
High-Throughput Screening for CEBPD-Modulating Compounds in THP-1-Derived Reporter Macrophages Identifies Anti-Inflammatory HDAC and BET Inhibitors



Abstract
:1. Introduction
2. Results
2.1. Screening Assay Development
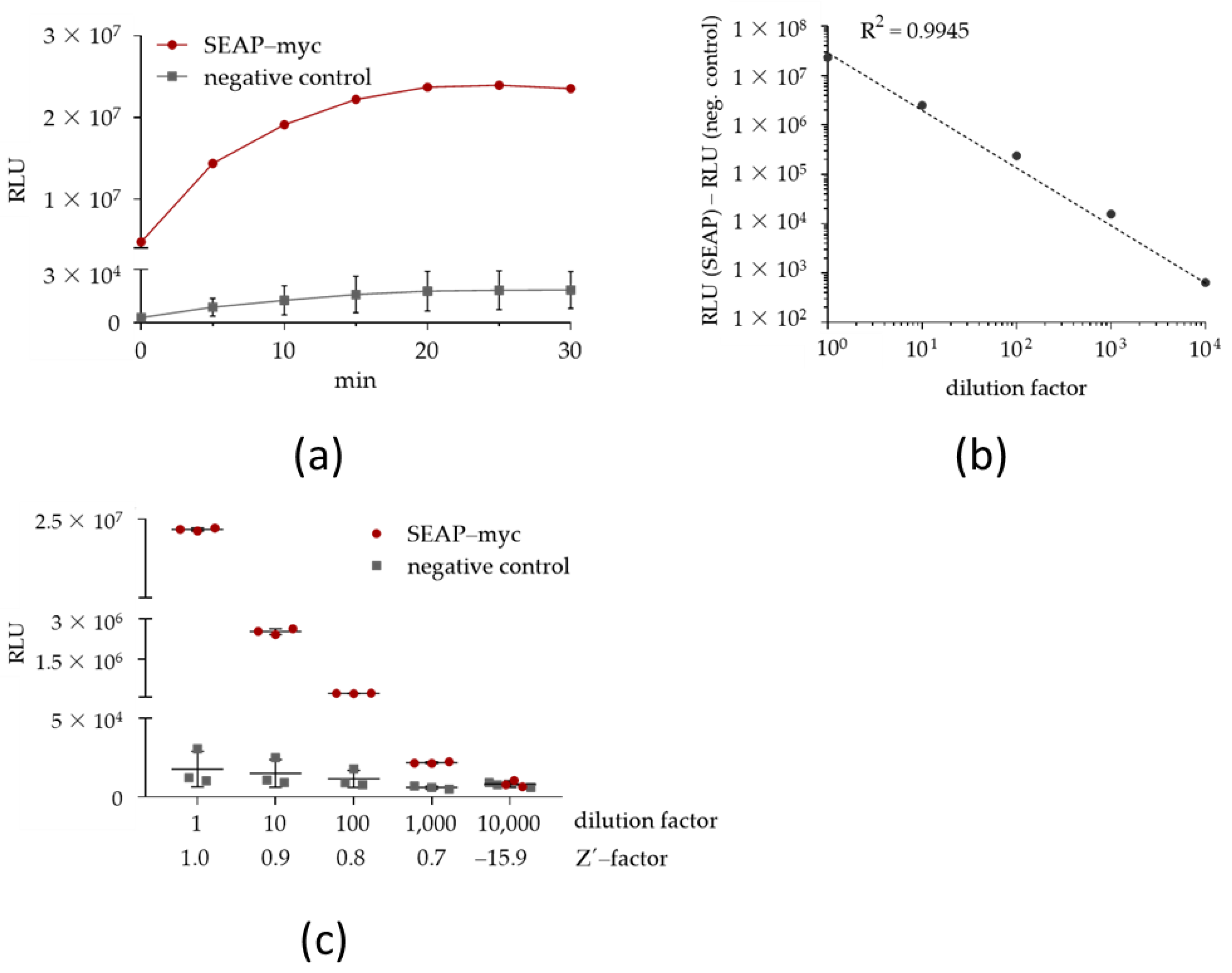
2.1.1. Characterization of the Chemiluminescent SEAP Assay
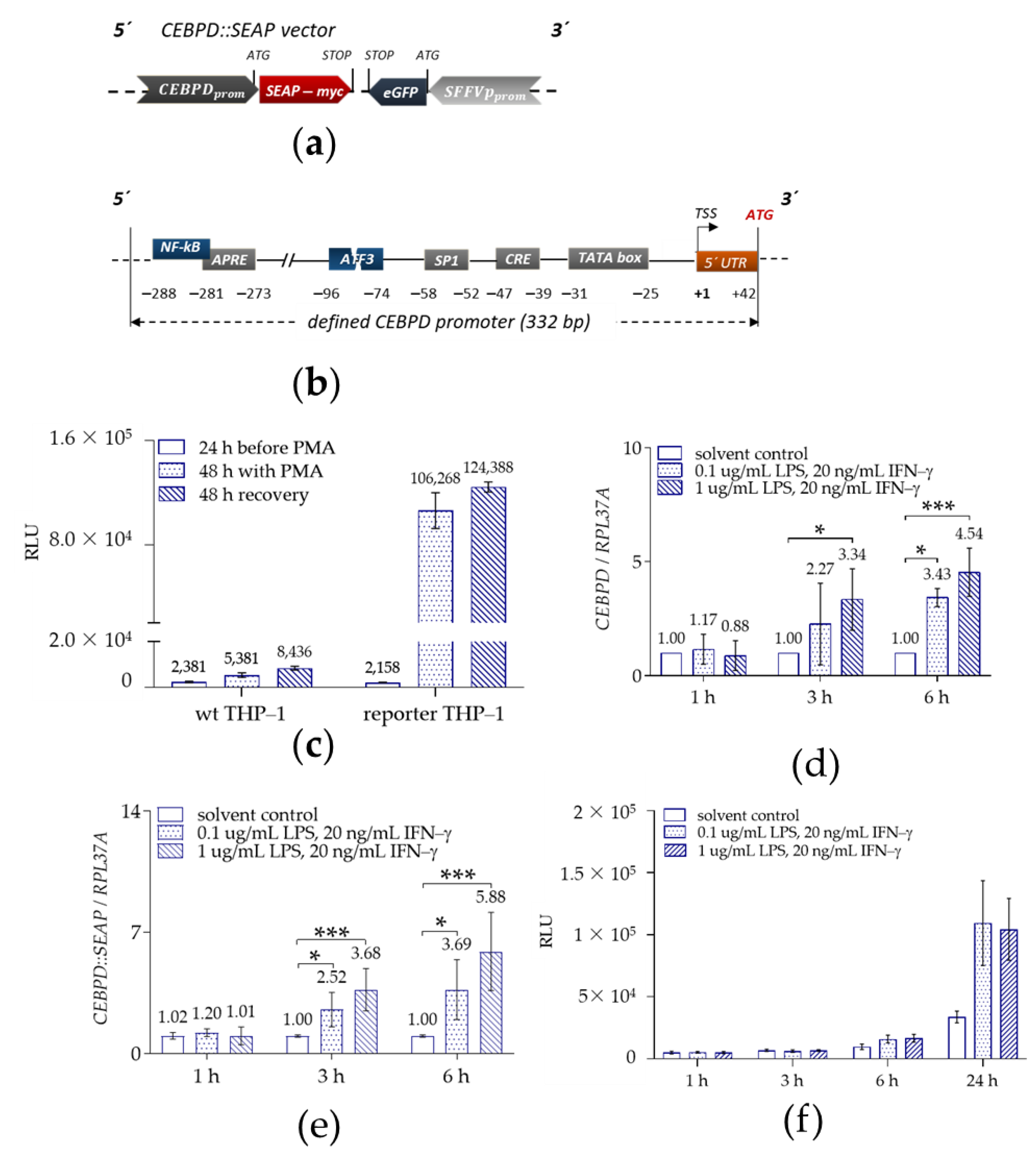
2.1.2. Validation of THP-1-Derived Mϕ Reporter Cell Line
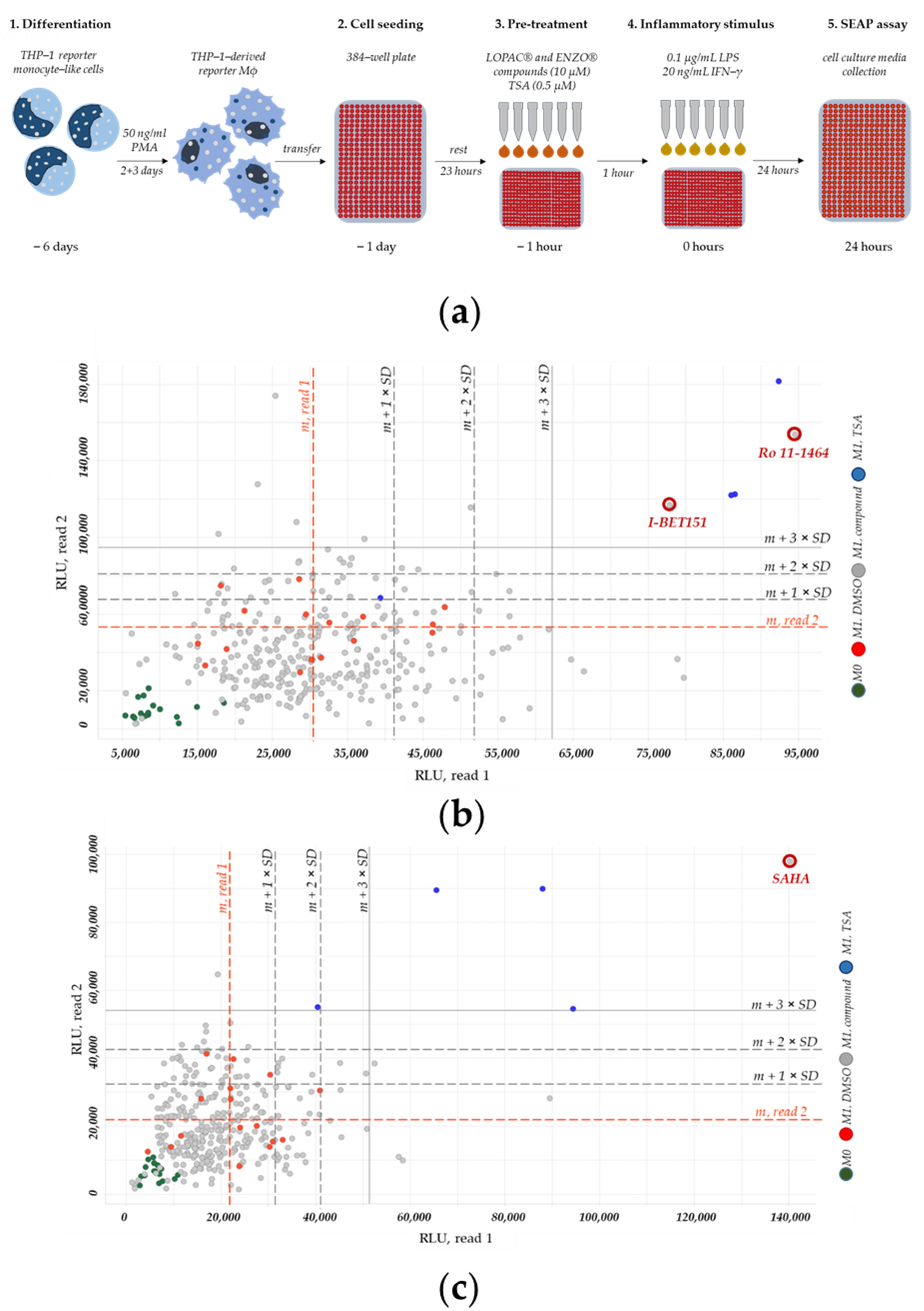
2.2. A High-Throughput Screening for CEBPD-Modulationg Compounds
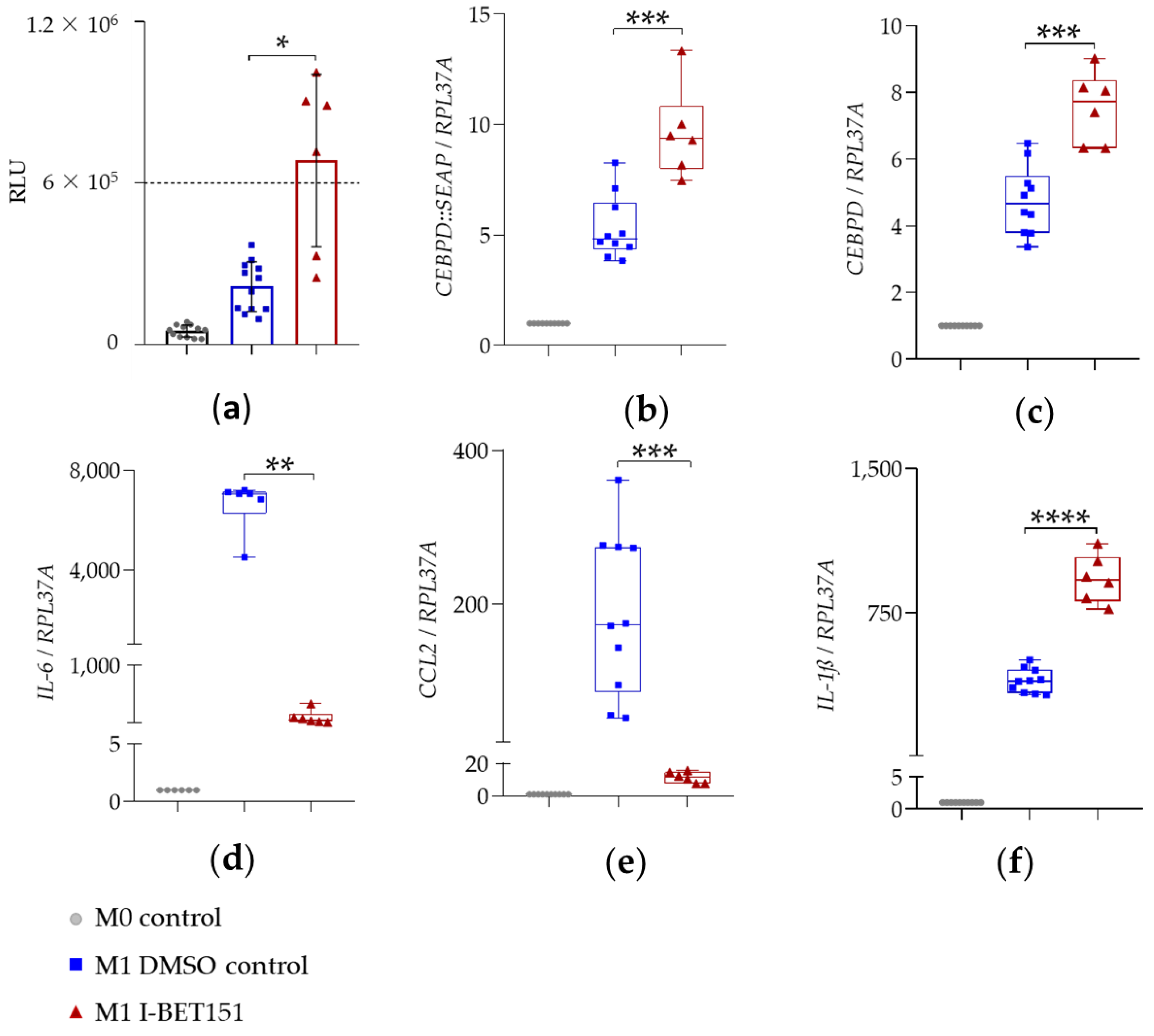
2.3. Analysis of Hit Compound Effects on Gene Expression
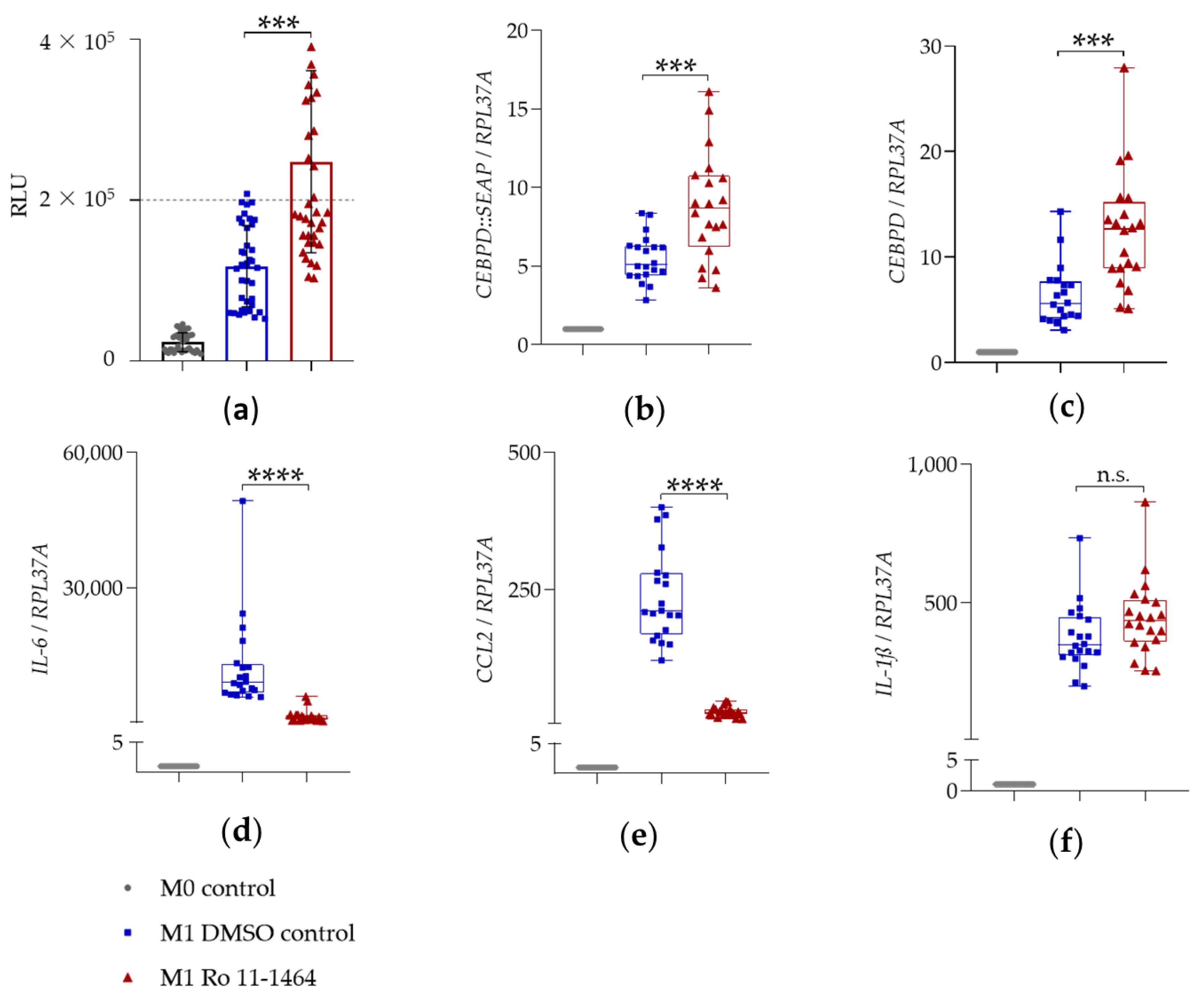
2.3.1. BET Inhibitors I-BET151 and Ro 11-1464
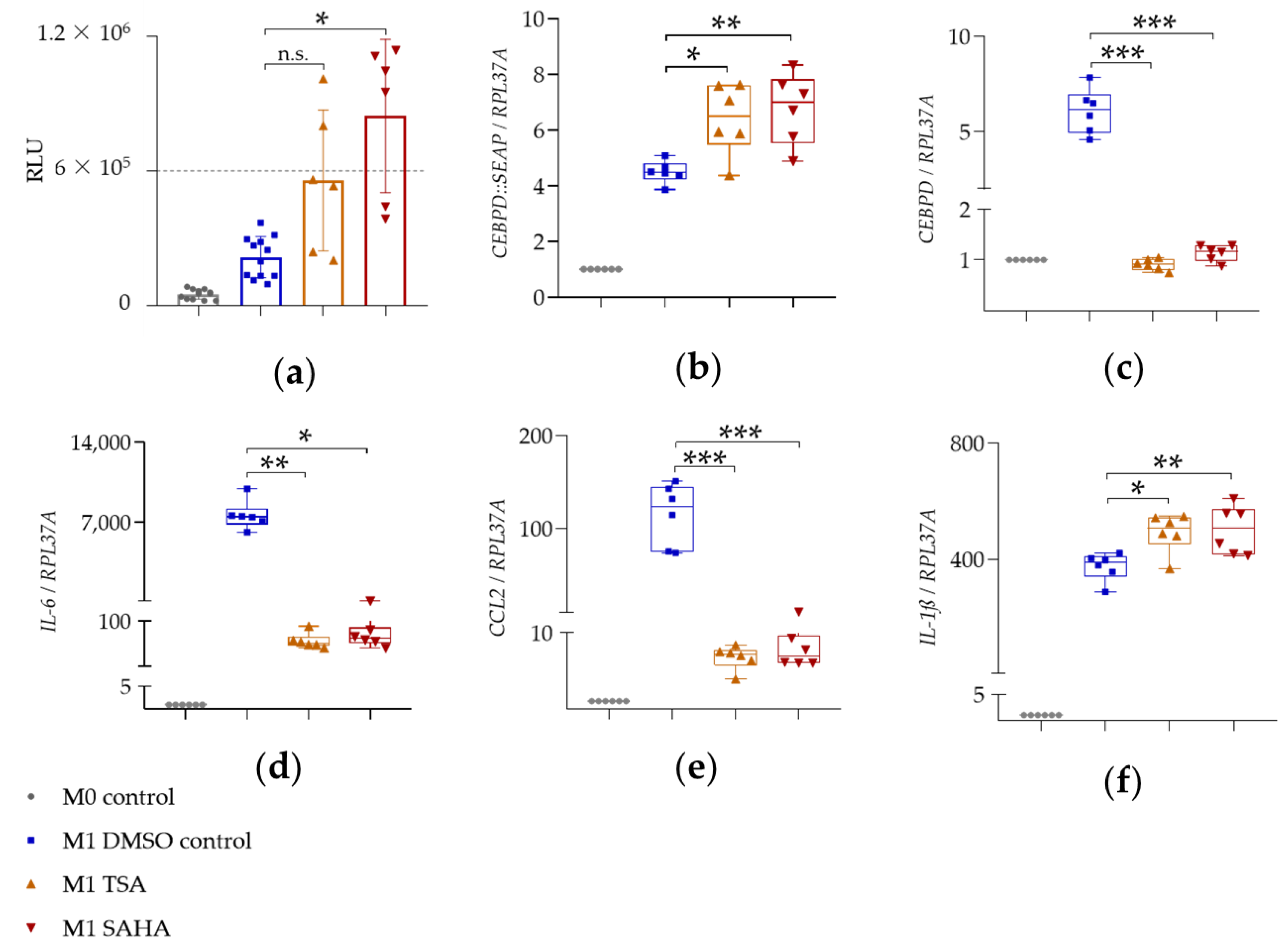
2.3.2. HDAC Inhibitors SAHA and TSA
3. Discussion
3.1. Hit Compounds Modulate Gene Expression in THP-1-Derived Reporter Mϕ
3.1.1. BET Inhibitor I-BET151
3.1.2. BET Inhibitor Ro 11-1464
3.1.3. HDAC Inhibitor SAHA
3.1.4. HDAC Inhibitor TSA
3.2. The Role of C/EBPδ TF in Gene Expression Regulation
4. Materials and Methods
4.1. Cloning
4.2. Cell Culture
4.3. Generation of HEK293T Reporter Cell Lines
4.4. Generation of THP-1 Reporter Cell Line
4.5. In Vitro PMA-Induced Differentiation
4.6. LPS and IFN-γ Treatment
4.7. Reagents and Compound Libraries
4.8. High-Throughput Screening
4.9. SEAP Assay
4.10. SEAP Assay Characterization Parameters
4.11. qRT-PCR Analysis
4.12. Live-Cell Imaging
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| apoA-I | apolipoprotein A-I |
| ATF3 | activating transcription factor 3 |
| BAC | bacterial artificial chromosome |
| BET | bromodomain and extraterminal domain |
| BRD | bromodomain |
| cAMP | cyclic adenosine monophosphate |
| CCL | CC-chemokine ligand |
| CEBPD | CCAAT/enhancer binding protein delta |
| CMV | cytomegalovirus |
| CREB | cAMP responsive element-binding protein |
| DMSO | dimethyl sulfoxide |
| HDAC | histone deacetylase |
| HTS | high-throughput screening |
| IFN | interferon |
| IL | interleukin |
| IRF-1 | interferon regulatory factor 1 |
| LPS | lipopolysaccharide |
| m | arithmetical mean |
| Mϕ | macrophages |
| n | number of individual experiments |
| NF-kB | nuclear factor-kappa B |
| PBS | phosphate buffered saline |
| PMA | phorbol 12-myristate 13-acetate |
| pTEFb | positive transcription elongation factor b |
| RLU | relative luminescence units |
| RPL37A | ribosomal protein L37a |
| SAHA | suberoylanilide hydroxamic acid |
| SD | standard deviation |
| SEAP | secreted alkaline phosphatase |
| SP1 | specificity protein 1 |
| STAT | signal transducer and activator of transcription |
| TLR | toll-like receptor |
| TNF | tumor necrosis factor |
| TSA | trichostatin A |
| TSS | transcription start site |
| UTR | untranslated region |
References
- Nau, G.J.; Richmond, J.F.; Schlesinger, A.; Jennings, E.G.; Lander, E.S.; Young, R.A. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. USA 2002, 99, 1503–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldrick, J.C.; Alizadeh, A.A.; Diehn, M.; Dudoit, S.; Liu, C.L.; Belcher, C.E.; Botstein, D.; Staudt, L.M.; Brown, P.O.; Relman, D.A. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc. Natl. Acad. Sci. USA 2002, 99, 972–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 9, 692–703. [Google Scholar] [CrossRef]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landschulz, W.H.; Johnson, P.F.; Adashi, E.Y.; Graves, B.J.; McKnight, S.L. Isolation of a recombinant copy of the gene encoding C/EBP. Genes Dev. 1988, 2, 786–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukada, J.; Yoshida, Y.; Kominato, Y.; Auron, P.E. The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine 2011, 54, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Pulido-Salgado, M.; Vidal-Taboada, J.M.; Saura, J. C/EBPβ and C/EBPδ transcription factors: Basic biology and roles in the CNS. Prog. Neurobiol. 2015, 132, 1–33. [Google Scholar] [CrossRef]
