
Thinking Small: Small Molecules as Potential Synergistic Adjuncts to Checkpoint Inhibition in Melanoma



Abstract
:1. Introduction
1.1. Current Trends in Melanoma Treatment
1.2. Small Molecule Drugs as Cancer Therapeutics
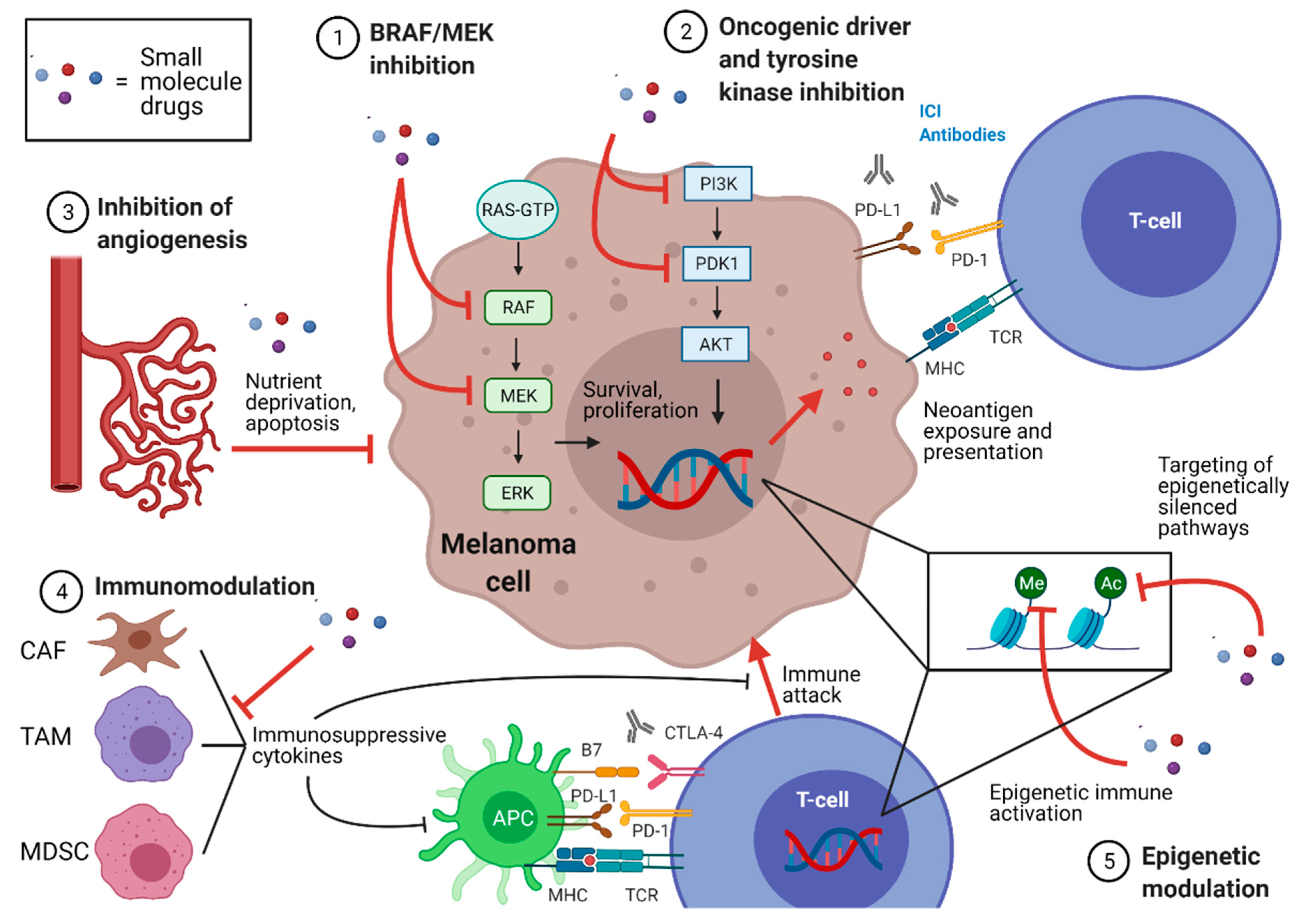
2. Small Molecule Drugs and Potential Synergy with Immune Checkpoint Inhibition
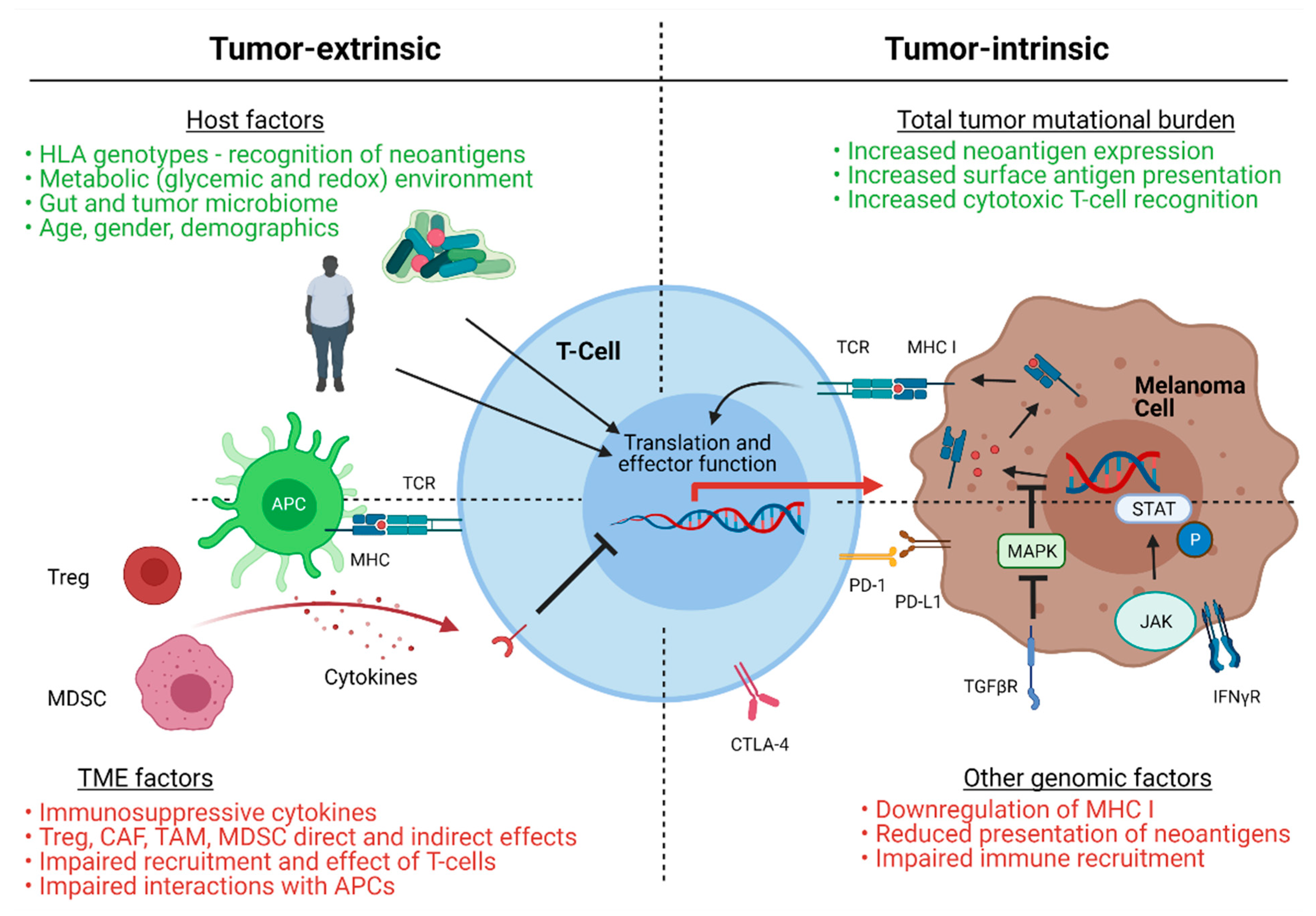
2.1. Mechanisms of Immunotherapy Resistance
2.2. Classification of Small Molecule Drugs
2.3. BRAF/MEK Inhibitors
2.4. Oncogenic Driver Inhibitors and Kinase Inhibitors
2.5. Anti-Angiogenic Molecules
2.6. Epigenetic Modifiers
2.7. Dual Immunomodulation
2.8. Novel Checkpoint Therapies on the Horizon
2.9. Other Potential Synergistic Therapies, Including Nutritive Therapies
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korn, E.L.; Liu, P.Y.; Lee, S.J.; Chapman, J.A.; Niedzwiecki, D.; Suman, V.J.; Moon, J.; Sondak, V.K.; Atkins, M.B.; Eisenhauer, E.A.; et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J. Clin. Oncol. 2008, 26, 527–534. [Google Scholar] [CrossRef]
- Swetter, S.M.; Thompson, J.A.; Albertini, M.R.; Barker, C.A.; Baumgartner, J.; Boland, G.; Coit, D.G.; Carson, W.E.; Contreras, C.; Daniels, G.A.; et al. Cutaneous Melanoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 367–402. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmink, B.A.; Gaudreau, P.O.; Wargo, J.A. Immune Checkpoint Blockade across the Cancer Care Continuum. Immunity 2018, 48, 1077–1080. [Google Scholar] [CrossRef] [Green Version]
- Seth, R.; Messersmith, H.; Kaur, V.; Kirkwood, J.M.; Kudchadkar, R.; McQuade, J.L.; Provenzano, A.; Swami, U.; Weber, J.; Alluri, K.C.; et al. Systemic Therapy for Melanoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3947–3970. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Larkin, J.; Minor, D.; D’Angelo, S.; Neyns, B.; Smylie, M.; Miller, W.H., Jr.; Gutzmer, R.; Linette, G.; Chmielowski, B.; Lao, C.D.; et al. Overall Survival in Patients With Advanced Melanoma Who Received Nivolumab Versus Investigator’s Choice Chemotherapy in CheckMate 037: A Randomized, Controlled, Open-Label Phase III Trial. J. Clin. Oncol. 2018, 36, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Del Vecchio, M.; Mandala, M.; Gogas, H.; Arance, A.M.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab versus ipilimumab in resected stage IIIB-C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1465–1477. [Google Scholar] [CrossRef]
- Menzies, A.M.; Amaria, R.N.; Rozeman, E.A.; Huang, A.C.; Tetzlaff, M.T.; Van de Wiel, B.A.; Lo, S.; Tarhini, A.A.; Burton, E.M.; Pennington, T.E.; et al. Pathological response and survival with neoadjuvant therapy in melanoma: A pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nat. Med. 2021, 27, 301–309. [Google Scholar] [CrossRef]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Lavanya, V.; Adil, M.; Ahmed, N.; Rishi, A.K.; Jamal, S. Small molecule inhibitors as emerging cancer therapeutics. Integr. Cancer Sci. Ther. 2014, 1, 39–46. [Google Scholar]
- Baker, M. Upping the ante on antibodies. Nat. Biotechnol. 2005, 23, 1065–1072. [Google Scholar] [CrossRef]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef]
