
If Virchow and Ehrlich Had Dreamt Together: What the Future Holds for KRAS-Mutant Lung Cancer



1
Dana-Farber Cancer Institute, Department of Medical Oncology, Harvard Medical School, Boston, MA 02215, USA
2
Belfer Center for Applied Cancer Sciences, Boston, MA 02215, USA
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(6), 3025; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063025
Submission received: 31 January 2021
/
Revised: 8 March 2021
/
Accepted: 11 March 2021
/
Published: 16 March 2021
(This article belongs to the Special Issue Precision Oncology in Non-small Cell Lung Cancer)
Abstract
:Non-small-cell lung cancer (NSCLC) with Kirsten rat sarcoma (KRAS) mutations has notoriously challenged oncologists and researchers for three notable reasons: (1) the historical assumption that KRAS is “undruggable”, (2) the disease heterogeneity and (3) the shaping of the tumor microenvironment by KRAS downstream effector functions. Better insights into KRAS structural biochemistry allowed researchers to develop direct KRAS(G12C) inhibitors, which have shown early signs of clinical activity in NSCLC patients and have recently led to an FDA breakthrough designation for AMG-510. Following the approval of immune checkpoint inhibitors for PDL1-positive NSCLC, this could fuel yet another major paradigm shift in the treatment of advanced lung cancer. Here, we review advances in our understanding of the biology of direct KRAS inhibition and project future opportunities and challenges of dual KRAS and immune checkpoint inhibition. This strategy is supported by preclinical models which show that KRAS(G12C) inhibitors can turn some immunologically “cold” tumors into “hot” ones and therefore could benefit patients whose tumors harbor subtype-defining STK11/LKB1 co-mutations. Forty years after the discovery of KRAS as a transforming oncogene, we are on the verge of approval of the first KRAS-targeted drug combinations, thus therapeutically unifying Paul Ehrlich’s century-old “magic bullet” vision with Rudolf Virchow’s cancer inflammation theory.
1. Introduction
In 1900, the German Nobel laureate Paul Ehrlich suggested a concept of “magic bullets” (“Zauberkugeln”) to specifically target invading microbes, a concept that was subsequently adapted to describe highly specific, oncogene-targeted cancer treatments [1]. More than 100 years later, non-small-cell lung cancer (NSCLC) with activating mutations of the Kirsten rat sarcoma (KRAS) oncogene—despite representing almost one-third of all lung cancer cases—remains a tumor entity for which no fully FDA- or EMA-approved oncogene-targeted therapies exist (for a broader overview of the frequencies of known oncogenic driver events in NSCLC we refer to [2,3,4,5,6]). Accordingly, affected patients still face a dismal prognosis [7,8,9,10]. In contrast to clinically approved oncogene-targeted therapies for various other malignancies, e.g., imatinib for BCR/ABL-positive chronic myeloid leukemia (CML) or EGFR and ALK inhibitors for EGFR-mutant and EML4/ALK-rearranged NSCLC, respectively [11,12,13,14], the development of a “magic bullet” against mutant KRAS has remarkably challenged scientists and physicians alike because it had long been considered “undruggable” due to biochemistry constraints [15].
Another reason for the aggressive behavior and difficulty in treating KRAS-mutant lung cancer is its highly inflammatory phenotype [16]. The first to postulate a connection between chronic inflammation and cancer in the 19th century was Rudolf Virchow, a pathologist at Berlin‘s Charité hospital. He had observed the presence of leucocytes (“lymphoreticular infiltrate”) in neoplastic tissues [17,18]. Unfortunately, in the century to follow, our mechanistic understanding of the bidirectional interaction between epithelial cancer and immune cells remained incomplete, and the molecular mechanisms that make inflammatory processes an important cofactor in carcinogenesis, tumor maintenance and metastasis have only recently begun to unravel [19,20].
2. KRAS-Mutant NSCLC: Therapeutically Challenging with a Plethora of Tumor Biologies
The transforming function of mutant RAS was first described by Mariano Barbacid and colleagues in 1982, and the case of a lung cancer patient with an activating KRAS mutation was published only two years later [21,22]. Since then, mutant KRAS has been identified as an important oncogenic driver for various types of solid malignancies (e.g., NSCLC, pancreatic and colorectal cancer) [23] that promotes cancer initiation, maintenance and progression in genetically engineered mouse models (GEMMs) [24,25,26]. With the general recognition of oncogene- over histology-driven tumor vulnerabilities in the early 2000s, pan-cancer sequencing efforts revealed a tissue-context-dependent distribution of mutational subtypes, with KRAS(G12C) being the most frequent mutation in NSCLC (45% of all RAS mutations), followed by KRAS(G12V) (~20%) and KRAS(G12D) (~10%) [23,27].
KRAS is a small GTPase that, if mutated, has a reduced ability to hydrolyze GTP or to interact with GTPase-activating proteins (GAPs). This locks KRAS in a GTP-bound, active state and promotes cancer cell growth and apoptosis resistance [28,29,30]. Overall, lung cancers with KRAS mutations are characterized by a marked disease heterogeneity: KRAS mutational isoforms differ in their biochemical properties to hydrolyze GTP and to activate downstream signaling pathways, which determines differences in their biological behavior and therapeutic vulnerabilities [31,32,33]. Furthermore, the presence of a wild-type KRAS allele affects the transforming potential of mutant KRAS through dimerization and impairs MEK inhibitor sensitivity [34]. Cancer cells and tumors also have variable degrees of KRAS dependency [35,36], and the effects of mutant KRAS on cellular reprogramming are tissue-context-dependent [37,38]. Finally, approximately 30% of KRAS-mutant tumors harbor subclass-defining co-mutations in TP53, STK11/LKB1 (the latter resulting in loss of LKB1 function) and other genes with emerging clinical and therapeutic relevance [39,40,41,42,43] (summarized in Table 1).
Etiologically, KRAS-mutant lung tumors are associated with a current or former smoking history (mostly KRAS(G12C)), but KRAS mutations are also found with a different mutational spectrum (mostly KRAS(G12D)) in up to 15% of never-smokers who develop lung cancer [44,45,46]. Most commonly, mutations are located in codon 12 (~86%), less frequently in codons 13 (~9%) and 61 (~5%) [7,47], and they occur in ~30% of adeno- and ~5% of squamous-cell carcinomas [40,48,49], even though more refined pathological analyses question the presence of KRAS mutations in tumors with pure squamous cell histology [50].
Despite some uncertainty regarding the prognostic impact of KRAS mutations due to the confounding effects of co-occurring genetic events (e.g., mutations in STK11 or KEAP1), many studies suggest a more aggressive tumor biology (frequent brain/central nervous system metastases), therapeutic resistance and poorer overall survival for affected lung cancer patients compared to those with other genotypes [7,8,9,10,42,51,52,53,54].
The major caveat of pharmacologically inhibiting mutant KRAS had long been its high intrinsic affinity for abundant cellular GTP and the limited spatial access for small molecules to inhibit the switch-II pocket in its “OFF” state [15]. Other reasons that render KRAS a challenging oncogene from a therapeutic point of view are its role as a nexus of multiple downstream (MAPK, PI3K/AKT/mTOR and CDK4/6-RB) and upstream (ErbB family members, FGFR, IGFR) signaling pathways as well as the high grade of adaptational plasticity between different effector pathways [55,56,57,58,59]. Past clinical trials that have focused on targeting these effector pathways were therefore largely unsuccessful. MEK inhibitors administered on an uninterrupted schedule exhibited gastrointestinal tract- and skin-related toxicities and showed poor antitumor activity in humans despite having some activity in preclinical models [60,61]. Abemaciclib—a CDK4/6 inhibitor—also had only limited single-agent activity [62], and MEK/PI3K inhibitor combinations caused significant toxicity in humans; dose-limiting toxicities included oral mucositis, acneiform rash, hypertension, diarrhea and liver enzyme changes [63,64]. Hence, for a long time, cytotoxic chemotherapy remained the mainstay of treatment that could achieve some, but mostly short-lived, tumor control [8,54]. Therapeutic efforts have recently focused more on ERK inhibitors (e.g., GDC0994 or LY3214996) or ERK-inhibitor-based drug combinations (e.g., combined with PI3K/mTOR or CDK4/6 inhibitors), since ERK1/2 proteins are considered to have a bottleneck function in transmitting mitogenic signals and preventing MAPK pathway feedback reactivation [65,66,67,68]. These drug combinations are effective in preclinical models if applied on intermittent treatment schedules, but future clinical trials will have to clarify if this approach can overcome therapeutic limitations and toxicities observed with continuous MEK inhibition.