- Ko, C.Y.; Chang, W.C.; Wang, J.M. Biological roles of CCAAT/Enhancer-binding protein delta during inflammation. J. Biomed. Sci. 2015, 22, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tengku-Muhammad, T.S.; Hughes, T.R.; Ranki, H.; Cryer, A.; Ramji, D.P. Differential regulation of macrophage CCAAT-enhancer binding protein isoforms by lipopolysaccharide and cytokines. Cytokine 2000, 12, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Litvak, V.; Ramsey, S.A.; Rust, A.G.; Zak, D.E.; Kennedy, K.A.; Lampano, A.E.; Nykter, M.; Shmulevich, I.; Aderem, A. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat. Immunol. 2009, 10, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, S.; Akira, S.; Kishimoto, T. A member of the C/EBP family, NF-IL6 beta, forms a heterodimer and transcriptionally synergizes with NF-IL6. Proc. Natl. Acad. Sci. USA 1992, 89, 1473–1476. [Google Scholar] [CrossRef] [Green Version]
- Alam, T.; An, M.R.; Papaconstantinou, J. Differential expression of three C/EBP isoforms in multiple tissues during the acute phase response. J. Biol. Chem. 1992, 267, 5021–5024. [Google Scholar] [CrossRef]
- Cardinaux, J.R.; Allaman, I.; Magistretti, P.J. Pro-inflammatory cytokines induce the transcription factors C/EBPbeta and C/EBPdelta in astrocytes. Glia 2000, 29, 91–97. [Google Scholar] [CrossRef]
- Granger, R.L.; Hughes, T.R.; Ramji, D.P. Stimulus- and cell-type-specific regulation of CCAAT-enhancer binding protein isoforms in glomerular mesangial cells by lipopolysaccharide and cytokines. Biochim. Biophys. Acta Mol. Basis Dis. 2000, 1501, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.; Yang, S.Q.; Lin, H.Z.; Lane, M.D.; Chatterjee, S.; Diehl, A.M. Tumor necrosis factor alpha promotes nuclear localization of cytokine-inducible CCAAT/enhancer binding protein isoforms in hepatocytes. J. Biol. Chem. 1996, 271, 17974–17978. [Google Scholar] [CrossRef] [Green Version]
- Cantwell, C.A.; Sterneck, E.; Johnson, P.F. Interleukin-6-specific activation of the C/EBPdelta gene in hepatocytes is mediated by Stat3 and Sp1. Mol. Cell Biol. 1998, 18, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, D.C.; DeWille, J.W. C/EBPdelta is a downstream mediator of IL-6 induced growth inhibition of prostate cancer cells. Prostate 2005, 63, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.Y.; Wang, W.L.; Wang, S.M.; Chu, Y.Y.; Chang, W.C.; Wang, J.M. Glycogen synthase kinase-3beta-mediated CCAAT/enhancer-binding protein delta phosphorylation in astrocytes promotes migration and activation of microglia/macrophages. Neurobiol. Aging 2014, 35, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.Y.; Chang, L.H.; Lee, Y.C.; Sterneck, E.; Cheng, C.P.; Chen, S.H.; Huang, A.M.; Tseng, J.T.; Wang, J.M. CCAAT/enhancer binding protein delta (CEBPD) elevating PTX3 expression inhibits macrophage-mediated phagocytosis of dying neuron cells. Neurobiol. Aging 2012, 33, 422.e11–422.e25. [Google Scholar] [CrossRef]
- MacDougald, O.A.; Cornelius, P.; Lin, F.T.; Chen, S.S.; Lane, M.D. Glucocorticoids reciprocally regulate expression of the CCAAT/enhancer-binding protein alpha and delta genes in 3T3-L1 adipocytes and white adipose tissue. J. Biol. Chem. 1994, 269, 19041–19047. [Google Scholar] [CrossRef]
- Yang, H.; Mammen, J.; Wei, W.; Menconi, M.; Evenson, A.; Fareed, M.; Petkova, V.; Hasselgren, P.O. Expression and activity of C/EBPbeta and delta are upregulated by dexamethasone in skeletal muscle. J. Cell Physiol. 2005, 204, 219–226. [Google Scholar] [CrossRef]