- Kluger, H.M.; Tawbi, H.A.; Ascierto, M.L.; Bowden, M.; Callahan, M.K.; Cha, E.; Chen, H.X.; Drake, C.G.; Feltquate, D.M.; Ferris, R.L.; et al. Defining tumor resistance to PD-1 pathway blockade: Recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J. Immunother. Cancer 2020, 8, e000398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, L.; Theodorescu, D. Determinants of Resistance to Checkpoint Inhibitors. Int. J. Mol. Sci 2020, 21, 1594. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keenan, T.E.; Burke, K.P.; Van Allen, E.M. Genomic correlates of response to immune checkpoint blockade. Nat. Med. 2019, 25, 389–402. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Lebbe, C.; Atkinson, V.; Mandala, M.; Nathan, P.D.; Arance, A.; Richtig, E.; Yamazaki, N.; Robert, C.; Schadendorf, D.; et al. Combined PD-1, BRAF and MEK inhibition in advanced BRAF-mutant melanoma: Safety run-in and biomarker cohorts of COMBI-i. Nat. Med. 2020, 26, 1557–1563. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. Pillars Article: IFNgamma and Lymphocytes Prevent Primary Tumour Development and Shape Tumour Immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Zelenay, S.; Van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.M.; Shabaneh, T.B.; Zhang, P.; Martyanov, V.; Li, Z.; Malik, B.T.; Wood, T.A.; Boni, A.; Molodtsov, A.; Angeles, C.V.; et al. Myeloid Cells That Impair Immunotherapy Are Restored in Melanomas with Acquired Resistance to BRAF Inhibitors. Cancer Res. 2017, 77, 1599–1610. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarhini, A.A.; Edington, H.; Butterfield, L.H.; Lin, Y.; Shuai, Y.; Tawbi, H.; Sander, C.; Yin, Y.; Holtzman, M.; Johnson, J.; et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE 2014, 9, e87705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e399. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.H.; et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.J.; Khullar, K.; Kim, S.; Yegya-Raman, N.; Malhotra, J.; Groisberg, R.; Crayton, S.H.; Silk, A.W.; Nosher, J.L.; Gentile, M.A.; et al. Effect of cyclo-oxygenase inhibitor use during checkpoint blockade immunotherapy in patients with metastatic melanoma and non-small cell lung cancer. J. Immunother. Cancer 2020, 8, e000889. [Google Scholar] [CrossRef] [PubMed]
- Elkrief, A.; Derosa, L.; Kroemer, G.; Zitvogel, L.; Routy, B. The negative impact of antibiotics on outcomes in cancer patients treated with immunotherapy: A new independent prognostic factor? Ann. Oncol. 2019, 30, 1572–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinsley, N.; Zhou, C.; Tan, G.; Rack, S.; Lorigan, P.; Blackhall, F.; Krebs, M.; Carter, L.; Thistlethwaite, F.; Graham, D.; et al. Cumulative Antibiotic Use Significantly Decreases Efficacy of Checkpoint Inhibitors in Patients with Advanced Cancer. Oncologist 2020, 25, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Hodi, F.S.; Callahan, M.; Konto, C.; Wolchok, J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 2013, 368, 1365–1366. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Lawson, D.H.; Salama, A.K.; Koon, H.B.; Guthrie, T., Jr.; Thomas, S.S.; O’Day, S.J.; Shaheen, M.F.; Zhang, B.; Francis, S.; et al. Phase II study of vemurafenib followed by ipilimumab in patients with previously untreated BRAF-mutated metastatic melanoma. J. Immunother. Cancer 2016, 4, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, R.J.; Hamid, O.; Gonzalez, R.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Lewis, K.D.; Tawbi, H.A.; Hernandez, G.; Wongchenko, M.J.; et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat. Med. 2019, 25, 929–935. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Kim, T.W.; Lee, C.B.; Goh, B.C.; Miller, W.H., Jr.; Oh, D.Y.; Jamal, R.; Chee, C.E.; Chow, L.Q.M.; Gainor, J.F.; et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann. Oncol. 2019, 30, 1134–1142. [Google Scholar] [CrossRef]
