
Hypoxia Inducible Factor 1A Supports a Pro-Fibrotic Phenotype Loop in Idiopathic Pulmonary Fibrosis

 , ,
, ,
Abstract
:1. Introduction
2. Results
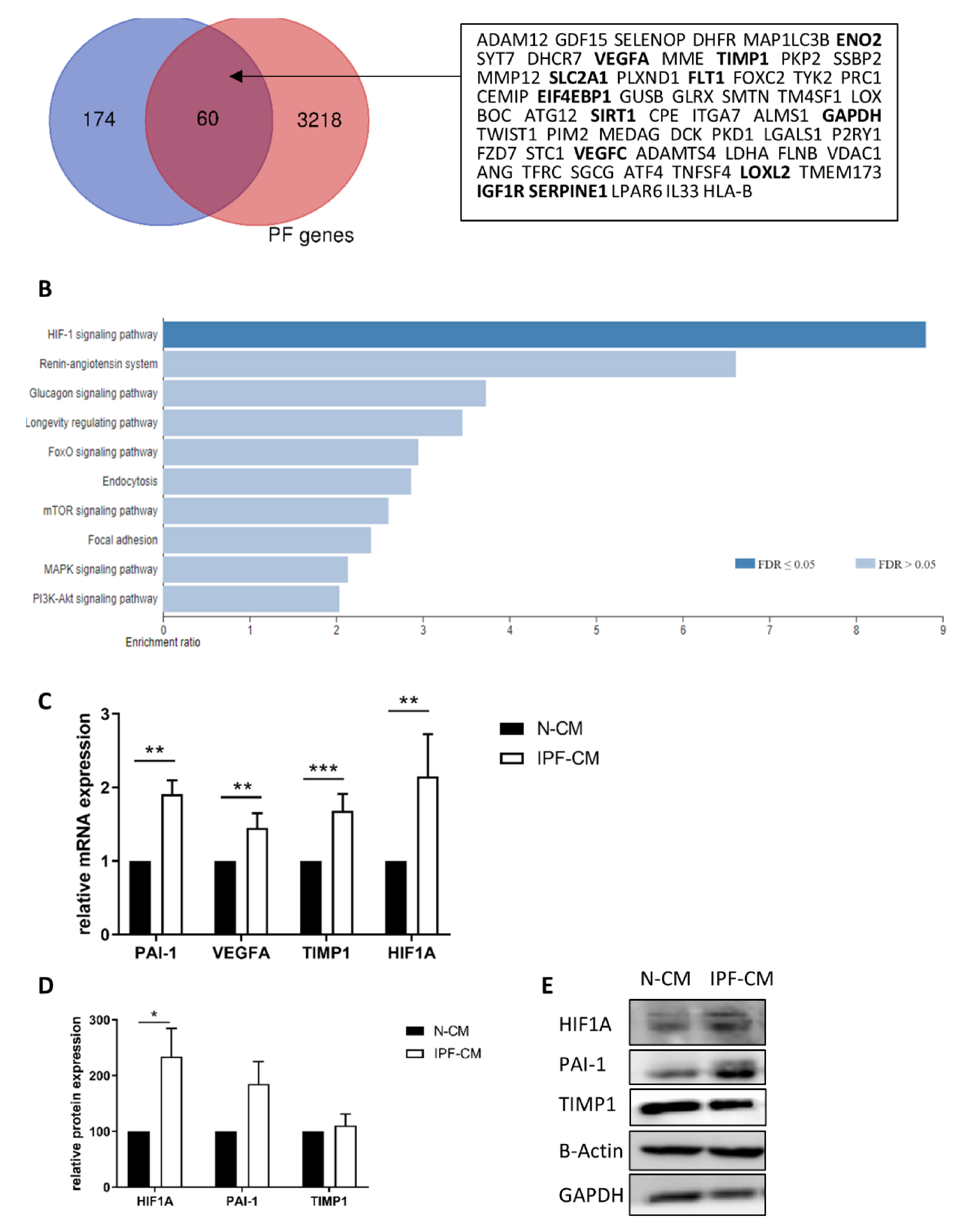
2.1. The HIF1A Pathway Is Over-Expressed in N-HLFs Exposed to IPF-CM
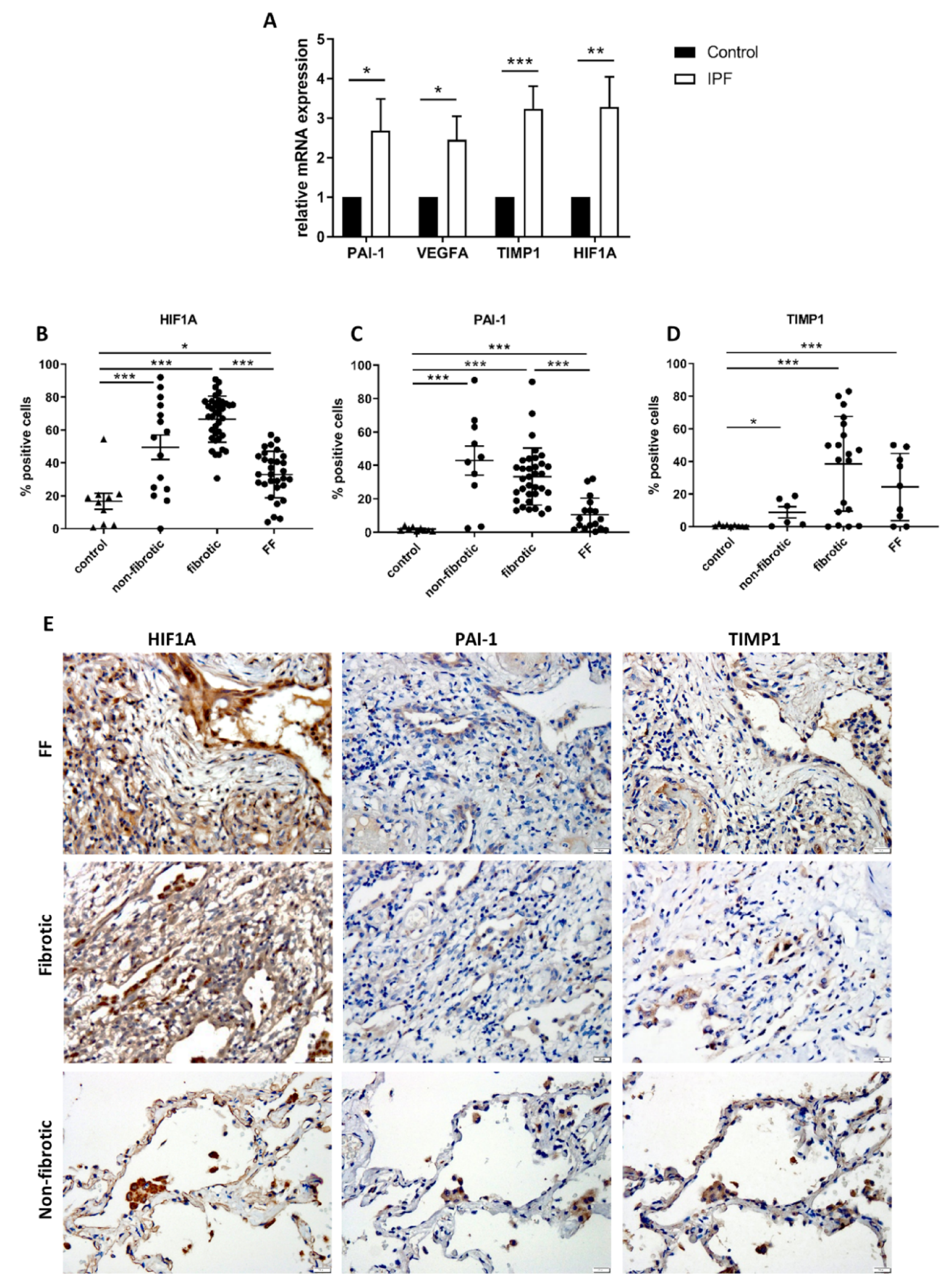
2.2. HIF1A-Related Signaling Is Upregulated in Tissue Samples from Patients with IPF
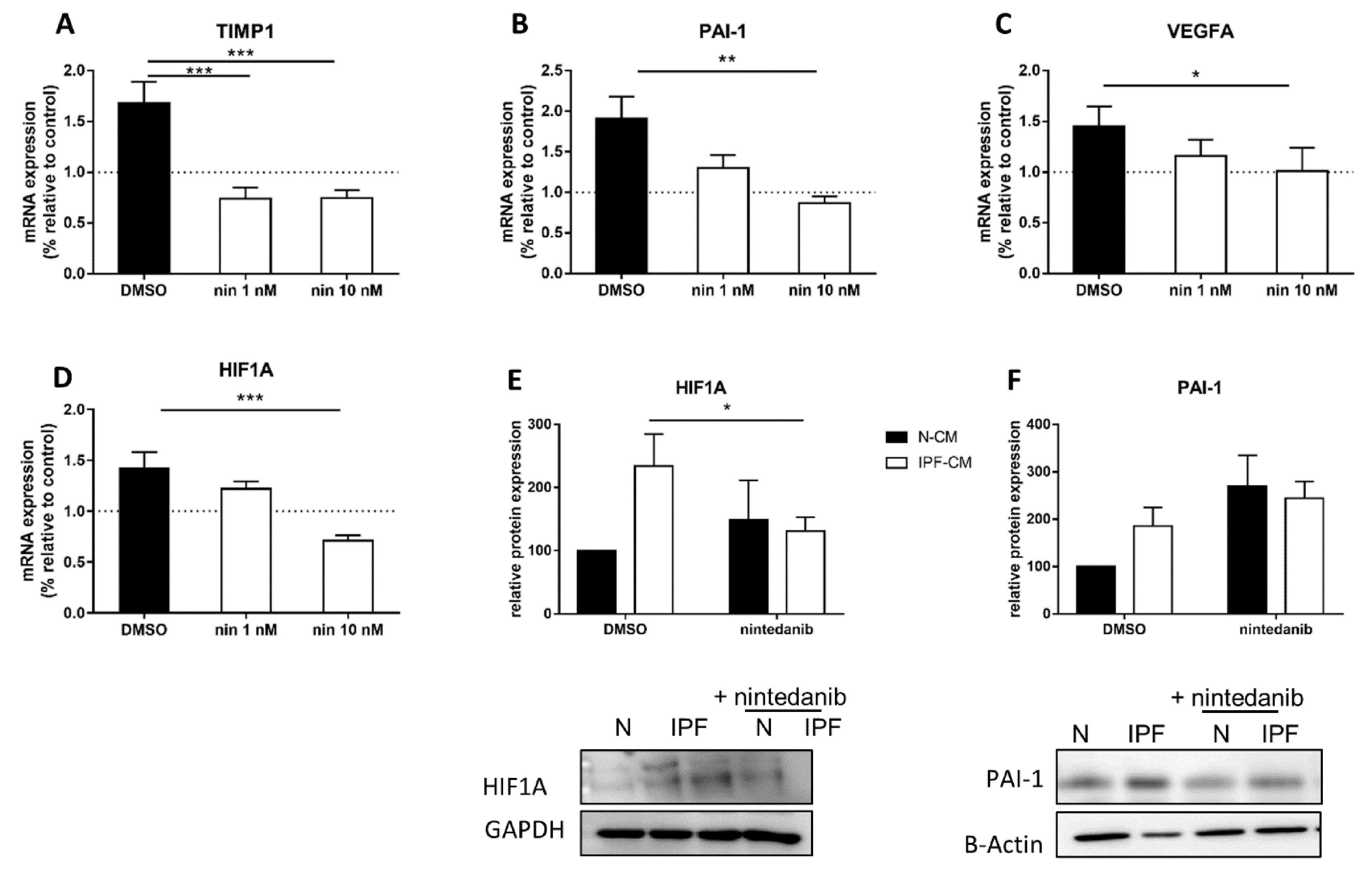
2.3. Nintedanib Prevents the Elevation of HIF1α-Related Targets
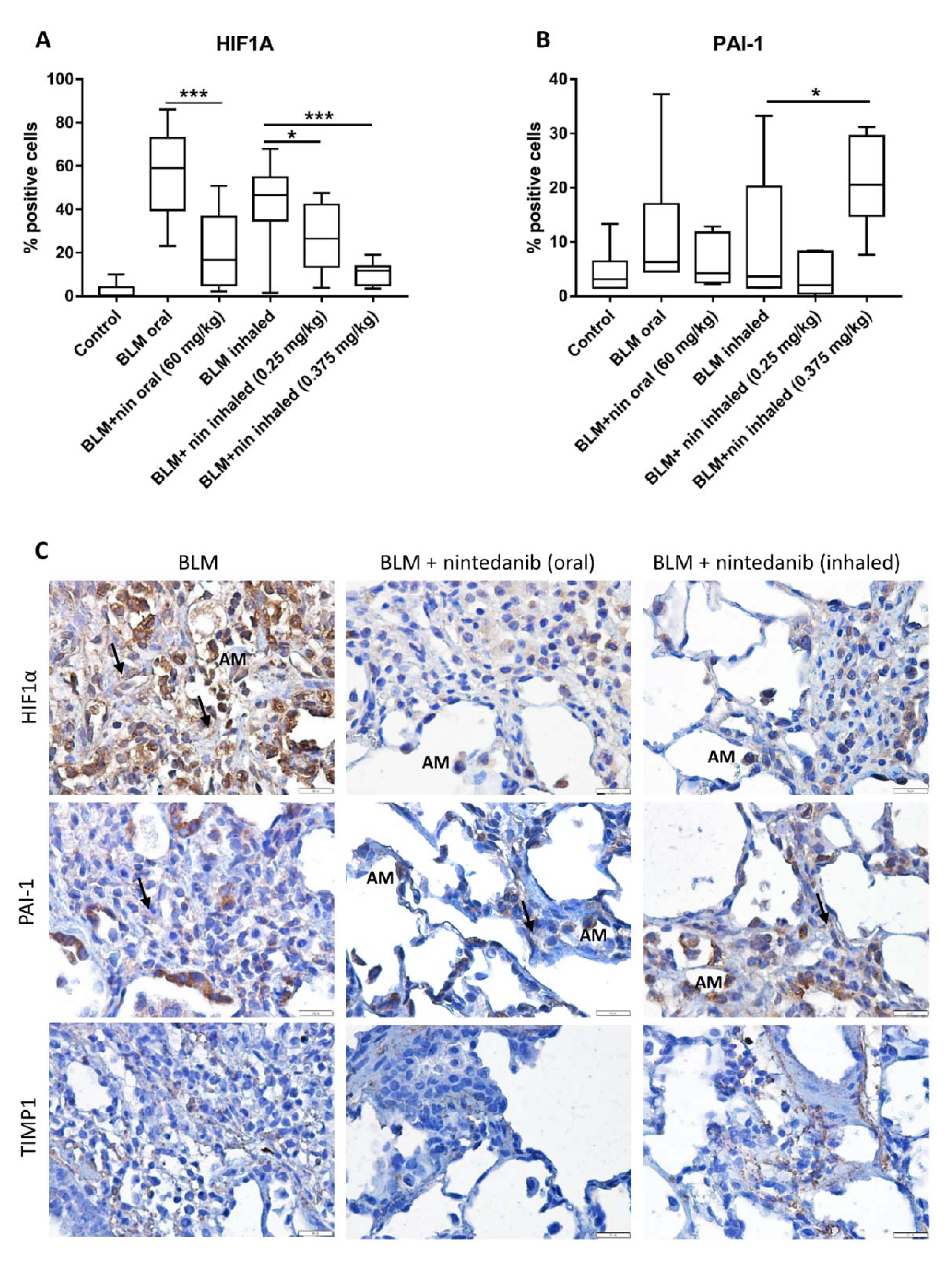
2.4. Rat Bleomycin Model Shows Elevated Levels of HIF1α within Fibrotic Tissue
3. Discussion
4. Materials and Methods
4.1. Primary HLF Culture
4.2. IPF-CM Model
4.3. Western Blot
4.4. Real Time Quantitative PCR (qPCR)
4.5. RNA-Sequencing
4.6. Rat Bleomycin Model
4.7. Immunohistochemistry (IHC)
4.8. Statistical Analysis
4.9. Ethics Statement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gribbin, J.; Hubbard, R.B.; Le Jeune, I.; Smith, C.J.; West, J.; Tata, L.J. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006, 61, 980–985. [Google Scholar] [CrossRef]
- Fielding, C.A.; Jones, G.W.; McLoughlin, R.M.; McLeod, L.; Hammond, V.J.; Uceda, J.; Williams, A.S.; Lambie, M.; Foster, T.L.; Liao, C.T.; et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity 2014, 40, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef]
- Epstein Shochet, G.; Wollin, L.; Shitrit, D. Fibroblast-matrix interplay: Nintedanib and pirfenidone modulate the effect of IPF fibroblast-conditioned matrix on normal fibroblast phenotype. Respirology 2018, 23, 756–763. [Google