- Hsiao, Y.-W.; Li, C.-F.; Chi, J.-Y.; Tseng, J.T.; Chang, Y.; Hsu, L.-J.; Lee, C.-H.; Chang, T.-H.; Wang, S.-M.; Wang, D.D.H.; et al. CCAAT/enhancer binding protein delta in macrophages contributes to immunosuppression and inhibits phagocytosis in nasopharyngeal carcinoma. Sci. Signal. 2013, 6, ra59. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Chang, W.; Centrella, M.; McCarthy, T.L. Activation domains of CCAAT enhancer binding protein delta: Regions required for native activity and prostaglandin E2-dependent transactivation of insulin-like growth factor I gene expression in rat osteoblasts. Mol. Endocrinol. 2003, 17, 1834–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Xie, N.; Cui, H.; Tan, Z.; Yang, S.; Icyuz, M.; Abraham, E.; Liu, G. MicroRNA let-7c regulates macrophage polarization. J. Immunol. 2013, 190, 6542–6549. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, V.V.; Mathison, J.C.; Schwamborn, K.; Mercurio, F.; Ulevitch, R.J. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J. Biol. Chem. 2003, 278, 26612–26619. [Google Scholar] [CrossRef] [Green Version]
- Balamurugan, K.; Sharan, S.; Klarmann, K.D.; Zhang, Y.; Coppola, V.; Summers, G.H.; Roger, T.; Morrison, D.K.; Keller, J.R.; Sterneck, E. FBXW7alpha attenuates inflammatory signalling by downregulating C/EBPdelta and its target gene Tlr4. Nat. Commun. 2013, 4, 1662. [Google Scholar] [CrossRef] [Green Version]
- Maitra, U.; Gan, L.; Chang, S.; Li, L. Low-dose endotoxin induces inflammation by selectively removing nuclear receptors and activating CCAAT/enhancer-binding protein δ. J. Immunol. 2011, 186, 4467–4473. [Google Scholar] [CrossRef]
- Poli, V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J. Biol. Chem. 1998, 273, 29279–29282. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.H.; Huang, H.S.; Wu, P.T.; Jou, I.M.; Pan, M.H.; Chang, W.C.; Wang, D.D.; Wang, J.M. Role of macrophage CCAAT/enhancer binding protein delta in the pathogenesis of rheumatoid arthritis in collagen-induced arthritic mice. PLoS ONE 2012, 7, e45378. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Ko, C.Y.; Chen, L.C.; Wang, W.L.; Chang, W.C. Functional role of NF-IL6beta and its sumoylation and acetylation modifications in promoter activation of cyclooxygenase 2 gene. Nucleic Acids Res. 2006, 34, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-W.; Chen, C.-C.; Tseng, H.-P.; Chang, W.-C. Lipopolysaccharide-induced transcriptional activation of interleukin-10 is mediated by MAPK-and NF-κB-induced CCAAT/enhancer-binding protein δ in mouse macrophages. Cell. Signal. 2006, 18, 1492–1500. [Google Scholar] [CrossRef]
- Balamurugan, K.; Sterneck, E. The many faces of C/EBPdelta and their relevance for inflammation and cancer. Int. J. Biol. Sci. 2013, 9, 917–933. [Google Scholar] [CrossRef] [Green Version]
- Moore, F.; Santin, I.; Nogueira, T.C.; Gurzov, E.N.; Marselli, L.; Marchetti, P.; Eizirik, D.L. The transcription factor C/EBP delta has anti-apoptotic and anti-inflammatory roles in pancreatic beta cells. PLoS ONE 2012, 7, e31062. [Google Scholar] [CrossRef] [Green Version]
- Rustenhoven, J.; Scotter, E.L.; Jansson, D.; Kho, D.T.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Faull, R.L.; Curtis, M.A.; Graham, S.E. An anti-inflammatory role for C/EBPδ in human brain pericytes. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Fu, Q.; Shah, S.K.; Melnyk, S.B.; Sterneck, E.; Hauer-Jensen, M.; Pawar, S.A. C/EBPδ protects from radiation-induced intestinal injury and sepsis by suppression of inflammatory and nitrosative stress. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Umek, R.M.; McKnight, S.L. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991, 5, 1538–1552. [Google Scholar] [CrossRef] [Green Version]
- Dearth, L.R.; DeWille, J. Posttranscriptional and posttranslational regulation of C/EBP delta in G0 growth-arrested mammary epithelial cells. J. Biol. Chem. 2003, 278, 11246–11255. [Google Scholar] [CrossRef] [Green Version]