- Gogas, H.; Dreno, B.; Larkin, J.; Demidov, L.; Stroyakovskiy, D.; Eroglu, Z.; Francesco Ferrucci, P.; Pigozzo, J.; Rutkowski, P.; Mackiewicz, J.; et al. Cobimetinib plus atezolizumab in BRAF(V600) wild-type melanoma: Primary results from the randomized phase III IMspire170 study. Ann. Oncol. 2021, 32, 384–394. [Google Scholar] [CrossRef]
- Minor, D.R.; Puzanov, I.; Callahan, M.K.; Hug, B.A.; Hoos, A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res. 2015, 28, 611–612. [Google Scholar] [CrossRef]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H., Jr.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Di Giacomo, A.M.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Stephens, R.; Svane, I.M.; et al. KEYNOTE-022 part 3: A randomized, double-blind, phase 2 study of pembrolizumab, dabrafenib, and trametinib in BRAF-mutant melanoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Rozeman, E.A.; Versluis, J.M.; Sikorska, K.; Lacroix, R.; Grijpink-Ongering, L.G.; Heeres, B.; Wiel, B.A.V.D.; Dimitriadis, P.; Sari, A.; Heijmink, S.; et al. The IMPemBra trial, a phase II study comparing pembrolizumab with intermittent/short-term dual MAPK pathway inhibition plus pembrolizumab in melanoma patients harboring the BRAFV600 mutation. J. Clin. Oncol. 2020, 38, 10021. [Google Scholar] [CrossRef]
- Burton, E.M.; Glitza, I.C.; Shephard, M.; Diab, A.; Milton, D.; Patel, S.; Mcquade, J.; Wong, M.; Hwu, P.; Wargo, J.; et al. Safety and Efficacy of TRIplet combination of Nivolumab (N) with Dabrafenib (D) and Trametinib (T) [TRIDeNT] in Patients (pts) with BRAF-mutated Metastatic Melanoma (MM): A Single Center Phase II Study. Ann. Oncol. 2019, 30, v534–v535. [Google Scholar] [CrossRef]
- Kreft, S.; Gesierich, A.; Eigentler, T.; Franklin, C.; Valpione, S.; Ugurel, S.; Utikal, J.; Haferkamp, S.; Blank, C.; Larkin, J.; et al. Efficacy of PD-1-based immunotherapy after radiologic progression on targeted therapy in stage IV melanoma. Eur. J. Cancer 2019, 116, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Mandala, M.; Ferrucci, P.F.; Rutkowski, M.; Guidoboni, A.M.; AranceFernandez, V.; Ferraresi, E.; Maiello, M.; Guida, M.; Del Vecchio, M.T.; et al. LBA45 First report of efficacy and safety from the phase II study SECOMBIT (SEquential COMBo Immuno and Targeted therapy study). Ann. Oncol. 2020, 31, S1173–S1174. [Google Scholar] [CrossRef]
- Ribas, A.; Algazi, A.; Ascierto, P.A.; Butler, M.O.; Chandra, S.; Gordon, M.; Hernandez-Aya, L.; Lawrence, D.; Lutzky, J.; Miller, W.H., Jr.; et al. PD-L1 blockade in combination with inhibition of MAPK oncogenic signaling in patients with advanced melanoma. Nat. Commun. 2020, 11, 6262. [Google Scholar] [CrossRef] [PubMed]
- Cobimetinib (Targeted Therapy) Plus Atezolizumab (Immunotherapy) in Participants With Advanced Melanoma Whose Cancer Has Worsened During or After Treatment With Previous Immunotherapy and Atezolizumab Monotherapy in Participants With Previously Untreated Advanced Melanoma. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03178851 (accessed on 2 February 2021).