Scholar] [CrossRef]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Kurundkar, A.; Kurundkar, D.; Bernard, K.; Sanders, Y.Y.; Ding, Q.; Antony, V.B.; Zhang, J.; Zmijewski, J.; Thannickal, V.J. Novel Mechanisms for the Antifibrotic Action of Nintedanib. Am. J. Respir. Cell Mol. Biol. 2016, 54, 51–59. [Google Scholar] [CrossRef]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [PubMed]
- Surber, M.W.; Beck, S.; Pham, S.; Marsden, A.T.; Gandi, S.K.; Baily, J.; McElroy, M.C. Inhaled nintedanib is well-tolerated and delivers key pharmacokinetic parameters required to treat bleomycin-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2020, 63, 101938. [Google Scholar] [CrossRef]
- Epstein-Shochet, G.; Pham, S.; Beck, S.; Naiel, S.; Mekhael, O.; Revill, S.; Hayat, A.; Vierhout, M.; Bardestein-Wald, B.; Shitrit, D.; et al. Inhalation: A means to explore and optimize nintedanib’s pharmacokinetic/pharmacodynamic relationship. Pulm. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef]
- Wang, Y.; Yella, J.K.; Ghandikota, S.; Cherukuri, T.C.; Ediga, H.H.; Madala, S.K.; Jegga, A.G. Pan-transcriptome-based Candidate Therapeutic Discovery for Idiopathic Pulmonary Fibrosis. bioRxiv 2020, 824367. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef]
- Yoshiji, H.; Kuriyama, S.; Miyamoto, Y.; Thorgeirsson, U.P.; Gomez, D.E.; Kawata, M.; Yoshii, J.; Ikenaka, Y.; Noguchi, R.; Tsujinoue, H.; et al. Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology 2000, 32, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Flower, V.A.; Pauling, J.D.; Millar, A.B. VEGF (Vascular Endothelial Growth Factor) and Fibrotic Lung Disease. Int. J. Mol. Sci. 2018, 19, 1269. [Google Scholar] [CrossRef]