- Balamurugan, K.; Wang, J.M.; Tsai, H.H.; Sharan, S.; Anver, M.; Leighty, R.; Sterneck, E. The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. EMBO J. 2010, 29, 4106–4117. [Google Scholar] [CrossRef] [Green Version]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Huang, Y.C.; Chang, W.C.; Su, J.G.; Cai, J.L.; Chen, C.C.; Hung, J.J.; Liu, Y.W. Peptidoglycan enhances transcriptional expression of CCAAT/enhancer-binding protein delta gene in mouse macrophages. J. Biomed. Sci. 2007, 14, 407–418. [Google Scholar] [CrossRef]
- Liu, Y.-W.; Chen, C.-C.; Wang, J.-M.; Chang, W.-C.; Huang, Y.-C.; Chung, S.-Y.; Chen, B.-K.; Hung, J.-J. Role of transcriptional factors Sp1, c-Rel, and c-Jun in LPS-induced C/EBPδ gene expression of mouse macrophages. Cell. Mol. Life Sci. 2007, 64, 3282–3294. [Google Scholar] [CrossRef]
- Sivko, G.S.; Sanford, D.C.; Dearth, L.D.; Tang, D.; DeWille, J.W. CCAAT/Enhancer binding protein delta (c/EBPdelta) regulation and expression in human mammary epithelial cells: II. Analysis of activating signal transduction pathways, transcriptional, post-transcriptional, and post-translational control. J. Cell Biochem. 2004, 93, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sif, S.; DeWille, J. The mouse C/EBPdelta gene promoter is regulated by STAT3 and Sp1 transcriptional activators, chromatin remodeling and c-Myc repression. J. Cell Biochem. 2007, 102, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Tobita, K.; Osada, S.; Nishihara, T.; Imagawa, M. CCAAT/enhancer-binding protein delta gene expression is mediated by APRF/STAT3. J. Biochem. 1997, 121, 731–738. [Google Scholar] [CrossRef]
- Ali, S.; Singh, N.N.; Yildirim, H.; Ramji, D.P. Requirement for nuclear factor kappa B signalling in the interleukin-1-induced expression of the CCAAT/enhancer binding protein-delta gene in hepatocytes. Int. J. Biochem. Cell Biol. 2010, 42, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.; Hauber, J.; Hauber, R.; Geiger, R.; Cullen, B.R. Secreted placental alkaline phosphatase: A powerful new quantitative indicator of gene expression in eukaryotic cells. Gene 1988, 66, 1–10. [Google Scholar] [CrossRef]
- Bettan, M.; Darteil, R.; Scherman, D. Secreted human placental alkaline phosphatase as a reporter gene for in vivo gene transfer. Anal. Biochem. 1999, 271, 187–189. [Google Scholar] [CrossRef]
- Schlatter, S.; Rimann, M.; Kelm, J.; Fussenegger, M. SAMY, a novel mammalian reporter gene derived from Bacillus stearothermophilus alpha-amylase. Gene 2002, 282, 19–31. [Google Scholar] [CrossRef]
- Cullen, B.R.; Malim, M.H. Secreted placental alkaline phosphatase as a eukaryotic reporter gene. Methods Enzymol. 1992, 216, 362–368. [Google Scholar] [PubMed]
- Sui, Y.; Wu, Z. Alternative statistical parameter for high-throughput screening assay quality assessment. J. Biomol. Screen. 2007, 12, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewings, D.S.; Wang, M.; Philpott, M.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Vuppusetty, C.; Marsden, B.; Knapp, S. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011, 54, 6761–6770. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Stefik, D.; Vranic, V.; Ivkovic, N.; Abazovic, D.; Maric, D.; Vojvodic, D.; Supic, G. An insight into osteoarthritis susceptibility: Integration of immunological and genetic background. Bosn. J. Basic Med. Sci. 2020, 1-8. [Google Scholar] [CrossRef]
- Latourte, A.; Kloppenburg, M.; Richette, P. Emerging pharmaceutical therapies for osteoarthritis. Nat. Rev. Rheumatol. 2020, 16, 673–688. [Google Scholar] [CrossRef]
- Monaco, S.; Nicholas, R.; Reynolds, R.; Magliozzi, R. Intrathecal Inflammation in Progressive Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 8217. [Google Scholar] [CrossRef]
- Wang, A.; Rojas, O.; Lee, D.; Gommerman, J.L. Regulation of neuroinflammation by B cells and plasma cells. Immunol. Rev. 2020, 299, 1–16. [Google Scholar] [CrossRef]