- Reilley, M.J.; Bailey, A.; Subbiah, V.; Janku, F.; Naing, A.; Falchook, G.; Karp, D.; Piha-Paul, S.; Tsimberidou, A.; Fu, S.; et al. Phase I clinical trial of combination imatinib and ipilimumab in patients with advanced malignancies. J. Immunother. Cancer 2017, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Yap, T.A.; Chung, H.C.; De Miguel, M.J.; Bang, Y.J.; Lin, C.C.; Su, W.C.; Italiano, A.; Chow, K.H.; Szpurka, A.M.; et al. Safety and Clinical Activity of a New Anti-PD-L1 Antibody as Monotherapy or Combined with Targeted Therapy in Advanced Solid Tumors: The PACT Phase Ia/Ib Trial. Clin. Cancer Res. 2020, 27, 1267–1277. [Google Scholar] [CrossRef]
- Sheng, X.; Yan, X.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Li, S.; Mao, L.; Lian, B.; Wang, X.; et al. Axitinib in Combination With Toripalimab, a Humanized Immunoglobulin G4 Monoclonal Antibody Against Programmed Cell Death-1, in Patients With Metastatic Mucosal Melanoma: An Open-Label Phase IB Trial. J. Clin. Oncol. 2019, 37, 2987–2999. [Google Scholar] [CrossRef] [PubMed]
- Arance Fernandez, A.M.; De la Cruz Merino, T.; Petrella, R.; Jamal, L.; Ny, A.; Carneiro, A.; Berrocal, I.; Márquez-Rodas, A.; Spreafico, V.; Victoria Atkinson, F.; et al. Long. Lenvatinib (len) plus pembrolizumab (pembro) for advanced melanoma (MEL) that progressed on a PD-1 or PD-L1 inhibitor: Initial results of LEAP-004. Ann. Oncol. 2020, 31, S1142–S1215. [Google Scholar]
- Jespersen, H.; Ullenhag, A.; Carneiro, H.; Helgadottir, I.; Ljuslinder, M.; Levin, C.; All-Eriksson, B.; Andersson, U.; Stierner, L.M.; Nilsson, J.A.; et al. Phase II multicenter open label study of pembrolizumab and entinostat in adult patients with metastatic uveal melanoma (PEMDAC study). Ann. Oncol. 2019, 30, v907. [Google Scholar] [CrossRef]
- Di Giacomo, A.M.; Covre, A.; Finotello, F.; Rieder, D.; Danielli, R.; Sigalotti, L.; Giannarelli, D.; Petitprez, F.; Lacroix, L.; Valente, M.; et al. Guadecitabine Plus Ipilimumab in Unresectable Melanoma: The NIBIT-M4 Clinical Trial. Clin. Cancer Res. 2019, 25, 7351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Zakharia Yousef, O.R.; Ward, J.H.; Joseph, J.; Drabick, M.F.; Shaheen, M.M.; Milhem, D.M.; Kennedy, E.P.; Nicholas, N.; Vahanian, C.J.; Link, R.R.; et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus checkpoint inhibition for the treatment of patients with advanced melanoma. J. Clin. Oncol. 2018, 36, 4015. [Google Scholar] [CrossRef]
- Postow, M.; Sullivan, R.; Cohen, E.; Gutierrez, M.; Hong, D.; Steuer, C.; McCarter, J.; Zizlsperger, N.; Kutok, J.; O’Connell, B.; et al. Updated clinical data from the melanoma expansion cohort of an ongoing Ph1/1b Study of eganelisib (formerly IPI-549) in combination with nivolumab. J. ImmunoTher. Cancer 2020, 8, 3. [Google Scholar] [CrossRef]
- Patel, S.P.; Hodi, F.S.; Gabrilovich, D.; Chin, M.; Gibney, G.; Goldsberry, A.; Gonzalez, R.; Hurt, J.; Markowitz, J.; Whitman, E.; et al. A phase 1b/2 study of omaveloxolone in combination with checkpoint inhibitors in patients with unresectable or metastatic melanoma. Ann. Oncol. 2017, 28, xi30–xi31. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Kwong, M.L.; Neyns, B.; Yang, J.C. Adoptive T-cell transfer therapy and oncogene-targeted therapy for melanoma: The search for synergy. Clin. Cancer Res. 2013, 19, 5292–5299. [Google Scholar] [CrossRef] [Green Version]
- Hughes, P.E.; Caenepeel, S.; Wu, L.C. Targeted Therapy and Checkpoint Immunotherapy Combinations for the Treatment of Cancer. Trends Immunol. 2016, 37, 462–476. [Google Scholar] [CrossRef] [PubMed]
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef] [Green Version]
- Yeon, M.; Kim, Y.; Jung, H.S.; Jeoung, D. Histone Deacetylase Inhibitors to Overcome Resistance to Targeted and Immuno Therapy in Metastatic Melanoma. Front. Cell Dev. Biol. 2020, 8, 486. [Google Scholar] [CrossRef]
- Feng, Y.; Duan, W.; Cu, X.; Liang, C.; Xin, M. Bruton’s tyrosine kinase (BTK) inhibitors in treating cancer: A patent review (2010–2018). Expert Opin. Ther. Pat. 2019, 29, 217–241. [Google Scholar] [CrossRef]
- Guo, L.; Qi, J.; Wang, H.; Jiang, X.; Liu, Y. Getting under the skin: The role of CDK4/6 in melanomas. Eur. J. Med. Chem. 2020, 204, 112531. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M. HGF/c-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, 3844. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Janostiak, R.; Wajapeyee, N. Transcriptional regulators and alterations that drive melanoma initiation and progression. Oncogene 2020, 39, 7093–7105. [Google Scholar] [CrossRef]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Nishino, M.; Giobbie-Hurder, A.; Ramaiya, N.H.; Hodi, F.S. Response assessment in metastatic melanoma treated with ipilimumab and bevacizumab: CT tumor size and density as markers for response and outcome. J. Immunother. Cancer 2014, 2, 40. [Google Scholar] [CrossRef]