{kind=link}
{kind=link}
Table 1.
Factors contributing to the disease heterogeneity of KRAS-mutant non-small-cell lung cancer (sources).
Table 1.
Factors contributing to the disease heterogeneity of KRAS-mutant non-small-cell lung cancer (sources).
|
3. Mutant KRAS Proteins Orchestrate the Tumor Microenvironment
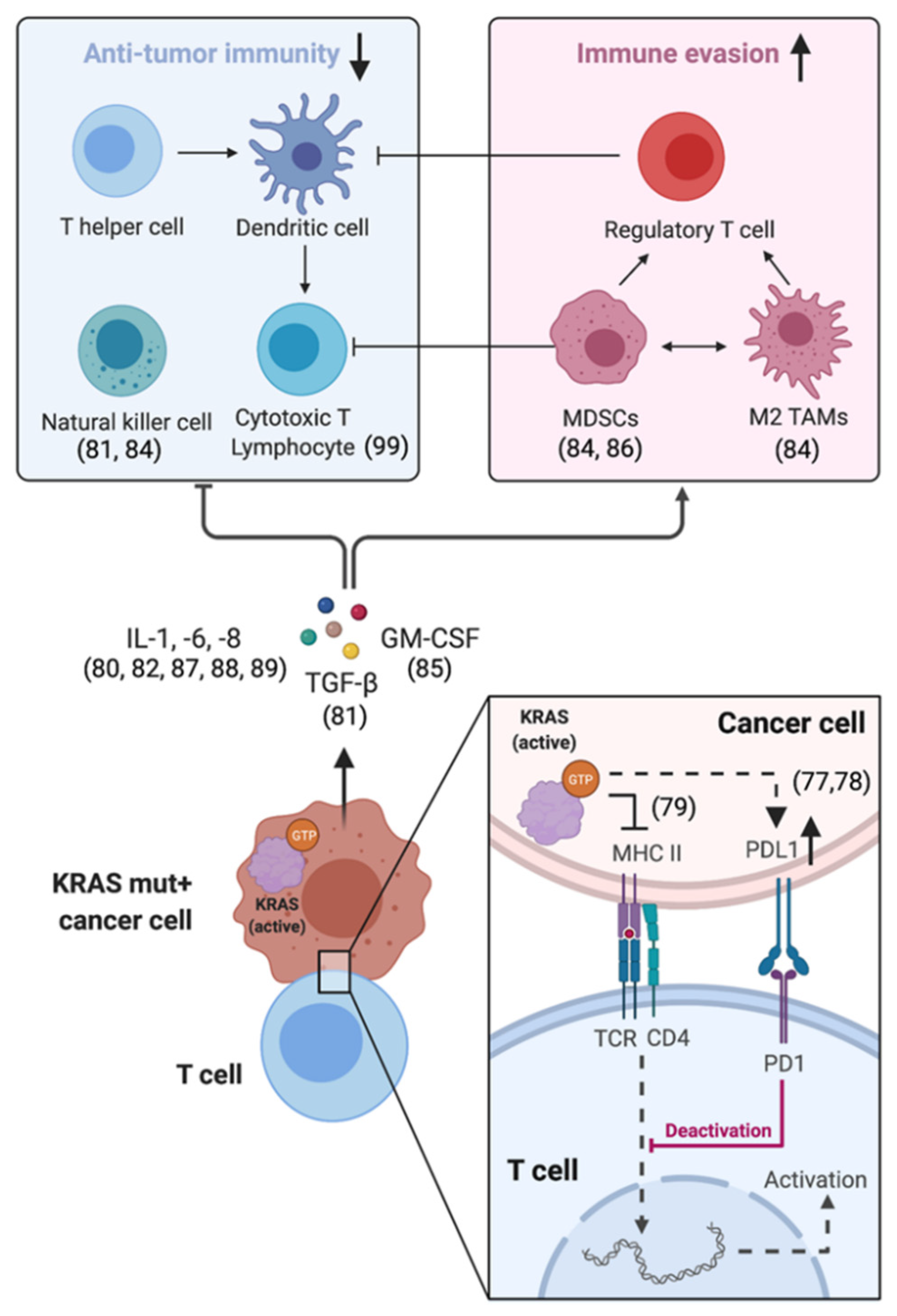
The abilities of cancer cells to promote local inflammation and to simultaneously escape immune-mediated elimination are important cancer hallmarks [76]. The tumor microenvironment (TME) represents an intricate ecosystem composed of multiple noncellular and cellular components including stroma and immune cells. Cancer cells actively shape the composition and functionality of the TME by direct cell-to-cell interactions and/or by chemokine secretion. Mutant KRAS proteins play a central role in this process. KRAS-dependent effector functions increase the expression of so-called immune checkpoints like programmed death ligand-1 (PDL1), which by binding to PD1 prevents T cells from killing cancer cells [77,78]. They also restrict cancer-cell-intrinsic MHC class II expression—essential for the recognition of cancer cells by T cells [79] and impair T-cell effector functions and antitumor immunity via cyto-/chemokine-mediated (e.g., IL-1, IL-6, IL-8, GM-CSF) induction of myeloid-derived suppressor cells (MDSCs), regulatory T cells and M2-differentiated tumor-associated macrophages (TAMs) [80,81,82,83,84,85,86,87,88,89] (Figure 1). Mutant KRAS also induces NF-kB and cooperates with MYC—two master regulators of inflammation and immunosuppression [90,91,92,93].
Immune checkpoint inhibitors (ICIs) block the PDL1–PD1 receptor interaction and thus can reinvigorate antitumor immune responses in some patients with so-called “hot” tumors. ICIs alone or in combination with chemotherapy have become standard-of-care treatment for NSCLC patients whose tumors express PDL1 and lack EGFR mutations or EML4/ALK rearrangements [94,95,96,97,98,99]. These immunologically “hot” tumors are characterized by the accumulation of proinflammatory cytokines, high PD-L1 expression and intratumoral accumulation of CD8+ tumor-infiltrating lymphocytes (TILs), which are required for ICIs to be effective [100]. In contrast, immunologically “cold” tumors are deprived of TILs. Interestingly, KRAS-mutant tumors with TP53 co-mutations are associated with a “hot” TME [73,74], whereas STK11/LKB1 co-mutated tumors exhibit lower expression of stimulator of interferon genes (STING) and of immune-related expression signatures than wild-type tumors (“cold” TME) and therefore are typically checkpoint-inhibitor-resistant [75,101]. Ongoing scientific efforts are seeking to decipher the mechanistic basis for this lack of ICI response in STK11/LKB1 co-mutated tumors with the aim of developing new strategies to turn “cold” tumors into “hot” ones and thus increase the benefit of immune checkpoint blockade for NSCLC patients with this prevalent genotype [102].
4. Efficacy of Direct KRAS(G12C) Inhibitors and Mechanisms of Resistance
The first evidence that inhibition of oncogenic KRAS is beneficial from a therapeutic viewpoint came from immunocompetent genetically engineered mouse models (GEMMs) of lung cancer in which lung tumors completely regressed upon genetic removal of mutant KRAS [24,25,26]. In the last decade, better biochemical insights into KRAS structural biology finally allowed researchers to develop direct KRAS(G12C) inhibitors like the preclinical compounds SML-8-73-1, compound 12 (“Shokat compound”), ARS-853 and ARS-1620, as well as the clinical compounds AMG-510 (Amgen), MRTX-849 (Mirati Therapeutics) and JNJ-74699157/ARS-3248 (Johnson & Johnson/Wellspring Biosciences) [103,104,105,106,107,108,109,110]. These inhibitors bind covalently to the cysteine residue in the switch-II pocket, which is newly created by the G12C mutation. Binding of the inhibitor shifts the relative nucleotide affinities to favor GDP over GTP binding and reduces the interactions between mutant KRAS and effector or regulatory molecules [107].