- Overed-Sayer, C.; Miranda, E.; Dunmore, R.; Liarte Marin, E.; Beloki, L.; Rassl, D.; Parfrey, H.; Carruthers, A.; Chahboub, A.; Koch, S.; et al. Inhibition of mast cells: A novel mechanism by which nintedanib may elicit anti-fibrotic effects. Thorax 2020, 75, 754–763. [Google Scholar] [CrossRef]
- Xiong, A.; Liu, Y. Targeting Hypoxia Inducible Factors-1alpha As a Novel Therapy in Fibrosis. Front. Pharmacol. 2017, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Huang, C.; Meng, X.M.; Song, Y.; Wu, X.Q.; Yang, Y.; Li, J. Hypoxia-inducible factor-1alpha in hepatic fibrosis: A promising therapeutic target. Biochimie 2015, 108, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Keogh, C.E. Respiratory gases and the regulation of transcription. Exp. Physiol. 2016, 101, 986–1002. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen-regulated transcription factors and their role in pulmonary disease. Respir. Res. 2000, 1, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Lokmic, Z.; Musyoka, J.; Hewitson, T.D.; Darby, I.A. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int. Rev. Cell Mol. Biol. 2012, 296, 139–185. [Google Scholar] [CrossRef]
- Goodwin, J.; Choi, H.; Hsieh, M.H.; Neugent, M.L.; Ahn, J.M.; Hayenga, H.N.; Singh, P.K.; Shackelford, D.B.; Lee, I.K.; Shulaev, V.; et al. Targeting Hypoxia-Inducible Factor-1alpha/Pyruvate Dehydrogenase Kinase 1 Axis by Dichloroacetate Suppresses Bleomycin-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 216–231. [Google Scholar] [CrossRef]