- Jour, G.; Ivan, D.; Aung, P.P. Angiogenesis in melanoma: An update with a focus on current targeted therapies. J. Clin. Pathol. 2016, 69, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C.; Jour, G.; Aung, P.P. Role of angiogenesis in melanoma progression: Update on key angiogenic mechanisms and other associated components. Semin. Cancer Biol. 2019, 59, 175–186. [Google Scholar] [CrossRef]
- Quaresmini, D.; Guida, M. Neoangiogenesis in Melanoma: An Issue in Biology and Systemic Treatment. Front. Immunol. 2020, 11, 584903. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Esteller, M.; Lee, S.H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10, 4278. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, S.J.; Shklovskaya, E.; Hersey, P. Epigenetic modulation in cancer immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 48–56. [Google Scholar] [CrossRef]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noonepalle, S.; Shen, S.; Ptacek, J.; Tavares, M.T.; Zhang, G.; Stransky, J.; Pavlicek, J.; Ferreira, G.M.; Hadley, M.; Pelaez, G.; et al. Rational Design of Suprastat: A Novel Selective Histone Deacetylase 6 Inhibitor with the Ability to Potentiate Immunotherapy in Melanoma Models. J. Med. Chem. 2020, 63, 10246–10262. [Google Scholar] [CrossRef] [PubMed]
- Gomez, S.; Tabernacki, T.; Kobyra, J.; Roberts, P.; Chiappinelli, K.B. Combining epigenetic and immune therapy to overcome cancer resistance. Semin. Cancer Biol. 2020, 65, 99–113. [Google Scholar] [CrossRef]
- Sun, W.; Lv, S.; Li, H.; Cui, W.; Wang, L. Enhancing the Anticancer Efficacy of Immunotherapy through Combination with Histone Modification Inhibitors. Genes 2018, 9, 633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osipov, A.; Saung, M.T.; Zheng, L.; Murphy, A.G. Small molecule immunomodulation: The tumor microenvironment and overcoming immune escape. J. Immunother. Cancer 2019, 7, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F.; Higgs, E.F. Immunotherapy with a sting. Science 2020, 369, 921–922. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Lacal, P.M.; Graziani, G.; Marini, S. On the Horizon: Targeting Next-Generation Immune Checkpoints for Cancer Treatment. Chemotherapy 2019, 64, 62–80. [Google Scholar] [CrossRef]
- Sasikumar, P.G.; Ramachandra, M. Small-Molecule Immune Checkpoint Inhibitors Targeting PD-1/PD-L1 and Other Emerging Checkpoint Pathways. BioDrugs 2018, 32, 481–497. [Google Scholar] [CrossRef]
- Li, K.; Tian, H. Development of small-molecule immune checkpoint inhibitors of PD-1/PD-L1 as a new therapeutic strategy for tumour immunotherapy. J. Drug Target. 2019, 27, 244–256. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis, E.S.C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e1014. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Roszik, J.; Cho, S.N.; Ogata, D.; Milton, D.R.; Peng, W.; Menter, D.G.; Ekmekcioglu, S.; Grimm, E.A. The COX2 Effector Microsomal PGE2 Synthase 1 is a Regulator of Immunosuppression in Cutaneous Melanoma. Clin. Cancer Res. 2019, 25, 1650–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- McQuade, J.L.; Daniel, C.R.; Helmink, B.A.; Wargo, J.A. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019, 20, e77–e91. [Google Scholar] [CrossRef]
- Van der Zanden, S.Y.; Luimstra, J.J.; Neefjes, J.; Borst, J.; Ovaa, H. Opportunities for Small Molecules in Cancer Immunotherapy. Trends Immunol. 2020, 41, 493–511. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Schalper, K.; Sosman, J. Targeted therapy and immunotherapy: Emerging biomarkers in metastatic melanoma. Pigment Cell Melanoma Res. 2020, 33, 390–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Chen, H.; Yang, M.; Wang, Q.; Song, F.; Li, X.; Chen, K. The new identified biomarkers determine sensitivity to immune check-point blockade therapies in melanoma. Oncoimmunology 2019, 8, 1608132. [Google Scholar] [CrossRef] [Green Version]
- Simmet, V.; Eberst, L.; Marabelle, A.; Cassier, P.A. Immune checkpoint inhibitor-based combinations: Is dose escalation mandatory for phase I trials? Ann. Oncol. 2019, 30, 1751–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozeman, E.A.; Hoefsmit, E.P.; Reijers, I.L.M.; Saw, R.P.M.; Versluis, J.M.; Krijgsman, O.; Dimitriadis, P.; Sikorska, K.; Van de Wiel, B.A.; Eriksson, H.; et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat. Med. 2021, 27, 256–263. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Small Molecule Agent | Molecular Targeting Mechanism | Immunotherapy Agent(s) | Cancer Type | Reference | Status | Phase | Outcome/Anticipated Date |
|---|---|---|---|---|---|---|---|
| Vemurafenib | BRAF inhibitor | Ipilimumab | Melanoma | NCT01400451 [51] | Terminated | 1 | Hepatotoxicity with concurrent dosing |
| Vemurafenib | BRAF inhibitor | Ipilimumab | Melanoma | NCT01673854 [52] | Completed | 2 | Median PFS 4.5 months, improved safety vs. concurrent administration |