The phase I/II CodeBreaK-100 trial (NCT03600883) investigating the clinically most advanced KRAS(G12C) inhibitor AMG-510 (international nonproprietary name (INN) sotorasib) reported a confirmed objective response rate (ORR) of ~32% and a disease control rate (DCR) of ~88% among lung cancer patients for the phase I trial part. Grade 3/4 toxicities and treatment discontinuation occurred in 11.6% and 7% of patients, respectively [111]. Based on this study, in December 2020, the FDA granted breakthrough therapy designation for sotorasib for patients with KRAS(G12C)-mutant, locally advanced or metastatic NSCLC following at least one prior systemic therapy. The phase II trial part with a data cutoff on 1 December 2020 and a median follow-up of 12.2 months validated the phase I results with a confirmed ORR of 37.1%, a DCR of 80.6% and a median duration of response of 10 months. The median progression-free survival was 6.8 months. Treatment-related adverse events (TRAEs) led to treatment discontinuation in 7.1% of patients. Most TRAEs were grade 1 and 2 and included diarrhea (31% any grade), nausea (19%), liver enzyme changes (15%) and fatigue (11%) (presented at the 2020 World Conference on Lung Cancer, 28–31 January 2021).
The median follow-up of the phase I/II KRYSTAL-1 trial (NCT03785249) for Mirati’s MRTX-849 compound (INN adagrasib) was still relatively short (9.6 months) at the last study update presented at the 32nd EORTC-NCI-AACR Symposium (24–25 October 2020), but the data reported for NSCLC patients appear slightly better than those for AMG-510, although longer follow-up is still required [112]. Of the evaluable patients, 45% achieved a partial response, and the DCR was 96%. TRAEs led to treatment discontinuation in 4.5% of patients and most commonly included nausea (54%), diarrhea (48%), vomiting (34%), fatigue (28%) and increased liver enzymes (23%). The only commonly reported grade 3/4 TRAE was hyponatremia (3%) (the clinical efficacy parameters are summarized in Table 2). Adagrasib has a longer half-life of about 24 h compared to sotorasib (half-life 6.5 h), which allows for continuous drug exposure and sustained KRAS target inhibition. Adagrasib also penetrates the blood–brain barrier in murine models and showed efficacy in one patient with NSCLC and active brain metastases, an important feature for this highly metastatic disease with frequent brain/central nervous system manifestation.
It is most likely, however, that allele-specific KRAS(G12C) inhibitors will be combined with other treatment modalities. Monotherapies presumably have limited long-term efficacy in terms of preventing adaptive resistance since (1) tumors with minor fractions of cancer cells that harbor non-G12C KRAS mutations will ultimately relapse due to selection of these subclones [113,114] and (2) several mechanisms have been proposed for how cancer cells can lose their KRAS dependency. Among others, these include YAP pathway activation [115] and increased KRAS(G12C) expression via EGFR or aurora kinase signaling [116]. Drug combinations can furthermore account for the fact that currently available KRAS(G12C) inhibitors only target the inactive, GDP-bound form of KRAS and thus rely on the residual intrinsic hydrolysis of GTP to revert KRAS into the GDP-bound state. This mechanism is vulnerable to adaptive responses that activate upstream signaling, e.g., via receptor tyrosine kinases (RTKs) like the ErbB family or FGFR [56,58,117]. These RTKs signal through SHP2 (encoded by PTPN11), increase the GTP-loaded “ON” form of KRAS and therefore reduce KRAS(G12C) inhibitor target engagement [118,119]. Inhibition of SHP2 reduces this conversion of GDP- into GTP-bound KRAS and overcomes adaptive resistance to MAPK-pathway-targeted agents including KRAS(G12C) inhibitors [120,121]. In a patient with NSCLC, a reduction of tumor volume has been observed when adagrasib was combined with the experimental SHP2 inhibitor TNO-155 (Novartis), and the corresponding phase I/II KRYSTAL-2 study is currently recruiting patients (NCT04330664). In another phase Ib clinical trial, the combination of sotorasib and the experimental SHP2 inhibitor RMC-4630 will be investigated [122,123]. Other drug combinations (adagrasib plus pan-ErbB inhibitor afatinib; ARS-1620 plus mTOR inhibitor (everolimus) or linsitinib) follow the same biological rationale of simultaneously inhibiting KRAS and the nucleotide exchange on KRAS via RTKs [124] (for a comprehensive overview of clinical trials investigating strategies to overcome resistance to drugs targeting the KRAS(G12C) mutation we refer to [125]).
5. Conclusions and Future Perspectives of Combination Therapeutic Approaches
More than a century after the pioneering scientific work of Rudolf Virchow and Paul Ehrlich [1,17,18], for non-small-cell lung cancer (NSCLC), the era of cancer immunotherapy—which harnesses the immune system to kill cancer cells—follows the era of groundbreaking discoveries in the field of oncogene-targeted therapies [19,20]. However, the progress made during the “targeted therapy revolution” for EGFR-mutant and EML4/ALK-rearranged lung cancers among other oncogene-addicted pulmonary malignancies (for a comprehensive overview of oncogene-directed therapies against NSCLC, e.g., with ROS1/NTRK, BRAF, MET and HER-2 aberrations, we refer to [126]) had largely spared KRAS-mutant NSCLC despite the anticipated efficacy of KRAS inhibitors for this highly prevalent but heterogeneous lung cancer subtype [11,12,14,24,25,26,127] (Table 1). After overcoming biochemistry constraints to directly inhibit KRAS(G12C), the most frequent mutational subtype in NSCLC, the historical assumption of KRAS as being an “undruggable” target needs to be irrevocably discarded [103,104,105,106,107,108,109,110]. First reports from phase I/II clinical trials investigating the direct KRAS(G12C) inhibitors sotorasib and adagrasib are very impressive considering this notoriously hard-to-treat patient subgroup. Response rates are slightly inferior to those observed with other oncogene-targeted therapies (e.g., against mutant EGFR or rearranged EML4/ALK) in pretreated patients, and differences in response rates could be due to the heterogeneity of KRAS-mutant lung tumors with multiple DNA-damage-associated genomic alterations [128,129,130,131]. Additional KRAS(G12C) inhibitors are continuing to emerge for which clinical efficacy parameters have yet to be reported (JNJ-74699157/ARS-3248: NCT04006301; GDC-6036: NCT04449874); for others, the clinical development has been stopped due to unexpected toxicities (LY-3499446: NCT04165031). Currently ongoing (CodeBreaK-200 for AMG-510: NCT04303780) and future randomized phase III trials will ultimately show the true benefit of direct KRAS(G12C) inhibitors in untreated patients and presumably establish KRAS(G12C) inhibitors as the frontline treatment for KRAS(G12C)-mutant lung cancers (the current status of clinical development of KRAS(G12C) inhibitors is summarized in Table 2).
To induce deeper initial tumor regressions and to prevent the emergence of resistant cancer cell clones, multidrug combinations (e.g., KRAS(G12C) inhibitors with SHP2 and pan-ErbB inhibitors) are currently being clinically evaluated. Historically, drug combinations targeting KRAS-dependent downstream pathways (e.g., continuous MEK and PI3K inhibition) have been limited by toxicities [63,64], but KRAS(G12C) inhibitors avoid wild-type KRAS reactivity and therefore are less prone to off-target effects that are believed to disturb tissue homeostasis [127]. The lack of dose-limiting toxicities observed with sotorasib and adagrasib is encouraging and seems to make them ideal partners for combination treatment strategies, including those that incorporate immunotherapy.
Sensitivity to immune checkpoint inhibitors (ICIs) has been associated with a high tumor mutational burden (TMB) [132,133,134], and therefore, the smoking-related etiology of KRAS(G12C)-mutant NSCLC with a high mutational burden predestines affected patients for immunotherapy [44,45,46,69,70,71,72,135]. ICIs have been established as standard-of-care treatment for NSCLC patients whose tumors express PDL1 and lack EGFR mutations or EML4/ALK rearrangements as a single agent or in combination with chemotherapy [94,95,96,97,134]. However, response rates to single-agent ICIs overall are modest, and strategies to overcome this limitation are urgently required. The clinical benefit of combined PD1/PDL1 and CTLA-4 inhibition remains controversial despite FDA approval of the ipilimumab plus pembrolizumab combination [136] and comes at the cost of an increased risk of serious immune-related adverse events compared to anti-PD-1 therapy alone [137].