- McDonough, J.E.; Ahangari, F.; Li, Q.; Jain, S.; Verleden, S.E.; Herazo-Maya, J.; Vukmirovic, M.; DeIuliis, G.; Tzouvelekis, A.; Tanabe, N.; et al. Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Philip, K.; Mills, T.W.; Davies, J.; Chen, N.Y.; Karmouty-Quintana, H.; Luo, F.; Molina, J.G.; Amione-Guerra, J.; Sinha, N.; Guha, A.; et al. HIF1A up-regulates the ADORA2B receptor on alternatively activated macrophages and contributes to pulmonary fibrosis. FASEB J. 2017, 31, 4745–4758. [Google Scholar] [CrossRef] [PubMed]
- Delbrel, E.; Soumare, A.; Naguez, A.; Label, R.; Bernard, O.; Bruhat, A.; Fafournoux, P.; Tremblais, G.; Marchant, D.; Gille, T.; et al. HIF-1alpha triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci. Rep. 2018, 8, 17939. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119. [Google Scholar] [CrossRef]
- Burman, A.; Kropski, J.A.; Calvi, C.L.; Serezani, A.P.; Pascoalino, B.D.; Han, W.; Sherrill, T.; Gleaves, L.; Lawson, W.E.; Young, L.R.; et al. Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes, revisited. Trends Genet. 2013, 29, 569–574. [Google Scholar] [CrossRef]
- Zhang, H.; Akman, H.O.; Smith, E.L.; Zhao, J.; Murphy-Ullrich, J.E.; Batuman, O.A. Cellular response to hypoxia involves signaling via Smad proteins. Blood 2003, 101, 2253–2260. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.K.; Hubchak, S.; Hayashida, T.; Runyan, C.E.; Schumacker, P.T.; Schnaper, H.W. Interdependence of HIF-1alpha and TGF-beta/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am. J. Physiol. Renal. Physiol. 2011, 300, F898–F905. [Google Scholar] [CrossRef]
