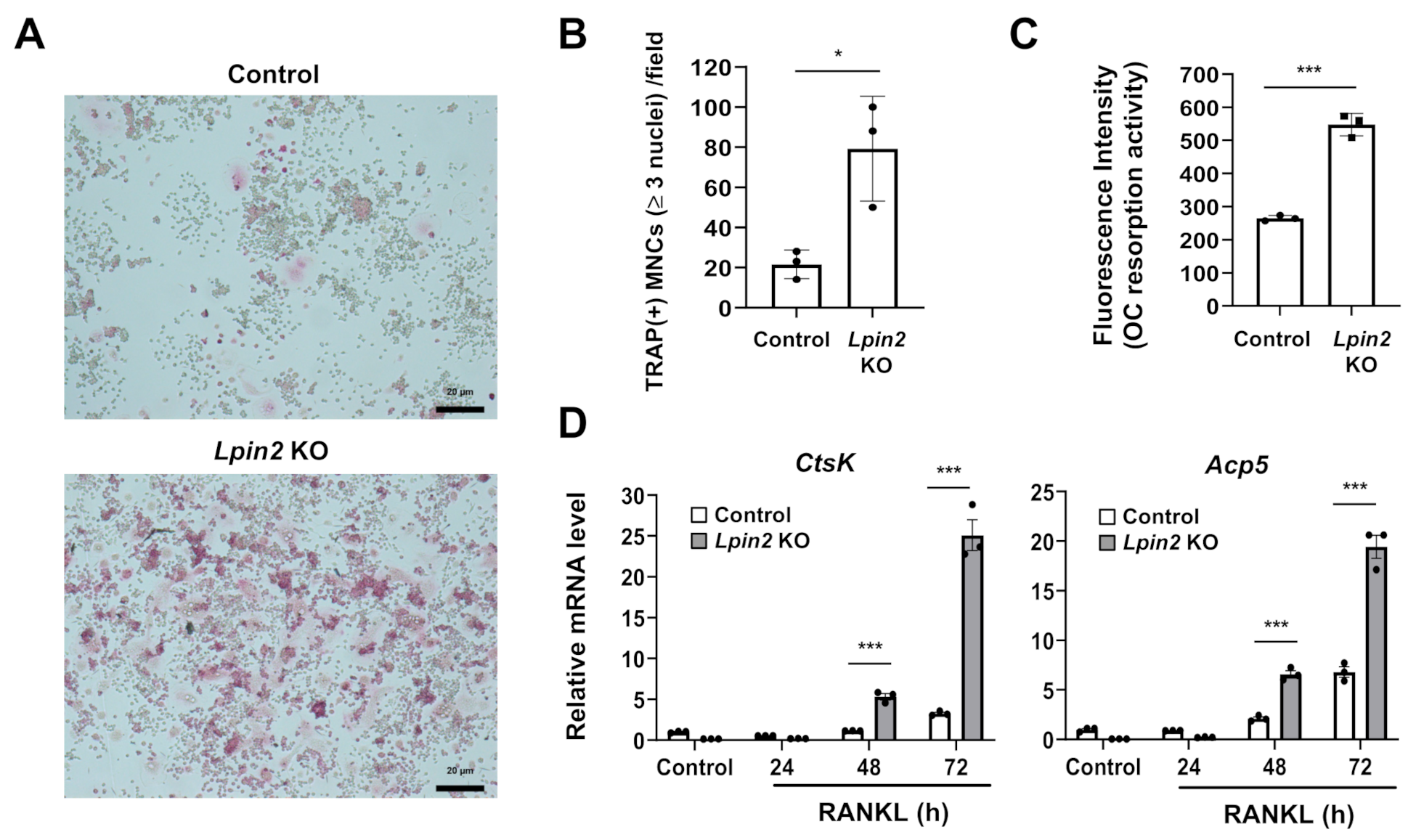
2.5. Lipin2 Deficiency Promotes RANKL-Dependent Osteoclastogenesis and Osteoclastic Resorption Activity in RAW-D Macrophages
2.6. Lipin2 Negatively Regulates NFATc1 Activity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies and Plasmids
4.3. Immunoblots and Immunoprecipitation
4.4. RT-PCR and Microarray Expression Analyses
4.5. Osteoclast Formation and Osteoclastic Resorption Activity Assays
4.6. Cellular Fractionation
4.7. Phosphatidic Acid (PA) Measurement
4.8. Luciferase Reporter Assay
4.9. Mass Spectrometry
4.10. Quantification and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Majeed, H.A.; Kalaawi, M.; Mohanty, D.; Teebi, A.S.; Tunjekar, M.F.; al-Gharbawy, F.; Majeed, S.A.; al-Gazzar, A.H. Congen-ital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with Sweet syndrome in two siblings. J. Pediatr. 1989, 115, 730–734. [Google Scholar] [CrossRef]
- Majeed, H.A.; Al-Tarawna, M.; El-Shanti, H.; Kamel, B.; Al-Khalaileh, F. The syndrome of chronic recurrent multifocal os-teomyelitis and congenital dyserythropoietic anaemia. Report of a new family and a review. Eur. J. Pediatr. 2001, 160, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, P.J.; Chen, S.; Tayeh, M.K.; Ochoa, L.; Leal, S.M.; Pelet, A.; Munnich, A.; Lyonnet, S.; Majeed, H.A.; El-Shanti, H. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and con-genital dyserythropoietic anaemia (Majeed syndrome). J. Med. Genet. 2005, 42, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Al-Mosawi, Z.S.; Al-Saad, K.K.; Ijadi-Maghsoodi, R.; El-Shanti, H.I.; Ferguson, P.J. A splice site mutation confirms the role of LPIN2 in Majeed syndrome. Arthritis Rheum. 2007, 56, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, X.Y.; Zhao, Z.P.; Guo, R.L.; Guo, J.; Li, W.; Hao, C.J.; Xu, B.P. Compound heterozygous LPIN2 pathogenic variants in a patient with Majeed syndrome with recurrent fever and severe neutropenia: Case report. BMC Med. Genet. 2019, 20, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herlin, T.; Fiirgaard, B.; Bjerre, M.; Kerndrup, G.; Hasle, H.; Bing, X.; Ferguson, P.J. Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann. Rheum. Dis. 2013, 72, 410–413. [Google Scholar] [CrossRef]
- Wang, H.; Airola, M.V.; Reue, K. How lipid droplets “TAG” along: Glycerolipid synthetic enzymes and lipid storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1131–1145. [Google Scholar] [CrossRef]
- Reue, K.; Wang, H. Mammalian lipin phosphatidic acid phosphatases in lipid synthesis and beyond: Metabolic and inflam-matory disorders. J. Lipid Res. 2019, 60, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balboa, M.A.; de Pablo, N.; Meana, C.; Balsinde, J. The role of lipins in innate immunity and inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Lorden, G.; Sanjuan-Garcia, I.; de Pablo, N.; Meana, C.; Alvarez-Miguel, I.; Perez-Garcia, M.T.; Pelegrin, P.; Balsinde, J.; Bal-boa, M.A. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J. Exp. Med. 2017, 214, 511–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdearcos, M.; Esquinas, E.; Meana, C.; Pena, L.; Gil-de-Gomez, L.; Balsinde, J.; Balboa, M.A. Lipin-2 reduces proinflamma-tory signaling induced by saturated fatty acids in macrophages. J. Biol. Chem. 2012, 287, 10894–10904. [Google Scholar] [CrossRef] [Green Version]