Due to the important function of KRAS in reducing cancer cell immunogenicity and inducing local immunosuppression (Figure 1), KRAS(G12C) inhibitors were expected to have profound effects on the tumor microenvironment (TME). In KRAS-mutant NSCLC, the TME is frequently characterized by a paucity, lack and/or dysfunction of tumor-infiltrating leukocytes (TILs), especially in the presence of co-occurring mutations in STK11/LKB1 [41,75,101]. This immunologically “cold” TME impairs the efficacy of ICIs [100], and therefore, strategies to turn “cold” tumors into “hot” ones are urgently needed. Indeed, similarly to MEK and SHP2 inhibition, sotorasib and adagrasib induced a more proinflammatory and TIL-infiltrated TME in mouse models (“reconditioning” effect) [103,121,138,139,140]. This translated into durable complete responses in combination with anti-PD-1 therapy. Mice that were cured with a combination of sotorasib and pembrolizumab subsequently rejected KRAS(G12C)-mutant CT26 tumors, suggesting that combining KRAS(G12C) inhibitors with immune checkpoint inhibitors could even drive an acquired immune response. Adaptive rather than innate immunity offers the greatest potential for durable, robust anticancer immune responses to prevent a tumor relapse and/or metastatic spread. However, these results from preclinical models have yet to be confirmed in human clinical trials. The combination of a KRAS(G12C) inhibitor and immunotherapy could specifically benefit those patients whose tumors harbor STK11/LKB1 co-mutations (~30% of KRAS(G12C)-mutant NSCLC). These tumors are linked to poor outcomes with immunotherapy and platinum-based chemotherapy [75,141,142]. Even though numerous strategies aimed at bolstering immunity against STK11-mutant tumors are currently under investigation (e.g., dual immune checkpoint inhibition with nivolumab and ipilimumab [143]), a broader spectrum of efficacious therapeutic options is urgently needed. Exploratory correlative analyses from the KRYSTAL-1 (presented at the 32nd EORTC-NCI-AACR Symposium, 24–25 October 2020) and CodeBreaK 100 (presented at the 2020 World Conference on Lung Cancer, 28–31 January 2021) trials in this context suggest higher response rates for single-agent adagrasib (64% versus 45%) and sotorasib (50% versus 42%) among patients whose tumors also harbored an STK11/LKB1 co-mutation. Even though these early findings need to be confirmed in larger clinical trials that combine sotorasib (NCT03600883) and adagrasib (NCT04613596, KRYSTAL-7) with the anti-PD-1 antibody pembrolizumab, they give us a glimpse of the extraordinary potential of these KRAS-targeted agents for this historically difficult-to-treat patient subgroup.
Other strategies to boost the immune response against KRAS-mutant cancers include STING agonists (ADU-S100, MK-1454) [101,144,145], as well as CAR-T cells (adoptive T-cell transfer) [146] and mRNA vaccine technology [147]. For the latter, the current worldwide first use of mRNA vaccine technology to fight the COVID-19 pandemic [148,149] could boost its development and acceptance in the field of oncology. In an ongoing phase I trial (V941-001), Moderna and Merck are testing mRNA-5671 alone and in combination with pembrolizumab in patients with KRAS-mutant cancers. mRNA-5671 is designed to generate and present the four most prevalent KRAS mutations (G12C, G12D, G12V and G13C) as neoantigens in host cells to the immune system to drive a more robust T-cell response (no efficacy data are publicly available yet).
A major caveat we still face today is the fact that so far no specific inhibitors of non-G12C mutations have entered clinical trials. In NSCLC, these mutations represent more than 50% of all KRAS mutations. A potential first-in-class inhibitor of KRAS(G12D), MRTX-1133, is currently in preclinical development by Mirati (to the best of our knowledge, there are no publicly available data on this compound yet). In an alternative approach, the son of sevenless 1 (SOS1) protein, which determines the nucleotide exchange on KRAS, has gained much attention as a therapeutic target to inhibit all major G12D/V/C and G13D variants. Boehringer Ingelheim’s BI-1701963 and Bayer’s BAY-293 “pan-KRAS inhibitors” selectively inhibit the SOS1–KRAS interaction [150,151], but unfortunately, apoptosis induction and tumor regressions were only observed when this drug class was combined with a MEK inhibitor. Phase I dose-finding studies are currently recruiting patients with solid tumors (BI-1701963 plus trametinib: NCT04111458) or are planned (BI-1701963 plus adagrasib). Other strategies to target non-G12C mutations include so-called switch I/II pocket inhibitors like BI-2852, which bind with nanomolar affinities to the active and inactive forms of KRAS [152,153], or mutant-selective “tricomplex” inhibitors, which sterically block interactions between KRAS and effector proteins such as RAF [154]. KRAS-targeting monobodies [155] and intrabodies [156] further add to the spectrum of therapeutic approaches currently under investigation.
From an oncologist’s perspective, the coming years in the field of KRAS-mutant NSCLC will be exciting and extremely laborious at the same time. Future clinical trials will have to teach us which drug combinations out of the multiple available therapeutic concepts (summarized in Figure 2) are the most efficacious ones and/or which treatment sequence is optimal from a tumor evolution perspective. The establishment of potent treatment predictors and systematic analysis of on-treatment longitudinal biopsies will hold the key to a better understanding of the biology of treatment resistance and guide clinical decision-making about rationally designed subsequent treatment combinations. To speed up the efficient development of drug combinations, the refinement of KRAS-mutant GEMMs to better recapitulate the genetic and immunologic complexity of human lung tumors with a high tumor mutational burden is absolutely desirable [157,158].
Despite some limitations, it is extremely exciting to see that more than a century after the groundbreaking scientific work of Paul Ehrlich and Rudolf Virchow, we are now on the verge of therapeutically unifying the concepts of both pioneers to harness synergistic effects between immune checkpoint inhibitors with “magic bullet” KRAS(G12C) inhibitors that have the potential to “recondition” the immunosuppressed tumor microenvironment. More than 40 years after the identification of RAS as a transforming oncogene, this approach will revolutionize paradigms for KRAS-mutant NSCLC. Therefore, after many discouraging therapeutic attempts in the past, we have every reason to look to the future with optimism.
Author Contributions
J.K. and P.A.J. wrote this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
J.K. received research support from Eli Lilly and Company and the German Cancer Aid Foundation. He previously served as a consultant and on advisory boards for Boehringer Ingelheim. P.A.J. reports consulting fees from AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, Loxo Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals, Silicon Therapeutics, Transcenta, Syndax, Nuvalent, Bayer, Esai and Biocartis; receiving postmarketing royalties from DFCI-owned intellectual property on EGFR mutations licensed to Lab Corp; sponsored research agreements with AstraZeneca, Daichi-Sankyo, PUMA, Boehringer Ingelheim, Eli Lilly and Company, Revolution Medicines and Astellas Pharmaceuticals; and stock ownership in Loxo Oncology and Gatekeeper Pharmaceuticals.