- Aquino-Galvez, A.; Gonzalez-Avila, G.; Jimenez-Sanchez, L.L.; Maldonado-Martinez, H.A.; Cisneros, J.; Toscano-Marquez, F.; Castillejos-Lopez, M.; Torres-Espindola, L.M.; Velazquez-Cruz, R.; Rodriguez, V.H.O.; et al. Dysregulated expression of hypoxia-inducible factors augments myofibroblasts differentiation in idiopathic pulmonary fibrosis. Respir. Res. 2019, 20, 130. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1alpha mediates TGF-beta-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef]
- Kaminski, N.; Rosas, I.O. Gene expression profiling as a window into idiopathic pulmonary fibrosis pathogenesis: Can we identify the right target genes? Proc. Am. Thorac. Soc. 2006, 3, 339–344. [Google Scholar] [CrossRef]
- Olman, M.A.; Mackman, N.; Gladson, C.L.; Moser, K.M.; Loskutoff, D.J. Changes in procoagulant and fibrinolytic gene expression during bleomycin-induced lung injury in the mouse. J. Clin. Investig. 1995, 96, 1621–1630. [Google Scholar] [CrossRef]
- Hamada, N.; Kuwano, K.; Yamada, M.; Hagimoto, N.; Hiasa, K.; Egashira, K.; Nakashima, N.; Maeyama, T.; Yoshimi, M.; Nakanishi, Y. Anti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in mice. J. Immunol. 2005, 175, 1224–1231. [Google Scholar] [CrossRef]
- Fehrenbach, H.; Kasper, M.; Haase, M.; Schuh, D.; Muller, M. Differential immunolocalization of VEGF in rat and human adult lung, and in experimental rat lung fibrosis: Light, fluorescence, and electron microscopy. Anat. Rec. 1999, 254, 61–73. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Anevlavis, S.; Bouros, D. Angiogenesis in interstitial lung diseases: A pathogenetic hallmark or a bystander? Respir. Res. 2006, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Shochet, G.E.; Pomerantz, A.; Shitrit, D.; Bardenstein-Wald, B.; Ask, K.; Surber, M.; Rabinowicz, N.; Levy, Y.; Benchetrit, S.; Edelstein, E.; et al. Galectin-3 levels are elevated following nintedanib treatment. Ther. Adv. Chronic Dis. 2020, 11, 2040622320968412. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant. Biol 2011, 76, 347–353. [Google Scholar] [CrossRef]
- Zhao, X.; Psarianos, P.; Ghoraie, L.S.; Yip, K.; Goldstein, D.; Gilbert, R.; Witterick, I.; Pang, H.; Hussain, A.; Lee, J.H.; et al. Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat. Metab. 2019, 1, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; Mutlu, G.M. A metabolic strategy to reverse fibrosis? Nat. Metab. 2019, 1, 12–13. [Google Scholar] [CrossRef]
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G.; Hu, J.Z.; et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751. [Google Scholar] [CrossRef]
- Andrianifahanana, M.; Hernandez, D.M.; Yin, X.; Kang, J.H.; Jung, M.Y.; Wang, Y.; Yi, E.S.; Roden, A.C.; Limper, A.H.; Leof, E.B. Profibrotic up-regulation of glucose transporter 1 by TGF-beta involves activation of MEK and mammalian target of rapamycin complex 2 pathways. FASEB J. 2016, 30, 3733–3744. [Google Scholar] [CrossRef]
- Epstein Shochet, G.; Brook, E.; Israeli-Shani, L.; Edelstein, E.; Shitrit, D. Fibroblast paracrine TNF-alpha signaling elevates integrin A5 expression in idiopathic pulmonary fibrosis (IPF). Respir. Res. 2017, 18, 122. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | FC | pV | # Reads |
|---|---|---|---|---|
| SLC2A1 | solute carrier family 2 member 1 | 1.456 | 0.00034 | 50–200 |
| ENO2 | Enolase 2 | 1.362 | 6.01E-06 | 300–800 |
| IGF1R | Insulin-like growth factor 1 receptor | 1.360 | 0.10716 | 30–80 |