| Vemurafenib + Cobimetinib | BRAF-MEK inhibitors | Nivolumab + Ipilimumab | Melanoma | NCT02968303 | Recruiting | 2 | 10/2023 |
| Vemurafenib + Cobimetinib | BRAF-MEK inhibitors | Nivolumab + Ipilimumab | Melanoma | NCT02968303 | Recruiting | 2 | 6/2020 |
| Vemurafenib + Cobimetinib | BRAF-MEK inhibitors | Prior first-line immunotherapy | Melanoma | NCT03224208 | Recruiting | 2 | 12/2022 |
| Vemurafenib + Cobimetinib | BRAF-MEK inhibitors | Pembrolizumab | Melanoma | NCT02818023 | Active, not recruiting | 1 | 5/2024 |
| Vemurafenib +/− Cobimetinib | BRAF-MEK inhibitors | Atezolizumab | Melanoma | NCT01656642 [53] | Completed | 1b | ORR 71.8%, substantial but manageable toxicity |
| Vemurafenib + Cobimetinib | BRAF-MEK inhibitors | Atezolizumab | Melanoma | NCT02902029 | Active, not recruiting | 2 | 6/2022 |
| Vemurabinib + Cobimetinib | BRAF-MEK inhibitors | Atezolizumab | Melanoma | NCT02908672—TRILOGY [54] | Active, not recruiting | 3 | Median PFS 15.1 vs. 10.6 months in triple therapy vs. without atezolizumab |
| Vemurafenib + Cobimetinib | BRAF-MEK inhibitors | Atezolizumab | Melanoma | NCT04722575 | Recruiting | 2 | 6/2027 |
| Vemurafenib + Cobimetinib | BRAF-MEK inhibitors | Atezolizumab | Melanoma | NCT02303951 | Terminated | 2 | Low recruitment |
| Vemurafenib + Cobimetinib | BRAF-MEK inhibitors | Atezolizumab | Melanoma | NCT03554083 | Recruiting | 2 | 6/2023 |
| Cobimetinib | MEK inhibitor | Atezolizumab | Advanced solid tumors | NCT01988896 [55] | Completed | 1 | ORR 41% in mixed BRAF mutant/WT population, median PFS 12 months |
| Cobimetinib | MEK inhibitor | Atezolizumab vs. Pembrolizumab | Melanoma | NCT03273153—IMspire 170 [56] | Active, not recruiting | 3 | 3/2025 Early results—median PFS 5.5 months for cobimetinib + atezolizumab, 5.3 months for Pem |
| Dabrafenib | BRAF inhibitor | Ipilimumab | Melanoma | NCT02200562 | Terminated | 1 | Support withdrawn |
| Dabrafenib +/− Trametinib +/− Nivolumab | BRAF-MEK inhibitors +/− αPD-1 | Ipilimumab | Melanoma | NCT01940809 | Active, not recruiting | 1 | 7/2020 |
| Dabrafenib +/− Trametinib | BRAF-MEK inhibitors | Ipilimumab | Melanoma | NCT01767454 [57] | Completed | 1 | Initially safe, 2/7 patients on triple therapy with colitis/perforation |
| Dabrafenib +/− Trametinib | BRAF-MEK inhibitors | Ipilimumab and Nivolumab | Melanoma | NCT02224781—DREAMSEQ | Recruiting | 3 | 10/2022 |
| Dabrafenib + Trametinib | BRAF-MEK inhibitors | Ipilimumab + Nivolumab | Melanoma | NCT02224781 | Recruiting | 3 | 10/2022 |
| Dabrafenib + Trametinib | BRAF-MEK inhibitors | Pembrolizumab | Melanoma | NCT02130466—KEYNOTE-022 [58,59,60] | Active, not recruiting | 2 | Median PFS 16 months on triple therapy vs. 10.3 months without, 73% ORR, 73% grade 3/4 AEs |
| Dabrafenib + Trametinib | BRAF-MEK inhibitors | Pembrolizumab | Melanoma | NCT02625337 [61] | Unknown | 2 | Pem and short-term/intermittent D/T—Median PFS 27.0 months vs. 10.6 months with Pem monotherapy |
| Dabrafenib + Trametinib | BRAF-MEK inhibitors | Spartalizumab | Melanoma | NCT04310397 | Recruiting | 2 | 2/2022 |
| Dabrafenib + Trametinib | BRAF-MEK inhibitors | PDR001 (anti PD-1) | Melanoma | NCT02967692—COMBI-I [29] | Active, not recruiting | 3 | 7/2023 ORR of 78%, including 44% complete responses (CRs) |
| Neoadjuvant Dabrafenib + Trametinib | BRAF-MEK inhibitors | Pembrolizumab | Melanoma | NCT02858921 | Recruiting | 2 | 11/2024 |
| Trametinib +/− Dabrafenib | BRAF-MEK inhibitors | Nivolumab | Melanoma | NCT02910700—TRIDeNT [62] | Recruiting | 2 | 12/2021 ORR 89%; PD1 refractory ORR 67% |
| Encorafenib + Binimetinib | BRAF-MEK inhibitors | Nivolumab | Melanoma with brain metastases | NCT04511013 | Recruiting | 2 | 6/2027 |
| Encorafenib + Binimetinib | BRAF-MEK inhibitors | Nivolumab + Ipilimumab | Advanced melanoma after progression on targeted therapy | NCT03235245 [63] | Recruiting | 2 | 2/2024 ORR 18.0% in the Nivo and 15.0% in the Ipi plus Nivo group |
| Encorafenib + Binimetinib | BRAF-MEK inhibitors | Nivolumab + Ipilimumab | Melanoma | NCT02631447 -SECOMBIT [64] | Active, not recruiting | 2 | 12/2021 ORR was highest at 82.6% in Arm A (encorafenib + binimetinib until disease progression followed by Ipi + Nivo) with lowest toxicity |
| Encorafenib + Binimetinib | BRAF-MEK inhibitors | Pembrolizumab | Melanoma | NCT02902042 | Active, not recruiting | 1/2 | 2/2021 |
| Encorafenib +/- Binimetinib | BRAF-MEK inhibitors | Nivolumab + Ipilimumab | Melanoma | NCT04655157 | Not yet recruiting | 1/2 | 7/2024 |
| LXH254 | BRAF/CRAF inhibitor | PDR001 (anti PD-1) | Advanced solid tumors | NCT02607813 | Active, not recruiting | 1 | 3/2021 |
| Dabrafenib + Trametinib | BRAF-MEK inhibitors | Durvalumab (anti PD-L1) | Melanoma | NCT02027961 [65] | Completed | 1 | ORR 69.2% |