References
- Schwartz, R.S. Paul Ehrlich’s magic bullets. N. Engl. J. Med. 2004, 350, 1079–1080. [Google Scholar] [CrossRef]
- Barlesi, F.; Mazieres, J.; Merlio, J.-P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.H.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic Landscape of Non-Small Cell Lung Cancer in Smokers and Never-Smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef] [Green Version]
- Lusk, C.M.; Watza, D.; Dyson, G.; Craig, D.B.; Ratliff, V.; Wenzlaff, A.S.; Lonardo, F.; Bollig-Fischer, A.; Bepler, G.; Purrington, K.S.; et al. Profiling the Mutational Landscape in Known Driver Genes and Novel Genes in African American Non–Small Cell Lung Cancer Patients. Clin. Cancer Res. 2019, 25, 4300–4308. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yang, H.; Teo, A.S.M.; Amer, L.B.; Sherbaf, F.G.; Tan, C.Q.; Alvarez, J.J.S.; Lu, B.; Lim, J.Q.; Takano, A.; et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet. 2020, 52, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- Izar, B.; Zhou, H.; Heist, R.S.; Azzoli, C.G.; Muzikansky, A.; Scribner, E.E.; Bernardo, L.A.; Dias-Santagata, D.; Iafrate, A.J.; Lanuti, M. The Prognostic Impact of KRAS, Its Codon and Amino Acid Specific Mutations, on Survival in Resected Stage I Lung Adenocarcinoma. J. Thorac. Oncol. 2014, 9, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Marabese, M.; Ganzinelli, M.; Garassino, M.C.; Shepherd, F.A.; Piva, S.; Caiola, E.; Macerelli, M.; Bettini, A.; Lauricella, C.; Floriani, I.; et al. KRAS mutations affect prognosis of non-small-cell lung cancer patients treated with first-line platinum containing chemotherapy. Oncotarget 2015, 6, 34014–34022. [Google Scholar] [CrossRef] [Green Version]
- Slebos, R.J.; Kibbelaar, R.E.; Dalesio, O.; Kooistra, A.; Stam, J.; Meijer, C.J.; Wagenaar, S.S.; Vanderschueren, R.G.; Van Zandwijk, N.; Mooi, W.J.; et al. K-rasOncogene Activation as a Prognostic Marker in Adenocarcinoma of the Lung. N. Engl. J. Med. 1990, 323, 561–565. [Google Scholar] [CrossRef]
- Nadal, E.; Chen, G.; Prensner, J.R.; Shiratsuchi, H.; Sam, C.; Zhao, L.; Kalemkerian, G.P.; Brenner, D.; Lin, J.; Reddy, R.M.; et al. KRAS-G12C Mutation Is Associated with Poor Outcome in Surgically Resected Lung Adenocarcinoma. J. Thorac. Oncol. 2014, 9, 1513–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in UntreatedEGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib Compared with Interferon and Low-Dose Cytarabine for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in AdvancedALK-Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Kitajima, S.; Thummalapalli, R.; Barbie, D.A. Inflammation as a driver and vulnerability of KRAS mediated oncogenesis. Semin. Cell Dev. Biol. 2016, 58, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Virchow, R. As Based upon Physiological and Pathological Histology. Nutr. Rev. 2009, 47, 23–25. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Kelly, P.N. The Cancer Immunotherapy Revolution. Science 2018, 359, 1344–1345. [Google Scholar] [CrossRef] [Green Version]
- Fink, P.J. The Cancer Immunotherapy Revolution: Mechanistic Insights. J. Immunol. 2018, 200, 371–372. [Google Scholar] [CrossRef]
- Reddy, E.P.; Reynolds, R.K.; Santos, E.; Barbacid, M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nat. Cell Biol. 1982, 300, 149–152. [Google Scholar] [CrossRef]
- Santos, E.; Martin-Zanca, D.; Reddy, E.P.; Pierotti, M.A.; Della Porta, G.; Barbacid, M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science 1984, 223, 661–664. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.; Mercer, K.; Greenbaum, D.; Bronson, R.T.; Crowley, D.; Tuveson, D.A.; Jacks, T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nat. Cell Biol. 2001, 410, 1111–1116. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic Kras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.H.; Wellen, S.L.; Klimstra, D.; Lenczowski, J.M.; Tichelaar, J.W.; Lizak, M.J.; Whitsett, J.A.; Koretsky, A.; Varmus, H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001, 15, 3249–3262. [Google Scholar] [CrossRef] [Green Version]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Gibbs, J.B.; Sigal, I.S.; Poe, M.; Scolnick, E.M. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc. Natl. Acad. Sci. USA 1984, 81, 5704–5708. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, Š. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta (BBA)-Bioenerg. 2007, 1773, 1177–1195. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-W.; Ambrogio, C.; Bera, A.K.; Li, Q.; Li, X.-X.; Li, L.; Son, J.; Gondi, S.; Li, J.; Campbell, E.; et al. KRASQ61H preferentially signals through MAPK in a RAF dimer-dependent manner in non-small cell lung cancer. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189–198. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Ambrogio, C.; Köhler, J.; Zhou, Z.-W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A Gene Expression Signature Associated with “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Nissley, D.V.; McCormick, F.; Stephens, R.M. ssGSEA score-based Ras dependency indexes derived from gene expression data reveal potential Ras addiction mechanisms with possible clinical implications. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Krasdriven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C. Metabolic Dependencies in RAS-Driven Cancers. Clin. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Brägelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-ras Mutation Subtypes in NSCLC and Associated Co-occuring Mutations in Other Oncogenic Pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nat. Cell Biol. 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.G.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef]
- Riely, G.J.; Kris, M.G.; Rosenbaum, D.; Marks, J.; Li, A.; Chitale, D.A.; Nafa, K.; Riedel, E.R.; Hsu, M.; Pao, W.; et al. Frequency and Distinctive Spectrum of KRAS Mutations in Never Smokers with Lung Adenocarcinoma. Clin. Cancer Res. 2008, 14, 5731–5734. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3,026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS-Mutant Cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slebos, R.J.C.; Hruban, R.H.; Dalesio, O.; Mooi, W.J.; Offerhaus, G.J.A.; Rodenhuis, S. Relationship Between K-ras Oncogene Activation and Smoking in Adenocarcinoma of the Human Lung. J. Natl. Cancer Inst. 1991, 83, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Vachtenheim, J.; Horáková, I.; Novotná, H.; Opáalka, P.; Roubková, H. Mutations of K-ras oncogene and absence of H-ras mutations in squamous cell carcinomas of the lung. Clin. Cancer Res. 1995, 1, 359–365. [Google Scholar]
- Rekhtman, N.; Paik, P.K.; Arcila, M.E.; Tafe, L.J.; Oxnard, G.R.; Moreira, A.L.; Travis, W.D.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M. Clarifying the Spectrum of Driver Oncogene Mutations in Biomarker-Verified Squamous Carcinoma of Lung: Lack of EGFR/KRAS and Presence of PIK3CA/AKT1 Mutations. Clin. Cancer Res. 2012, 18, 1167–1176. [Google Scholar] [CrossRef] [Green Version]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and Outcomes of Patients with Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2019, 14, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Y.; Sun, H.; Zhou, J.-Y.; Jie, G.-L.; Xie, Z.; Shao, Y.; Zhang, X.; Ye, J.-Y.; Chen, C.-X.; Zhang, X.-C.; et al. Clinical characteristics and prognostic value of the KRAS G12C mutation in Chinese non-small cell lung cancer patients. Biomark. Res. 2020, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wilkerson, M.D.; Shah, U.; Yin, X.; Wang, A.; Hayward, M.C.; Roberts, P.; Lee, C.B.; Parsons, A.M.; Thorne, L.B.; et al. Alterations of LKB1 and KRAS and risk of brain metastasis: Comprehensive characterization by mutation analysis, copy number, and gene expression in non-small-cell lung carcinoma. Lung Cancer 2014, 86, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Hames, M.L.; Chen, H.; Iams, W.; Aston, J.; Lovly, C.M.; Horn, L. Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer. Lung Cancer 2016, 92, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Hobor, S.; Bertotti, A.; Zecchin, D.; Huang, S.; Galimi, F.; Cottino, F.; Prahallad, A.; Grernrum, W.; Tzani, A.; et al. Intrinsic Resistance to MEK Inhibition in KRAS Mutant Lung and Colon Cancer through Transcriptional Induction of ERBB3. Cell Rep. 2014, 7, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruspig, B.; Monteverde, T.; Neidler, S.; Hock, A.; Kerr, E.; Nixon, C.; Clark, W.; Hedley, A.; Laing, S.; Coffelt, S.B.; et al. The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci. Transl. Med. 2018, 10, eaao2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Arcas, M.; Hancock, D.C.; Sheridan, C.; Kumar, M.S.; Downward, J. Coordinate Direct Input of Both KRAS and IGF1 Receptor to Activation of PI3 kinase in KRAS-Mutant Lung Cancer. Cancer Discov. 2013, 3, 548–563. [Google Scholar] [CrossRef] [Green Version]