| EIF4EBP1 | Eukaryotic translation initiation factor 4E-binding protein 1 | 1.355 | 0.14223 | 20–40 |
| TFRC | Transferrin receptor | 1.13 | 0.03398 | 570–800 |
| VEGFA | vascular endothelial growth factor A | 1.281 | 0.00117 | 1440–3400 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase | 1.219 | 0.00016 | 9–13K |
| SERPINE1 | serpin family E member 1 (PAI-1) | 1.160 | 0.02021 | 2000–4200 |
| TIMP1 | Tissue metallopeptidase inhibitor 1 | 1.133 | 0.00372 | 12–25K |
| FLT1 | Fms related tyrosine kinase 1 | 2.27 | 0.02767 | 3–12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Epstein Shochet, G.; Bardenstein-Wald, B.; McElroy, M.; Kukuy, A.; Surber, M.; Edelstein, E.; Pertzov, B.; Kramer, M.R.; Shitrit, D. Hypoxia Inducible Factor 1A Supports a Pro-Fibrotic Phenotype Loop in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 3331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073331
Epstein Shochet G, Bardenstein-Wald B, McElroy M, Kukuy A, Surber M, Edelstein E, Pertzov B, Kramer MR, Shitrit D. Hypoxia Inducible Factor 1A Supports a Pro-Fibrotic Phenotype Loop in Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences. 2021; 22(7):3331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073331
Chicago/Turabian StyleEpstein Shochet, Gali, Becky Bardenstein-Wald, Mary McElroy, Andrew Kukuy, Mark Surber, Evgeny Edelstein, Barak Pertzov, Mordechai Reuven Kramer, and David Shitrit. 2021. "Hypoxia Inducible Factor 1A Supports a Pro-Fibrotic Phenotype Loop in Idiopathic Pulmonary Fibrosis" International Journal of Molecular Sciences 22, no. 7: 3331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073331