- Laria, A.; Lurati, A.; Marrazza, M.; Mazzocchi, D.; Re, K.A.; Scarpellini, M. The macrophages in rheumatic diseases. J. Inflamm. Res. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X. Impact of Macrophages in Atherosclerosis. Curr. Med. Chem. 2016, 23, 1926–1937. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Hanke, T.; Merk, D.; Steinhilber, D.; Geisslinger, G.; Schubert-Zsilavecz, M. Small molecules with anti-inflammatory properties in clinical development. Pharmacol. Ther. 2016, 157, 163–187. [Google Scholar] [CrossRef]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef]
- Saad, J.; Pellegrini, M.V. Nonsteroidal Anti-Inflammatory Drugs (NSAID) Toxicity. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2019. [Google Scholar]
- Taniguchi, Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seal, J.; Lamotte, Y.; Donche, F.; Bouillot, A.; Mirguet, O.; Gellibert, F.; Nicodeme, E.; Krysa, G.; Kirilovsky, J.; Beinke, S. Identification of a novel series of BET family bromodomain inhibitors: Binding mode and profile of I-BET151 (GSK1210151A). Bioorg. Med. Chem. Lett. 2012, 22, 2968–2972. [Google Scholar] [CrossRef] [PubMed]
- Mirguet, O.; Lamotte, Y.; Donche, F.; Toum, J.; Gellibert, F.; Bouillot, A.; Gosmini, R.; Nguyen, V.-L.; Delannée, D.; Seal, J. From ApoA1 upregulation to BET family bromodomain inhibition: Discovery of I-BET151. Bioorg. Med. Chem. Lett. 2012, 22, 2963–2967. [Google Scholar] [CrossRef] [PubMed]
- de Bont, N.; Netea, M.G.; Rovers, C.; Smilde, T.; Demacker, P.N.; van der Meer, J.W.; Stalenhoef, A.F. LPS-induced cytokine production and expression of LPS-receptors by peripheral blood mononuclear cells of patients with familial hypercholesterolemia and the effect of HMG-CoA reductase inhibitors. Atherosclerosis 1998, 139, 147–152. [Google Scholar] [CrossRef]
- Barrett, E.; Brothers, S.; Wahlestedt, C.; Beurel, E. I-BET151 selectively regulates IL-6 production. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1549–1555. [Google Scholar] [CrossRef] [Green Version]
- Klein, K.; Kabala, P.A.; Grabiec, A.M.; Gay, R.E.; Kolling, C.; Lin, L.-L.; Gay, S.; Tak, P.P.; Prinjha, R.K.; Ospelt, C. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann. Rheum. Dis. 2016, 75, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Maksylewicz, A.; Bysiek, A.; Lagosz, K.B.; Macina, J.M.; Kantorowicz, M.; Bereta, G.; Sochalska, M.; Gawron, K.; Chomyszyn-Gajewska, M.; Potempa, J. BET bromodomain inhibitors suppress inflammatory activation of gingival fibroblasts and epithelial cells from periodontitis patients. Front. Immunol. 2019, 10, 933. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.H.; Fang, C.; Qiao, Y.; Yarilina, A.; Prinjha, R.K.; Ivashkiv, L.B. BET bromodomain inhibition suppresses transcriptional responses to cytokine-Jak-STAT signaling in a gene-specific manner in human monocytes. Eur. J. Immunol. 2015, 45, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Belkina, A.C.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 beta—A friend or foe in malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgila, K.; Vyrla, D.; Drakos, E. Apolipoprotein AI (ApoA-I), immunity, inflammation and cancer. Cancers 2019, 11, 1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempen, H. Use of a Thienotriazolodiazephine to Increase Apolipoprotein A-I Levels. International Patent Application No. WO1997009048A1, 29 December 1998. [Google Scholar]
- Kempen, H.J.; Bellus, D.; Fedorov, O.; Nicklisch, S.; Filippakopoulos, P.; Picaud, S.; Knapp, S. Stimulation of Hepatic Apolipoprotein AI Production by Novel Thieno-Triazolodiazepines: Roles of the Classical Benzodiazepine Receptor, PAF Receptor, and Bromodomain Binding; SAGE Publications: London, UK, 2013. [Google Scholar]
- Chen, W.; Wu, Y.; Lu, Q.; Wang, S.; Xing, D. Endogenous ApoA-I expression in macrophages: A potential target for protection against atherosclerosis. Clin. Chim. Acta 2020, 505, 55–59. [Google Scholar] [CrossRef]