| Cobimetinib | MEK inhibitor | Atezolizumab | Melanoma, progressive disease on anti-PD-1 | NCT03178851 [66] | Completed | 1 | ORR 36.4%, DCR 54.4%, DOR 12.7%, PFS 9.3% with cobimetinib prior to atezolizumab |
| Cobimetinib | MEK inhibitor | Atezolizumab + Bevacizumab | Melanoma with brain metastases | NCT03175432 | Recruiting | 2 | 6/2021 |
| Binimetinib | MEK inhibitor | Nivolumab | Melanoma | NCT04375527 | Recruiting | 2 | 6/2023 |
| TAK-580 | Pan-RAF inhibitor | Nivolumab | Melanoma | NCT02723006 | Terminated | 1 | Futility met |
| Oncogenic Driver and Tyrosine Kinase Inhibitors | |||||||
| Imatinib | Multiple TKI | Ipilimumab | Melanoma, GIST | NCT01738139 [67] | Recruiting | 1 | Safe, without clear signal for synergy |
| Imatinib | Multiple TKI | Pembrolizumab | Melanoma w/ C-KIT mutation | NCT02812693 | Withdrawn | 1/2 | Poor accrual |
| BMS-908662 | RAF kinase inhibitor | Ipilimumab | Melanoma | NCT01245556 | Completed | 1 | 7/2012 |
| Ibrutinib | BTK inhibitor | Pembrolizumab | Melanoma | NCT03021460 | Recruiting | 1 | 2/2021 |
| ARRY-614 | p38 MAPK and Tie2 inhibitor | Nivolumab + Ipilimumab | Melanoma | NCT04074967 | Recruiting | 1/2 | 11/2021 |
| Capmatinib + Robociclib | MET inhibitor, CyclinD1/CDK4/6 inhibitor | PDR001 (anti PD-1) | Melanoma | NCT03484923 | Recruiting | 2 | 6/2022 |
| Abemaciclib, Merestinib | CDK4/6 inhibitor, MET inhibitor | LY3300054 (anti PD-1), LY3321367 (Anti TIM-3) | Advanced solid tumors | NCT02791334 [68] | Active, not recruiting | 1 | 12/2021 Early results—dose limiting hepatotoxicity, one patient PR |
| Sonidegib | Hedgehog Pathway inhibitor | Pembrolizumab | Melanoma, Advanced solid tumors | NCT04007744 | Recruiting | 1 | 7/2022 |
| Avadomide | Cereblon inhibitor | Nivolumab | Melanoma | NCT03834623 | Recruiting | 2 | 5/2023 |
| Ceralasertib | Ataxia telangiectasia and rad3 inhibitor | Durvalumab | Melanoma | NCT03780608 | Active, not recruiting | 2 | 12/2022 |
| APG-115 | MDM2 inhibitor | Pembrolizumab | Melanoma, Advanced solid tumors | NCT03611868 | Recruiting | 1b/2 | 2/1/2022 |
| Anti-angiogenic Molecules | |||||||
| Axitinib | VEGFR 1-3, C-KIT, PDGFR | Nivolumab | Melanoma | NCT04493203 | Suspended | 2 | |
| Axitinib | VEGFR 1-3, C-KIT, PDGFR | Toripalimab (anti-PD-1) | Melanoma | NCT03941795 | Recruiting | 2 | 12/2022 |
| Axitinib | VEGFR 1-3, C-KIT, PDGFR | Toripalimab (anti-PD-1) | Melanoma | NCT03086174 [69] | Completed | 1b | 48.3% ORR, median PFS 7.5 |
| Lenvatinib | Multiple TKI—VEGFR1-2, FGFR1-4, PDGFR, KIT, RET | Pembrolizumab, Quavonlimab (anti CTLA-4) | Melanoma | NCT04700072 | Not yet recruiting | 1/2 | 4/2020 |
| Lenvatinib | Multiple TKI—VEGFR1-2, FGFR1-4, PDGFR, KIT, RET | Pembrolizumab, Quavonlimab (anti CTLA-4), Vibostolimab (anti-TIGIT) | Melanoma | NCT04305041 | Recruiting | 1/2 | 4/2030 |
| Lenvatinib | Multiple TKI—VEGFR1-2, FGFR1-4, PDGFR, KIT, RET | Pembrolizumab, Quavonlimab (anti CTLA-4), Vibostolimab (anti-TIGIT) | Melanoma | NCT04305054 | Recruiting | 1/2 | 4/2030 |
| Lenvatinib | Multiple TKI—VEGFR1-2, FGFR1-4, PDGFR, KIT, RET | Pembrolizumab | Melanoma | NCT03776136 | Active, not recruiting | 2 | 6/1/2021 |
| Lenvatinib | Multiple TKI—VEGFR1-2, FGFR1-4, PDGFR, KIT, RET | Pembrolizumab | Melanoma | NCT03820986 [70] | Recruiting | 3 | 8 responses, DCR 67.4%, Median PFS 4.2 months |
| Lenvatinib | Multiple TKI—VEGFR1-2, FGFR1-4, PDGFR, KIT, RET | Pembrolizumab | Melanoma | NCT04207086 | Recruiting | 2 | 3/2024 |
| Cabozantinib | Multiple TKI—MET, VEGFR2, RET | Nivolumab | Advanced cancers and HIV | NCT04514484 | Recruiting | 1 | 11/2025 |
| Apatinib | VEGFR-2 TKI | SHR1210 (anti PD-1) | Melanoma | NCT03986515 | Recruiting | 2 | 5/2022 |
| Apatinib | VEGFR-2 TKI | SHR1210 (anti PD-1) | Acral Melanoma | NCT03955354 | Recruiting | 2 | 4/2021 |
| Apatinib Temozolomide | VEGFR-2 TKI, DNA alkylating agent | SHR1210 (anti PD-1) | Acral Melanoma | NCT04397770 | Not yet recruiting | 2 | 2/2023 |
| Anlotinib | VEGFR-2 TKI, other TKI | TQB2450 (anti PD-L1) | Acral Melanoma | NCT03991975 | Recruiting | 1b | 12/2021 |
| Trebananib | Angiopoietin-2 inhibitor | Pembrolizumab | Advanced solid tumors | NCT03239145 | Recruiting | 1b | 8/2024 Results only in colorectal cohort |
| ENB-003 | ETBR inhibitor | Pembrolizumab | Solid tumors | NCT04205227 | Not yet recruiting | 1/2a | 11/2023 |
| Epigenetic Modifiers | |||||||
| Entinostat | HDAC inhibitor | Pembrolizumab | Melanoma | NCT03765229 | Recruiting | 2 | 6/2023 |
| Entinostat | HDAC inhibitor | Pembrolizumab | Metastatic uveal melanoma | NCT02697630 [71] | Active, not recruiting | 2 | 8/2023 (PR) observed in 3 patients resulting in an ORR of 10%; OS 11.5 months |
| Entinostat | HDAC inhibitor | Pembrolizumab | NSLC, expansion cohort in melanoma | NCT02437136 | Unknown | 1b/2 | 8/2019 |
| Panobinostat | HDAC inhibitor | Ipilimumab | Melanoma | NCT02032810 | Active, not recruiting | 1 | 12/2021 |
| Domatinostat | HDAC inhibitor | Pembrolizumab | Melanoma | NCT03278665 | Recruiting | 1b/2 | 12/2022 |