- El-Shanti, H.I.; Ferguson, P.J. Chronic recurrent multifocal osteomyelitis: A concise review and genetic update. Clin. Orthop. Relat. Res. 2007, 462, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, C.M.; Hofmann, S.R.; Pablik, J.; Morbach, H.; Girschick, H.J. Autoinflammatory bone disorders with special focus on chronic recurrent multifocal osteomyelitis (CRMO). Pediatr. Rheumatol. Online J. 2013, 11, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuyan, F.; de Jesus, A.A.; Mitchell, J.; Leikina, E.; VanTries, R.; Herzog, R.; Onel, K.B.; Oler, A.; Montealegre Sanchez, G.A.; Johnson, K.A.; et al. Novel Majeed syndrome causing LPIN2 mutations link bone inflamma tion to inflammatory M2 macrophages and accelerated osteoclastogenesis. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef]
- Kukita, T.; Wada, N.; Kukita, A.; Kakimoto, T.; Sandra, F.; Toh, K.; Nagata, K.; Iijima, T.; Horiuchi, M.; Matsusaki, H.; et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J. Exp. Med. 2004, 200, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Kukita, T.; Kukita, A.; Wada, N.; Toh, K.; Nagata, K.; Nomiyama, H.; Iijima, T. Direct stimulation of osteo-clastogenesis by MIP-1alpha: Evidence obtained from studies using RAW264 cell clone highly responsive to RANKL. J. Endocrinol. 2004, 180, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csaki, L.S.; Dwyer, J.R.; Fong, L.G.; Tontonoz, P.; Young, S.G.; Reue, K. Lipins, lipinopathies, and the modulation of cellular lipid storage and signaling. Prog. Lipid Res. 2013, 52, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Gottipati, S.; Rao, N.L.; Fung-Leung, W.P. IRAK1: A critical signaling mediator of innate immunity. Cell Signal 2008, 20, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Kaczanowska, S.; Davila, E. IL-1 Receptor-Associated Kinase Signaling and Its Role in Inflammation, Cancer Pro-gression, and Therapy Resistance. Front Immunol. 2014, 5, 553. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Zhu, J.; Zhuang, X.; Zhang, X.; Luo, T.; Esser, K.A.; Ren, H. Lipin1 Regulates Skeletal Muscle Differentia-tion through Extracellular Signal-regulated Kinase (ERK) Activation and Cyclin D Complex-regulated Cell Cycle With-drawal. J. Biol. Chem. 2015, 290, 23646–23655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finck, B.N.; Gropler, M.C.; Chen, Z.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, J.C., Jr.; Kelly, D.P. Lipin 1 is an in-ducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006, 4, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, Y.K.; Lee, M.Y.; Kim, J.W.; Kim, M.; Moon, J.S.; Lee, Y.J.; Ahn, Y.H.; Kim, K.S. Lipin1 is a key factor for the maturation and maintenance of adipocytes in the regulatory network with CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma 2. J. Biol. Chem. 2008, 283, 34896–34906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.B.; Kumar, A.; Wang, L.; Liu, G.H.; Keller, S.R.; Lawrence, J.C., Jr.; Finck, B.N.; Harris, T.E. Lipin 1 represses NFATc4 transcriptional activity in adipocytes to inhibit secretion of inflammatory factors. Mol. Cell Biol. 2010, 30, 3126–3139. [Google Scholar] [CrossRef] [Green Version]