- Belkina, A.C.; Nikolajczyk, B.S.; Denis, G.V. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J. Immunol. 2013, 190, 3670–3678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogilenko, D.A.; Orlov, S.V.; Trulioff, A.S.; Ivanov, A.V.; Nagumanov, V.K.; Kudriavtsev, I.V.; Shavva, V.S.; Tanyanskiy, D.A.; Perevozchikov, A.P. Endogenous apolipoprotein AI stabilizes ATP-binding cassette transporter A1 and modulates Toll-like receptor 4 signaling in human macrophages. FASEB J. 2012, 26, 2019–2030. [Google Scholar] [CrossRef]
- Bursill, C.A.; Castro, M.L.; Beattie, D.T.; Nakhla, S.; van der Vorst, E.; Heather, A.K.; Barter, P.J.; Rye, K.-A. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1773–1778. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, S.; Chen, J.; Yu, Z. Histone deacetylases (HDACs) guided novel therapies for T-cell lymphomas. Int. J. Med. Sci. 2019, 16, 424. [Google Scholar] [CrossRef] [Green Version]
- Leoni, F.; Zaliani, A.; Bertolini, G.; Porro, G.; Pagani, P.; Pozzi, P.; Dona, G.; Fossati, G.; Sozzani, S.; Azam, T.; et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. USA 2002, 99, 2995–3000. [Google Scholar] [CrossRef] [Green Version]
- Lohman, R.J.; Iyer, A.; Fairlie, T.J.; Cotterell, A.; Gupta, P.; Reid, R.C.; Vesey, D.A.; Sweet, M.J.; Fairlie, D.P. Differential Anti-inflammatory Activity of HDAC Inhibitors in Human Macrophages and Rat Arthritis. J. Pharmacol. Exp. Ther. 2016, 356, 387–396. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Weiss, U.; Möller, M.; Husseini, S.A.; Manderscheid, C.; Häusler, J.; Geisslinger, G.; Niederberger, E. Inhibition of HDAC Enzymes Contributes to Differential Expression of Pro-Inflammatory Proteins in the TLR-4 Signaling Cascade. Int. J. Mol. Sci. 2020, 21, 8943. [Google Scholar] [CrossRef] [PubMed]
- Matsusaka, T.; Fujikawa, K.; Nishio, Y.; Mukaida, N.; Matsushima, K.; Kishimoto, T.; Akira, S. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc. Natl. Acad. Sci. USA 1993, 90, 10193–10197. [Google Scholar] [CrossRef] [Green Version]
- Weiss, H.J.; O’Neill, L.A. Bridging the gap—A new role for STAT3 in TLR4-mediated metabolic reprogramming. Immunol. Cell Biol. 2021, 99, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, R.; Bres, V.; Ng, R.W.; Coudart, M.P.; El Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Choo, Q.Y.; Ho, P.C.; Tanaka, Y.; Lin, H.S. Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology 2010, 49, 1447–1460. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, K.; Park, D.; Lee, E.; Lee, H.; Lee, Y.S.; Choe, J.; Jeoung, D. Histone deacetylase 3 mediates allergic skin inflammation by regulating expression of MCP1 protein. J. Biol. Chem. 2012, 287, 25844–25859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Telles, E.; Karl, M.; Cheng, F.; Luetteke, N.; Sotomayor, E.M.; Miller, R.H.; Seto, E. Loss of HDAC11 ameliorates clinical symptoms in a multiple sclerosis mouse model. Life Sci. Alliance 2018, 1, e201800039. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Luesch, H. Discovery and mechanism of natural products as modulators of histone acetylation. Curr. Drug Targets 2012, 13, 1029–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar] [PubMed]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. Biomed. Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Han, S.B.; Lee, J.K. Anti-inflammatory effect of Trichostatin-A on murine bone marrow-derived macrophages. Arch. Pharm. Res. 2009, 32, 613–624. [Google Scholar] [CrossRef]
- Aung, H.T.; Schroder, K.; Himes, S.R.; Brion, K.; van Zuylen, W.; Trieu, A.; Suzuki, H.; Hayashizaki, Y.; Hume, D.A.; Sweet, M.J.; et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 2006, 20, 1315–1327. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, F.; Li, X.; Wang, L.; Chu, X.; Zhang, H.; Gong, Z. Trichostatin A inhibits inflammation in phorbol myristate acetateinduced macrophages by regulating the acetylation of histone and/or nonhistone proteins. Mol. Med. Rep. 2016, 13, 845–852. [Google Scholar] [CrossRef]