- Manchado, E.; Weissmueller, S.; Morris, J.P.; Chen, C.-C.; Wullenkord, R.; Lujambio, A.; De Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nat. Cell Biol. 2016, 534, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Haines, E.; Chen, T.; Kommajosyula, N.; Chen, Z.; Herter-Sprie, G.S.; Cornell, L.; Wong, K.K.; Shapiro, G.I. Palbociclib resistance confers dependence on an FGFR-MAP kinase-mTOR-driven pathway in KRAS-mutant non-small cell lung cancer. Oncotarget 2018, 9, 31572–31589. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Van Den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib Plus Docetaxel Compared with Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [Green Version]
- Blumenschein, G.R., Jr.; Smit, E.F.; Planchard, D.; Kim, D.W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.J.; Hanna, N.H.; et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, R.K.; Von Hoff, D.D.; Eskens, F.; Blumenschein, G.; Richards, D.; Genvresse, I.; Reschke, S.; Granvil, C.; Skubala, A.; Peña, C.; et al. Phase Ib Trial of the PI3K Inhibitor Copanlisib Combined with the Allosteric MEK Inhibitor Refametinib in Patients with Advanced Cancer. Target Oncol. 2020, 15, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol. Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Kohler, J.; Catalano, M.; Ambrogio, C. Back to the Bench? MEK and ERK Inhibitors for the Treatment of KRAS Mutant Lung Adenocarcinoma. Curr. Med. Chem. 2018, 25, 558–574. [Google Scholar] [CrossRef]
- Köhler, J.; Zhao, Y.; Li, J.; Gokhale, P.C.; Tiv, H.L.; Knott, A.R.; Wilkens, M.K.; Soroko, K.M.; Lin, M.; Ambrogio, C.; et al. ERK Inhibitor LY3214996-Based Treatment Strategies for RAS-Driven Lung Cancer. Mol. Cancer Ther. 2021. [Google Scholar] [CrossRef]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 2017, 12, e0185862. [Google Scholar] [CrossRef] [Green Version]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Kadara, H.; Choi, M.; Zhang, J.; Parra, E.; Rodriguez-Canales, J.; Gaffney, S.; Zhao, Z.; Behrens, C.; Fujimoto, J.; Chow, C.; et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann. Oncol. 2017, 28, 75–82. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; De Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; Van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nat. Cell Biol. 2019, 575, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Assoun, S.; Theou-Anton, N.; Nguenang, M.; Cazes, A.; Danel, C.; Abbar, B.; Pluvy, J.; Gounant, V.; Khalil, A.; Namour, C.; et al. Association of TP53 mutations with response and longer survival under immune checkpoint inhibitors in advanced non-small-cell lung cancer. Lung Cancer 2019, 132, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.A.; Trécesson, S.D.C.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099.e6. [Google Scholar] [CrossRef] [Green Version]
- Neuwelt, A.J.; Kimball, A.K.; Johnson, A.M.; Arnold, B.W.; Bullock, B.L.; Kaspar, R.E.; Kleczko, E.K.; Kwak, J.W.; Wu, M.-H.; Heasley, L.E.; et al. Cancer cell-intrinsic expression of MHC II in lung cancer cell lines is actively restricted by MEK/ERK signaling and epigenetic mechanisms. J. Immunother. Cancer 2019, 8, e000441. [Google Scholar] [CrossRef]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras–Mutant Lung Cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, P.D.; Guimarães, C.F.; Cardoso, A.P.; Mendonça, S.; Costa, Â.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Ancrile, B.; Lim, K.-H.; Counter, C.M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007, 21, 1714–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, G.D.; McLeod, L.; Alhayyani, S.; Miller, A.; Russell, P.A.; Ferlin, W.; Rose-John, S.; Ruwanpura, S.M.; Jenkins, B.J. IL6 Trans-signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res. 2016, 76, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Guo, J.; Weng, L.; Tang, W.; Jin, S.; Ma, W. Myeloid-derived suppressor cells—new and exciting players in lung cancer. J. Hematol. Oncol. 2020, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Sunaga, N.; Imai, H.; Shimizu, K.; Shames, D.S.; Kakegawa, S.; Girard, L.; Sato, M.; Kaira, K.; Ishizuka, T.; Gazdar, A.F.; et al. Oncogenic KRAS-induced interleukin-8 overexpression promotes cell growth and migration and contributes to aggressive phenotypes of non-small cell lung cancer. Int. J. Cancer 2011, 130, 1733–1744. [Google Scholar] [CrossRef]
- Kumar, M.S.; Hancock, D.C.; Molina-Arcas, M.; Steckel, M.; East, P.; Diefenbacher, M.; Armenteros-Monterroso, E.; Lassailly, F.; Matthews, N.; Nye, E.; et al. The GATA2 Transcriptional Network Is Requisite for RAS Oncogene-Driven Non-Small Cell Lung Cancer. Cell 2012, 149, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef]
- Basseres, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniluk, J.; Liu, Y.; Deng, D.; Chu, J.; Huang, H.; Gaiser, S.; Cruz-Monserrate, Z.; Wang, H.; Ji, B.; Logsdon, C.D. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Investig. 2012, 122, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Swigart, L.B.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Şenler, F.Ç.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhang, Y.; Guo, G.; Cai, X.; Yu, H.; Cai, Y.; Zhang, B.; Hong, S.; Zhang, L. Nivolumab plus ipilimumab versus pembrolizumab as chemotherapy-free, first-line treatment for PD-L1-positive non-small cell lung cancer. Clin. Transl. Med. 2020, 10, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Gonzalez-Cao, M. Cemiplimab monotherapy in advanced non-squamous and squamous non-small cell lung cancer. Lancet 2021, 397, 557–559. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Faculty Opinions recommendation of Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, M.; Lu, J.; Li, L.; Feru, F.; Quan, C.; Gero, T.W.; Ficarro, S.B.; Xiong, Y.; Ambrogio, C.; Paranal, R.M.; et al. Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12C. Cell Chem. Biol. 2017, 24, 1005–1016.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nat. Cell Biol. 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.C.; Gurbani, D.; Ficarro, S.B.; Carrasco, M.A.; Lim, S.M.; Choi, H.G.; Xie, T.; Marto, J.A.; Chen, Z.; Gray, N.S.; et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. USA 2014, 111, 8895–8900. [Google Scholar] [CrossRef] [Green Version]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.; Rybkin, I.; Spira, A.; Riely, G.; Papadopoulos, K.; Sabari, J.; Johnson, M.; Heist, R.; Bazhenova, L.; Barve, M.; et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Advanced/ Metastatic Non–Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. Eur. J. Cancer 2020, 138, S1–S2. [Google Scholar] [CrossRef]
- McGranahan, N.; Favero, F.; De Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.I.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra54. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, D.; Arima, K.; Yokoyama, N.; Chikamoto, A.; Taki, K.; Inoue, R.; Kaida, T.; Higashi, T.; Nitta, H.; Ohmuraya, M.; et al. Heterogeneity of KRAS Mutations in Pancreatic Ductal Adenocarcinoma. Pancreas 2016, 45, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; De Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nat. Cell Biol. 2020, 577, 421–425. [Google Scholar] [CrossRef]
- Moll, H.P.; Pranz, K.; Musteanu, M.; Grabner, B.; Hruschka, N.; Mohrherr, J.; Aigner, P.; Stiedl, P.; Brcic, L.; Laszlo, V.; et al. Afatinib restrains K-RAS–driven lung tumorigenesis. Sci. Transl. Med. 2018, 10, eaao2301. [Google Scholar] [CrossRef] [Green Version]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; De Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef]
- Lou, K.; Steri, V.; Alex, Y.G.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal. 2019, 12, eaaw9450. [Google Scholar] [CrossRef]
- Adamopoulos, C.; Ahmed, T.A.; Karoulia, Z.; Wu, X.; Sachidanandam, R.; Aaronson, S.A.; Poulikakos, P.I. Abstract LB-119: SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep. 2019, 26, 65–78. [Google Scholar] [CrossRef]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A.; et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Bendell, J.; Ulahannan, S.; Koczywas, M.; Brahmer, J.; Capasso, A.; Eckhardt, S.; Gordon, M.; McCoach, C.; Nagasaka, M.; Ng, K.; et al. Intermittent dosing of RMC-4630, a potent, selective inhibitor of SHP2, combined with the MEK inhibitor cobimetinib, in a phase 1b/2 clinical trial for advanced solid tumors with activating mutations of RAS signaling. Eur. J. Cancer 2020, 138, S8–S9. [Google Scholar] [CrossRef]