| Domatinostat | HDAC inhibitor | Nivolumab + Ipilimumab | Melanoma | NCT04133948 | Recruiting | 1b | 3/2024 |
| Tinostamustine | HDAC inhibitor | Nivolumab | Melanoma | NCT03903458 | Recruiting | 1b | 3/2024 |
| Chidamide | HDAC inhibitor | Nivolumab | Melanoma | NCT04674683 | Recruiting | 3 | 10/2025 |
| Abexinostat | pan-HDAC inhibitor | Pembrolizumab | Advanced solid tumors | NCT03590054 | Recruiting | 1b | 4/2022 |
| ACY-241 | HDAC inhibitor | Nivolumab and Ipilimumab | Melanoma | NCT02935790 | Completed | 1 | 12/2017 |
| BMS-986158 | BET inhibitor | Nivolumab | Advanced solid tumors | NCT02419417 | Recruiting | 1 | 12/2023 |
| Guadecitabine | DNA hypomethylating agent | Ipilimumab | Melanoma | NCT02608437 [72] | Unknown | 1 | ORR 26%, disease control rate 42% |
| Evofosfamide | Alkylating agent | Ipilimumab | Melanoma, other solid tumors | NCT03098160 | Unknown | 1 | 4/2019 |
| Olaparib | PARP inhibitor | Pembrolizumab | Melanoma | NCT04633902 | Not yet recruiting | 2 | 12/2024 |
| Azacitidine | Cytidine nucleoside analog | Pembrolizumab | Melanoma | NCT02816021 | Recruiting | 2 | 2/2026 |
| Immunomodulators | |||||||
| Epacadostat | IDO inhibitor | Pembrolizumab, INCAGN01876 (GITR inhibitor) | Advanced malignancies | NCT03277352 | Completed | 1/2 | 6/2020 |
| Epacadostat | IDO inhibitor | Pembrolizumab | Melanoma | NCT02752074 [73] | Completed | 3 | Median PFS 4.7 vs. 4.9 in Pem only—no significance |
| INCB001158, Epacadostat | Arginase inhibitor, IDO inhibitor | Pembrolizumab | Advanced solid tumors | NCT03361228 | Terminated | 1/2 | Based on emerging data with epacadostat and Pem |
| BMS-986205 | IDO 1 inhibitor | Nivolumab +/− Ipilimumab | Melanoma | NCT04007588 | Withdrawn, slow accrual | 2 | Slow accrual |
| BMS-986205 | IDO 1 inhibitor | Nivolumab | Melanoma | NCT03329846 | Active, not recruiting | 3 | 8/2020 |
| Indoximod | IDO 1 inhibitor | Nivolumab, Pembrolizumab, Ipilimumab | Melanoma | NCT02073123 | Completed [74] | 1/2 | ORR 55.7% (39/70, 36 confirmed) with CR of 18.6% (13/70, all confirmed). Median PFS 12.4 months |
| Duvelisib | PI3K inhibitor | Nivolumab | Melanoma | NCT04688658 | Not yet recruiting | 1/2 | 6/2022 |
| Eganelisib | PI3K inhibitor | Nivolumab | Advanced solid tumors | NCT02637531 [75] | Completed | 1 | Early results SITC 2020—22% ORR in patients who had been refractory to ICI |
| PLX3397 | CSF-1 receptor inhibitor | Pembrolizumab | Melanoma, other solid tumors | NCT02452424 | Terminated | 1 2 | Insufficient evidence of clinical efficacy |
| ARRY-382 | CSF-1 receptor inhibitor | Pembrolizumab | Melanoma, other advanced solid tumors | NCT02880371 | Completed | 1b/2 | 10/2019 |
| SX-682 | CXCR1/2—MDSC recruitment | Pembrolizumab | Melanoma | NCT03161431 | Recruiting | 1/2 | 12/2021 |
| RTA 408 (Omaveloxolone) | NRF2 activator | Nivolumab +/− Ipilimumab | Melanoma | NCT02259231 | Completed [76] | 1/2 | ORR 27%, 6 partial response and 2 complete response |
| Aspirin | COX-2 inhibitor | Ipilimumab, Pembrolizumab | Melanoma | NCT03396952 | Active, not recruiting | 2 | 6/2024 |
| L-NMMA | INOS inhibitor | Pembrolizumab | Melanoma, solid tumors | NCT03236935 | Recruiting | 1b | 3/2021 |
| Itacitinib | JAK1 inhibitor | Pembrolizumab | Advanced solid tumors | NCT02646748 | Active, not recruiting | 1b | 12/2021 |
| RGX-104 | LXR/ApoE inhibitor | Nivolumab, Pembrolizumab, Ipilimumab | Advanced solid malignancies and lymphoma | NCT02922764 | Recruiting | 2 | 3/2021 |
| MIW815 | STING agonist | PDR001 (anti PD-1) | Advanced solid tumors | NCT03172936 | Completed | 1b | 12/2020 |
| Other | |||||||
| CB-839 | Glutaminase inhibitor | Nivolumab | Melanoma, RCC, NSCLC | NCT02771626 | Completed | 1/2 | 6/2020 |
| Trigriluzole | Glutamate release inhibitor | Nivolumab, Pembrolizumab | Metastatic solid malignancies or lymphoma | NCT03229278 | Completed | 1 | 1/2020 |
| Etrumadenant | Adenosine receptor antagonist | Zimberelimab | Advanced malignancies | NCT03629756 | Active, not recruiting | 1 | 9/2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chacon, A.C.; Melucci, A.D.; Qin, S.S.; Prieto, P.A. Thinking Small: Small Molecules as Potential Synergistic Adjuncts to Checkpoint Inhibition in Melanoma. Int. J. Mol. Sci. 2021, 22, 3228. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063228
Chacon AC, Melucci AD, Qin SS, Prieto PA. Thinking Small: Small Molecules as Potential Synergistic Adjuncts to Checkpoint Inhibition in Melanoma. International Journal of Molecular Sciences. 2021; 22(6):3228. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063228
Chicago/Turabian StyleChacon, Alexander C., Alexa D. Melucci, Shuyang S. Qin, and Peter A. Prieto. 2021. "Thinking Small: Small Molecules as Potential Synergistic Adjuncts to Checkpoint Inhibition in Melanoma" International Journal of Molecular Sciences 22, no. 6: 3228. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063228