- Manmontri, B.; Sariahmetoglu, M.; Donkor, J.; Bou Khalil, M.; Sundaram, M.; Yao, Z.; Reue, K.; Lehner, R.; Brindley, D.N. Glucocorticoids and cyclic AMP selectively increase hepatic lipin-1 expression, and insulin acts antagonistically. J. Lipid Res. 2008, 49, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 2012, 33, 522–530. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef]
- Yamazaki, K.; Gohda, J.; Kanayama, A.; Miyamoto, Y.; Sakurai, H.; Yamamoto, M.; Akira, S.; Hayashi, H.; Su, B.; Inoue, J. Two mechanistically and temporally distinct NF-kappaB activation pathways in IL-1 signaling. Sci. Signal 2009, 2, ra66. [Google Scholar] [CrossRef]
- Fan, Y.; Yu, Y.; Shi, Y.; Sun, W.; Xie, M.; Ge, N.; Mao, R.; Chang, A.; Xu, G.; Schneider, M.D.; et al. Lysine 63-linked polyubiq-uitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J. Biol. Chem. 2010, 285, 5347–5360. [Google Scholar] [CrossRef] [Green Version]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Okamura, H.; Aramburu, J.; Garcia-Rodriguez, C.; Viola, J.P.; Raghavan, A.; Tahiliani, M.; Zhang, X.; Qin, J.; Hogan, P.G.; Rao, A. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell 2000, 6, 539–550. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. In-duction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Donkor, J.; Zhang, P.; Wong, S.; O’Loughlin, L.; Dewald, J.; Kok, B.P.; Brindley, D.N.; Reue, K. A conserved serine residue is required for the phosphatidate phosphatase activity but not the transcriptional coactivator functions of lipin-1 and lipin-2. J. Biol. Chem. 2009, 284, 29968–29978. [Google Scholar] [CrossRef] [Green Version]
- Yamin, T.T.; Miller, D.K. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphor-ylation. J. Biol. Chem. 1997, 272, 21540–21547. [Google Scholar] [CrossRef] [Green Version]
- Longo, P.A.; Kavran, J.M.; Kim, M.S.; Leahy, D.J. Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol. 2013, 529, 227–240. [Google Scholar]
- Watahiki, A.; Shimizu, K.; Hoshikawa, S.; Chiba, M.; Kitamura, H.; Egusa, H.; Fukumoto, S.; Inuzuka, H. Lipin-2 degrada-tion elicits a proinflammatory gene signature in macrophages. Biochem. Biophys. Res. Commun. 2020, 524, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, Y.; Watanabe, E.; Umeyama, T.; Nakajima, D.; Hattori, M.; Honda, K.; Ohara, O. Optimization of Da-ta-Independent Acquisition Mass Spectrometry for Deep and Highly Sensitive Proteomic Analysis. Int. J. Mol. Sci. 2019, 20, 5932. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watahiki, A.; Hoshikawa, S.; Chiba, M.; Egusa, H.; Fukumoto, S.; Inuzuka, H. Deficiency of Lipin2 Results in Enhanced NF-κB Signaling and Osteoclast Formation in RAW-D Murine Macrophages. Int. J. Mol. Sci. 2021, 22, 2893. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062893
Watahiki A, Hoshikawa S, Chiba M, Egusa H, Fukumoto S, Inuzuka H. Deficiency of Lipin2 Results in Enhanced NF-κB Signaling and Osteoclast Formation in RAW-D Murine Macrophages. International Journal of Molecular Sciences. 2021; 22(6):2893. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062893
Chicago/Turabian StyleWatahiki, Asami, Seira Hoshikawa, Mitsuki Chiba, Hiroshi Egusa, Satoshi Fukumoto, and Hiroyuki Inuzuka. 2021. "Deficiency of Lipin2 Results in Enhanced NF-κB Signaling and Osteoclast Formation in RAW-D Murine Macrophages" International Journal of Molecular Sciences 22, no. 6: 2893. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062893