- Hu, X.L.; Zhang, X.; Li, Q.; Qiu, S.F.; Mei, R.H. Effects of trichostatin A on the expressions of inflammatory cytokines and toll-like receptor 4 and the acetylation of nuclear factor-κB induced by lipopolysaccharide in macrophage. Acta Physiol. Sin. 2012, 64, 651–656. [Google Scholar]
- Laribee, R.N.; Klemsz, M.J. Loss of PU.1 expression following inhibition of histone deacetylases. J. Immunol. 2001, 167, 5160–5166. [Google Scholar] [CrossRef] [Green Version]
- Hepp, M.I.; Escobar, D.; Farkas, C.; Hermosilla, V.E.; Alvarez, C.; Amigo, R.; Gutierrez, J.L.; Castro, A.F.; Pincheira, R. A Trichostatin A (TSA)/Sp1-mediated mechanism for the regulation of SALL2 tumor suppressor in Jurkat T cells. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 623–636. [Google Scholar] [CrossRef]
- Hu, H.-M.; Baer, M.; Williams, S.C.; Johnson, P.F.; Schwartz, R.C. Redundancy of C/EBPα,-β, and-δ in supporting the lipopolysaccharide-induced transcription of IL-6 and monocyte chemoattractant protein-1. J. Immunol. 1998, 160, 2334–2342. [Google Scholar]
- Descombes, P.; Schibler, U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 1991, 67, 569–579. [Google Scholar] [CrossRef]
- Parkin, S.E.; Baer, M.; Copeland, T.D.; Schwartz, R.C.; Johnson, P.F. Regulation of CCAAT/enhancer-binding protein (C/EBP) activator proteins by heterodimerization with C/EBPgamma (Ig/EBP). J. Biol. Chem. 2002, 277, 23563–23572. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Habener, J.F. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992, 6, 439–453. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.L.; Lee, Y.C.; Yang, W.M.; Chang, W.C.; Wang, J.M. Sumoylation of LAP1 is involved in the HDAC4-mediated repression of COX-2 transcription. Nucleic Acids Res. 2008, 36, 6066–6079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chang, L.; Chen, C.; Zhang, M.; Luo, Y.; Hamblin, M.; Villacorta, L.; Xiong, J.W.; Chen, Y.E.; Zhang, J.; et al. Rad GTPase inhibits cardiac fibrosis through connective tissue growth factor. Cardiovasc. Res. 2011, 91, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezy, O.; Elabd, C.; Cochet, O.; Petersen, R.K.; Kristiansen, K.; Dani, C.; Ailhaud, G.; Amri, E.Z. Delta-interacting protein A, a new inhibitory partner of CCAAT/enhancer-binding protein beta, implicated in adipocyte differentiation. J. Biol. Chem. 2005, 280, 11432–11438. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Kim, I.; Lye, E.; Shen, F.; Suzuki, N.; Suzuki, S.; Gerondakis, S.; Akira, S.; Gaffen, S.L.; Yeh, W.C.; et al. Differential role for c-Rel and C/EBPbeta/delta in TLR-mediated induction of proinflammatory cytokines. J. Immunol. 2009, 182, 7212–7221. [Google Scholar] [CrossRef]
- Yan, C.; Johnson, P.F.; Tang, H.; Ye, Y.; Wu, M.; Gao, H. CCAAT/enhancer-binding protein delta is a critical mediator of lipopolysaccharide-induced acute lung injury. Am. J. Pathol. 2013, 182, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Slofstra, S.H.; Groot, A.P.; Obdeijn, M.H.P.; Reitsma, P.H.; Cate, H.T.; Spek, C.A. Gene expression profiling identifies C/EBPdelta as a candidate regulator of endotoxin-induced disseminated intravascular coagulation. Am. J. Respir. Crit. Care Med. 2007, 176, 602–609. [Google Scholar] [CrossRef]
- Gatla, H.R.; Muniraj, N.; Thevkar, P.; Yavvari, S.; Sukhavasi, S.; Makena, M.R. Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Knethen, A.; Brüne, B. Histone deacetylation inhibitors as therapy concept in sepsis. Int. J. Mol. Sci. 2019, 20, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osoegawa, K.; Woon, P.Y.; Zhao, B.; Frengen, E.; Tateno, M.; Catanese, J.J.; de Jong, P.J. An improved approach for construction of bacterial artificial chromosome libraries. Genomics 1998, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, H.; Feinweber, C.; Luckhardt, S.; Linke, B.; Resch, E.; Geisslinger, G.; Weigert, A.; Parnham, M.J. THP-1 and human peripheral blood mononuclear cell-derived macrophages differ in their capacity to polarize in vitro. Mol. Immunol. 2017, 88, 58–68. [Google Scholar] [CrossRef]