- Ou, S.; Koczywas, M.; Ulahannan, S.; Janne, P.; Pacheco, J.; Burris, H.; McCoach, C.; Wang, J.; Gordon, M.; Haura, E.; et al. A12 The SHP2 Inhibitor RMC-4630 in Patients with KRAS-Mutant Non-Small Cell Lung Cancer: Preliminary Evaluation of a First-in-Man Phase 1 Clinical Trial. J. Thorac. Oncol. 2020, 15, S15–S16. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Moore, C.; Rana, S.; van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.-S.; Janes, M.R.; et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 2019, 11, eaaw7999. [Google Scholar] [CrossRef]
- Jiao, D.; Yang, S. Overcoming Resistance to Drugs Targeting KRAS(G12C) Mutation. Innovation 2020, 1, 100035. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.; Greenbaum, D.; Cichowski, K.; Mercer, K.; Murphy, E.; Schmitt, E.; Bronson, R.T.; Umanoff, H.; Edelmann, W.; Kucherlapati, R.; et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997, 11, 2468–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.; Wu, P.; Zhang, H. Comparison of gefitinib as first- and second-line therapy for advanced lung adenocarcinoma patients with positive exon 21 or 19 del epidermal growth factor receptor mutation. Cancer Manag. Res. 2017, 9, 243–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garassino, M.C.; Martelli, O.; Broggini, M.; Farina, G.; Veronese, S.; Rulli, E.; Bianchi, F.; Bettini, A.; Longo, F.; Moscetti, L.; et al. Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): A randomised controlled trial. Lancet Oncol. 2013, 14, 981–988. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Ando, M.; Asami, K.; Okano, Y.; Fukuda, M.; Nakagawa, H.; Ibata, H.; Kozuki, T.; Endo, T.; Tamura, A.; et al. Randomized Phase III Trial of Erlotinib Versus Docetaxel as Second- or Third-Line Therapy in Patients with Advanced Non–Small-Cell Lung Cancer: Docetaxel and Erlotinib Lung Cancer Trial (DELTA). J. Clin. Oncol. 2014, 32, 1902–1908. [Google Scholar] [CrossRef]
- Blackhall, F.; Camidge, D.R.; Shaw, A.T.; Soria, J.-C.; Solomon, B.J.; Mok, T.; Hirsh, V.; Jänne, P.A.; Shi, Y.; Yang, P.-C.; et al. Final results of the large-scale multinational trial PROFILE 1005: Efficacy and safety of crizotinib in previously treated patients with advanced/metastatic ALK-positive non-small-cell lung cancer. ESMO Open 2017, 2, e000219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Porta, M.; Crous-Bou, M.; Wark, P.A.; Vineis, P.; Real, F.X.; Malats, N.; Kampman, E. Cigarette smoking and K-ras mutations in pancreas, lung and colorectal adenocarcinomas: Etiopathogenic similarities, differences and paradoxes. Mutat. Res. Mutat. Res. 2009, 682, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Caro, R.B.; Zurawski, B.; Kim, S.-W.; Costa, E.C.; Park, K.; Alexandru, A.; Lupinacci, L.; De la Mora, J.E.; et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Sznol, M.; Melero, I. Revisiting anti-CTLA-4 antibodies in combination with PD-1 blockade for cancer immunotherapy. Ann. Oncol. 2021, 32, 295–297. [Google Scholar] [CrossRef]
- Briere, D.M.; Calinisan, A.; Aranda, R.; Sudhakar, N.; Hargis, L.; Gatto, S.; Fernandez-Banet, J.; Pavlicek, A.; Engstrom, L.D.; Hallin, J.; et al. The KRAS(G12C) inhibitor MRTX849 reconditions the tumor immune microenvironment and leads to durable complete responses in combination with anti-PD-1 therapy in a syngeneic mouse model. Mol. Cancer Ther. 2019, 18, 615. [Google Scholar]
- Choi, H.; Deng, J.; Li, S.; Silk, T.; Dong, L.; Brea, E.J.; Houghton, S.; Redmond, D.; Zhong, H.; Boiarsky, J.; et al. Pulsatile MEK Inhibition Improves Anti-tumor Immunity and T Cell Function in Murine Kras Mutant Lung Cancer. Cell Rep. 2019, 27, 806–819 e805. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Zhang, Y.; Eoh, K.J.; Sharma, R.; Sanmamed, M.F.; Wu, J.; Choi, J.; Park, H.S.; Iwasaki, A.; Kaftan, E.; et al. The Combination of MEK Inhibitor With Immunomodulatory Antibodies Targeting Programmed Death 1 and Programmed Death Ligand 1 Results in Prolonged Survival in Kras/p53-Driven Lung Cancer. J. Thorac. Oncol. 2019, 14, 1046–1060. [Google Scholar] [CrossRef]
- Chen, Z.; Cheng, K.; Walton, Z.; Wang, Y.; Ebi, H.; Shimamura, T.; Liu, Y.; Tupper, T.; Ouyang, J.; Li, J.; et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nat. Cell Biol. 2012, 483, 613–617. [Google Scholar] [CrossRef]
- Skoulidis, F.; Arbour, K.C.; Hellmann, M.D.; Patil, P.D.; Marmarelis, M.E.; Awad, M.M.; Murray, J.C.; Hellyer, J.; Gainor, J.F.; Dimou, A.; et al. Association of STK11/LKB1 genomic alterations with lack of benefit from the addition of pembrolizumab to platinum doublet chemotherapy in non-squamous non-small cell lung cancer. J. Clin. Oncol. 2019, 37, 102. [Google Scholar] [CrossRef]
- Stein, M.K.; Prouet, P.; Martin, M.G. Dual Checkpoint Inhibition: An Approach for STK11 and KRAS Co-Mutated Lung Adenocarcinoma? JCO Precis. Oncol. 2019, 1-3, 1–3. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Sen, T.; Gay, C.M.; Ramkumar, K.; Diao, L.; Cardnell, R.J.; Rodriguez, B.L.; Stewart, C.A.; Papadimitrakopoulou, V.A.; Gibson, L.; et al. STING Pathway Expression Identifies NSCLC With an Immune-Responsive Phenotype. J. Thorac. Oncol. 2020, 15, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Brody, J.; Ingham, M.; Strauss, J.; Cemerski, S.; Wang, M.; Tse, A.; Khilnani, A.; Marabelle, A.; Golan, T. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann. Oncol. 2018, 29, viii712. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ma, J.-A.; Zhang, H.-X.; Jiang, Y.-N.; Luo, W.-H. Cancer vaccines: Targeting KRAS-driven cancers. Expert Rev. Vaccines 2020, 19, 163–173. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, D.; Gmachl, M.; Ramharter, J.; Teh, J.; Fu, S.-C.; Trapani, F.; Kessler, D.; Rumpel, K.; Botesteanu, D.-A.; Ettmayer, P.; et al. Abstract 1091: BI-3406 and BI 1701963: Potent and selective SOS1::KRAS inhibitors induce regressions in combination with MEK inhibitors or irinotecan. Tumor Biol. 2020, 80, 1091. [Google Scholar] [CrossRef]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J.; et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.H.; Alexander, P.; Dharmaiah, S.; Agamasu, C.; Nissley, D.V.; McCormick, F.; Esposito, D.; Simanshu, D.K.; Stephen, A.G.; Balius, T.E. The small molecule BI-2852 induces a nonfunctional dimer of KRAS. Proc. Natl. Acad. Sci. USA 2020, 117, 3363–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, R.; Schulze, C.; Bermingham, A.; Choy, T.; Cregg, J.; Kiss, G.; Marquez, A.; Reyes, D.; Saldajeno-Concar, M.; Weller, C.; et al. A06 Tri-complex Inhibitors of the Oncogenic, GTP-Bound Form of KRASG12C Overcome RTK-Mediated Escape Mechanisms and Drive Tumor Regressions in Preclinical Models of NSCLC. J. Thorac. Oncol. 2020, 15, S13–S14. [Google Scholar] [CrossRef]
- Spencer-Smith, R.; Koide, A.; Zhou, Y.; Eguchi, R.R.; Sha, F.; Gajwani, P.; Santana, D.; Gupta, A.; Jacobs, M.; Herrero-Garcia, E.; et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. [Google Scholar] [CrossRef]
- Tanaka, T.; Rabbitts, T.H. Intrabodies based on intracellular capture frameworks that bind the RAS protein with high affinity and impair oncogenic transformation. EMBO J. 2003, 22, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Salehi-Rad, R.; Li, R.; Tran, L.M.; Lim, R.J.; Abascal, J.; Momcilovic, M.; Park, S.J.; Ong, S.L.; Shabihkhani, M.; Huang, Z.L.; et al. Novel Kras-mutant murine models of non-small cell lung cancer possessing co-occurring oncogenic mutations and increased tumor mutational burden. Cancer Immunol. Immunother. 2021, 1–12. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; Halliwill, K.D.; To, M.D.; Rashid, M.; Rust, A.G.; Keane, T.M.; DelRosario, R.; Jen, K.-Y.; Gurley, K.E.; Kemp, C.J.; et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nat. Cell Biol. 2015, 517, 489–492. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Simplified overview of mutant-KRAS-dependent effects on the surrounding tumor microenvironment via direct cell-to-cell interactions and/or paracrine secretion of interleukins, GM-CSF and TGFβ. These paracrine signals induce the accumulation of myeloid-derived suppressor cells (MDSCs), M2-differentiated tumor-associated macrophages (TAMs) and regulatory T cells, which impair antitumor immunity by suppressing T-cell effector functions. References are displayed in brackets.
Figure 1.
Simplified overview of mutant-KRAS-dependent effects on the surrounding tumor microenvironment via direct cell-to-cell interactions and/or paracrine secretion of interleukins, GM-CSF and TGFβ. These paracrine signals induce the accumulation of myeloid-derived suppressor cells (MDSCs), M2-differentiated tumor-associated macrophages (TAMs) and regulatory T cells, which impair antitumor immunity by suppressing T-cell effector functions. References are displayed in brackets.

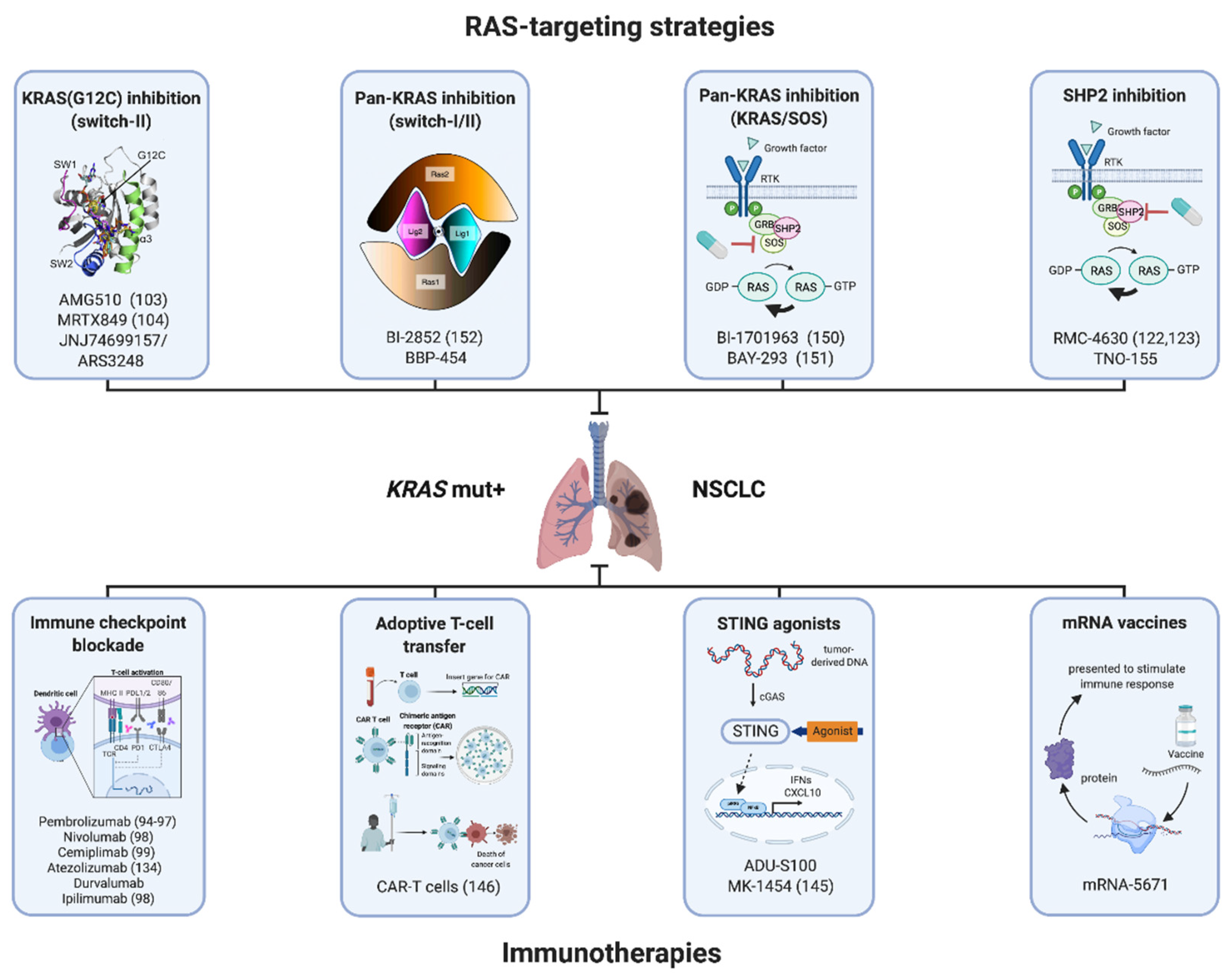
Figure 2.
Overview of therapeutic concepts for patients with KRAS-mutant NSCLC in different stages of clinical development that target mutant KRAS itself or the surrounding tumor immune microenvironment. Future clinical trials are required to decipher the optimal strategy of simultaneously or sequentially combining these treatment strategies. References if applicable are displayed in brackets. Created with biorender.com (accessed on 3 March 2021).
Figure 2.
Overview of therapeutic concepts for patients with KRAS-mutant NSCLC in different stages of clinical development that target mutant KRAS itself or the surrounding tumor immune microenvironment. Future clinical trials are required to decipher the optimal strategy of simultaneously or sequentially combining these treatment strategies. References if applicable are displayed in brackets. Created with biorender.com (accessed on 3 March 2021).

Table 2.
Summary of ongoing clinical trials investigating KRAS(G12C) inhibitors alone or in combination with other treatment modalities in NSCLC. ORR = objective response rate, PFS = progression-free survival, TRAE = treatment-related adverse events.
Table 2.
Summary of ongoing clinical trials investigating KRAS(G12C) inhibitors alone or in combination with other treatment modalities in NSCLC. ORR = objective response rate, PFS = progression-free survival, TRAE = treatment-related adverse events.
| Drug | Trial # | Clinical Phase | Efficacy (%) | Median PFS | Reported Toxicity Any Grade in % (Grade 3–4 in %) | Data Cutoff |
|---|---|---|---|---|---|---|
| Sotorasib (AMG510) −/+pembrolizumab | NCT03600883 CodeBreak 100 | Phase 1/2 recruiting | ORR 37.1 Complete response 2.4 Partial response 34.7 Stable disease 43.5Progressive disease 16.1 Disease control rate 80.6 | 6.8 months | Diarrhea 69.8 (19.8) Nausea 19.0 (0) ALT increase 15.1 (6.3) AST increase 15.1 (5.6) Fatigue 11.1 (0) Vomiting 7.9 (0) Rash 5.6 (0) Treatment discontinuation in 7.1% | 1 December 2020 with a median follow-up time of 12.2 months |
| Sotorasib (AMG510) +MEK inhibitor or anti-PD1 | NCT04185883 CodeBreak 101 | Phase 1b recruiting | - | - | - | - |
| Sotorasib (AMG510) in subjects of Chinese descent | NCT04380753 CodeBreak 105 | Phase 1 recruiting | - | - | - | - |
| Sotorasib (AMG510) vs. docetaxel | NCT04303780 CodeBreak 200 | Phase 3 recruiting | - | - | - | - |
| Adagrasib (MRTX849) −/+pembrolizumab −/+afatinib | NCT03785249 KRYSTAL-1 | Phase 1/2 recruiting | ORR 45 Complete response 0 Partial response 45 Stable disease 51 Progressive disease 2 Disease control rate 96 | - | Nausea 54 (2) Diarrhea 51 (2) Vomiting 35 (2) Fatigue 32 (6) Increased ALT 20 (5) Increased AST 17 (5) Increased creatinine 15 (0) Decreased appetite 15 (0) QT prolongation 14 (3) Anemia 13 (2) Grade 5 TRAEs in two patients (pneumonitis, cardiac failure) Discontinuation due to TRAEs in 4.5% | 30 August 2020 with a median follow-up time of 9.6 months |
| Adagrasib (MRTX849) +TNO155 | NCT04330664 KRYSTAL-2 | Phase 1/2 recruiting | - | - | - | - |
| Adagrasib (MRTX849) +pembrolizumab | NCT04613596 KRYSTAL-7 | Phase 2 recruiting | - | - | - | - |
| GDC-6036 −/+atezolizumab −/+bevacizumab −/+erlotinib | NCT04449874 | Phase 1a/1b recruiting | - | - | - | - |
| LY3499446 +/− abemaciclib/ erlotinib vs. docetaxel | NCT04165031 | Phase 1 terminated due to toxicity | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Köhler, J.; Jänne, P.A. If Virchow and Ehrlich Had Dreamt Together: What the Future Holds for KRAS-Mutant Lung Cancer. Int. J. Mol. Sci. 2021, 22, 3025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063025
AMA Style
Köhler J, Jänne PA. If Virchow and Ehrlich Had Dreamt Together: What the Future Holds for KRAS-Mutant Lung Cancer. International Journal of Molecular Sciences. 2021; 22(6):3025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063025
Chicago/Turabian StyleKöhler, Jens, and Pasi A. Jänne. 2021. "If Virchow and Ehrlich Had Dreamt Together: What the Future Holds for KRAS-Mutant Lung Cancer" International Journal of Molecular Sciences 22, no. 6: 3025. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063025
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.