- Maess, M.B.; Sendelbach, S.; Lorkowski, S. Selection of reliable reference genes during THP-1 monocyte differentiation into macrophages. BMC Mol. Biol. 2010, 11, 90. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Intra-Assay CV (%) | Inter-Assay CV (%) |
|---|---|---|
| SEAP-myc, undiluted | 2.00 | 0.84 |
| SEAP-myc, 1:10-diluted | 3.33 | 4.16 |
| SEAP-myc, 1:100-diluted | 2.67 | 2.62 |
| SEAP-myc, 1:1000-diluted | 5.33 | 2.71 |
| SEAP-myc, 1:10,000-diluted | 15.67 | 24.41 |
| Gene | Condition | 4 h, Mean ± SD |
|---|---|---|
| CEBPD::SEAP | M1 DMSO | 5.3 ± 1.4 |
| M1 I-BET151 | 9.6 ± 2.0 | |
| CEBPD | M1 DMSO | 4.8 ± 1.0 |
| M1 I-BET151 | 7.5 ± 1.1 | |
| IL-6 | M1 DMSO | 6625.0 ± 1045.0 |
| M1 I-BET151 | 102.3 ± 118.1 | |
| CCL2 | M1 DMSO | 187.7 ± 105.4 |
| M1 I-BET151 | 11.5 ± 3.3 | |
| IL-1ß | M1 DMSO | 396.0 ± 62.4 |
| M1 I-BET151 | 927.1 ± 124.2 |
| Gene | Condition | 4 h, Mean ± SD |
|---|---|---|
| CEBPD::SEAP | M1 DMSO | 5.5 ± 1.5 |
| M1 Ro 11-1464 | 8.8 ± 3.4 | |
| CEBPD | M1 DMSO | 6.3 ± 2.8 |
| M1 Ro 11-1464 | 12.4 ± 5.5 | |
| IL-6 | M1 DMSO | 12,432.0 ± 10092.0 |
| M1 Ro 11-1464 | 1364.0 ± 1426.0 | |
| CCL2 | M1 DMSO | 237.2 ± 82.8 |
| M1 Ro 11-1464 | 26.6 ± 9.0 | |
| IL-1ß | M1 DMSO | 344.8 ± 119.0 |
| M1 Ro 11-1464 | 445.3 ± 138.6 |
| Gene | Condition | 4 h, Mean ± SD |
|---|---|---|
| CEBPD::SEAP | M1 DMSO | 4.5 ± 0.4 |
| M1 TSA | 6.4 ± 1.3 | |
| M1 SAHA | 6.8 ± 1.3 | |
| CEBPD | M1 DMSO | 6.1 ± 1.2 |
| M1 TSA | 0.9 ± 0.1 | |
| M1 SAHA | 1.1 ± 0.2 | |
| IL-6 | M1 DMSO | 7617.0 ± 1258.0 |
| M1 TSA | 58.1 ± 16.1 | |
| M1 SAHA | 68.7 ± 20.1 | |
| CCL2 | M1 DMSO | 114.9 ± 33.4 |
| M1 TSA | 6.8 ± 1.5 | |
| M1 SAHA | 7.6 ± 1.9 | |
| IL-1ß | M1 DMSO | 374.3 ± 47.9 |
| M1 TSA | 492.5 ± 67.1 | |
| M1 SAHA | 501.9 ± 83.6 |
| Gene Name | GenBank Accession Number | Primer Sequences | Reference |
|---|---|---|---|
| CEBPD | NM_005195.4 | Forward: 5′-CAG CAA CGA CCC ATA CCT CA-3′ | this study |
| Reverse: 5′-TCT TTG CGC TCC TAT GTC CC-3′ | |||
| CEBPD::SEAP | U89937 | Forward: 5′-GAG ATG AGT TTT TGT TCA CCC G-3′ | this study |
| Reverse: 5′-GAC CTT CAT AGC GCA CGT CA-3′ | |||
| RPL37A | NM_000998.5 | Forward: 5′-CTC GTC CGC CTA ATA CCG C-3′ | this study |
| Reverse: 5′-TAC CGA CGA TCC CGA CTT TC-3′ | |||
| IL-6 | NM_000600.5 | Forward: 5′-GTG TGA AAG CAG CAA AGA GGC-3′ | [120] |
| Reverse: 5′-TCT GTT CTG GAG GTA CTC TAG GTA T-3′ | |||
| IL-1ß | NM_000576.3 | Forward: 5′-GTG GCA ATG AGG ATG ACT TGT TCT-3′ | [120] |
| Reverse: 5′-TGT AGT GGT GGT CGG AGA TTC G-3′ | |||
| CCL2 | NM_002982.4 | Forward: 5′-AAA CTG AAG CTC GCA CTC TCG C-3′ | [120] |
| Reverse: 5′-AGG TGA CTG GGG CAT TGA TTG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullmann, T.; Luckhardt, S.; Wolf, M.; Parnham, M.J.; Resch, E. High-Throughput Screening for CEBPD-Modulating Compounds in THP-1-Derived Reporter Macrophages Identifies Anti-Inflammatory HDAC and BET Inhibitors. Int. J. Mol. Sci. 2021, 22, 3022. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063022
Ullmann T, Luckhardt S, Wolf M, Parnham MJ, Resch E. High-Throughput Screening for CEBPD-Modulating Compounds in THP-1-Derived Reporter Macrophages Identifies Anti-Inflammatory HDAC and BET Inhibitors. International Journal of Molecular Sciences. 2021; 22(6):3022. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063022
Chicago/Turabian StyleUllmann, Tatjana, Sonja Luckhardt, Markus Wolf, Michael J. Parnham, and Eduard Resch. 2021. "High-Throughput Screening for CEBPD-Modulating Compounds in THP-1-Derived Reporter Macrophages Identifies Anti-Inflammatory HDAC and BET Inhibitors" International Journal of Molecular Sciences 22, no. 6: 3022. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063022