
Participation of ABCA1 Transporter in Pathogenesis of Chronic Obstructive Pulmonary Disease


Department of Nursing, Ryazan State Medical University, 390026 Ryazan, Russia
Int. J. Mol. Sci. 2021, 22(7), 3334; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073334
Submission received: 3 March 2021
/
Revised: 19 March 2021
/
Accepted: 22 March 2021
/
Published: 24 March 2021
(This article belongs to the Special Issue High-Density Lipoproteins in Non-cardiovascular Diseases)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Chronic obstructive pulmonary disease (COPD) is the important medical and social problem. According to modern concepts, COPD is a chronic inflammatory disease, macrophages play a key role in its pathogenesis. Macrophages are heterogeneous in their functions, which is largely determined by their immunometabolic profile, as well as the features of lipid homeostasis, in which the ATP binding cassette transporter A1 (ABCA1) plays an essential role. The objective of this work is the analysis of the ABCA1 protein participation and the function of reverse cholesterol transport in the pathogenesis of COPD. The expression of the ABCA1 gene in lung tissues takes the second place after the liver, which indicates the important role of the carrier in lung function. The participation of the transporter in the development of COPD consists in provision of lipid metabolism, regulation of inflammation, phagocytosis, and apoptosis. Violation of the processes in which ABCA1 is involved may be a part of the pathophysiological mechanisms, leading to the formation of a heterogeneous clinical course of the disease.
1. Introduction
Chronic obstructive pulmonary disease (COPD) is one of the most widespread diseases, it has great medical significance due to the high frequency of temporary and persistent disability and mortality. Social and economic burden of the disease is becoming more evident for health care systems as well as patients [1,2,3]. COPD is characterized by a steadily progressive course, it has heterogeneous clinical manifestations and it is often associated with a wide range of comorbid diseases, among which cardiovascular diseases take the key position [4]. The inflammation, which is due to prolonged exposure to smoking, is the basis of the development and progression of the disease [4,5]. Despite this, there are relatively few research works describing the triggering events and inadequate regulatory mechanisms, corresponding to them, as well as the subsequent cellular and molecular processes that lead to tissue damage and remodeling.
Inflammation in COPD is characterized by the participation of many cells and humoral factors and is believed to have local and systemic components that can be the basis for the development of comorbid diseases. Comorbidity is a part of the extrapulmonary clinical heterogeneity of COPD and in many respects it determines the nature of the course and prognosis.
Macrophages—key participants in the innate link of the immune system, as indicated by a significant increase in their number in the lungs, take an important place in the pathogenesis of COPD [6,7,8]. It is impossible not to note the role of lipid metabolism disorders besides the innate immune system, to the involvement of which in the development of bronchial inflammation many works are devoted. Macrophages are actively involved in the processes under consideration, and immunometabolic investigations that have been developed in recent years show the importance of lipid metabolism disorders in the basis of dysregulation of the immune response [9]. Cross-links and their disruptions in the regulation of lipid metabolism, the innate immune system, and inflammation may be the key to understanding the pathophysiological bases of COPD.
In this regard, information about the participation of cholesterol and its metabolic pathways in the immune response and inflammation is of considerable clinical interest. The objective of this article is the analysis of the ATP binding cassette transporter A1 (ABCA1 transporter) participation and the function of reverse cholesterol transport in the pathogenesis of COPD. The ABCA1 transporter has a lot of functions, the key to which is participation in the provision of lipid homeostasis of cells. The transport function, the nature of the transported substrates and the cellular localization of the protein have determined its role in many processes and mechanisms underlying the pathogenesis of COPD.
2. Disorders of Lipid Metabolism in the Development and Progression of COPD
Despite the active development of medicine, the current understanding of the natural course of COPD is incomplete and often contradictory [10]. It is known that the development and progression of the disease can be very different in different patients, and its onset is not possible to establish. The molecular mechanisms underlying the processes that determine the clinical picture of COPD in different patients or even in one patient at different times, are largely unclear.
Negative dynamics of lung function remains the main criterion for the diagnostics of COPD and a key predictor of the prognosis. The data about an association between a decrease in the airflow parameters and the level of high-density lipoprotein (HDL) cholesterol are interesting. Oelsner E. et al. showed on the basis of a seven-year analysis of a large sample, that higher HDL cholesterol was associated with a higher rate of decrease in FEV1 (p < 0.0001) and FEV1/FVC (p < 0.0001). The magnitude of indicated effect was similar to the 10-year increase in the pack-years index [11]. Previously the risk of emphysema by 0.4% for every 10 mg/dl increase in HDL cholesterol has already been stated in the MESA LungStudy [12].
The role of high HDL level in the reduction of lung function and in the progression of emphysema has been described previously [12,13,14,15], and it has already been associated with the participation of apolipoprotein M in the previous studies [14,15]. However, there are other data available from the literature indicating a positive association of high HDL levels and pulmonary function. A decrease in HDL in patients with COPD with more severe stages is also noted [15,16,17,18]. It is demonstrated in the recent study that the best predictor of pulmonary function in patients with COPD can be the ratio of lymphocytes to HDL [19]. It is also shown that HDL levels decrease in patients with COPD, who have undergone lung transplantation [20].
In general, these ambiguous data do not correspond to the protective role generally accepted for HDL, according to which they are well known in the pathogenesis of atherosclerosis [21,22]. The results are of more interest if we take into account that COPD and atherosclerosis are often combined and have similar trends of progression.
The presented data allow to suggest that the processes associated with the formation and function of HDL play a significant role in lung function. The differences shown in the data can be related to the clinical heterogeneity of COPD patients, which is associated with individual features of pathophysiological mechanisms and lipid metabolism.
It is necessary to emphasize that the high levels of HDL in the presented studies corresponded to the development of the emphysematous phenotype of COPD. It is known that patients belonging to another, the so-called bronchitic type of COPD, suffer from concomitant cardiovascular diseases, which is associated with an increased predisposition to the development of atherosclerosis in this category of patients. The prevalence of cardiovascular disease and metabolic syndrome in patients with lower body mass index and emphysema is generally relatively low [23,24].
Although the exact mechanisms of the relationship of lung function and the development of emphysema with HDL levels are not clear, various explanations are proposed, for example, the intake of glucocorticosteroids by patients with more severe course of COPD [25].
Thus, the obvious involvement of lipid metabolism in the pathogenesis of COPD, demonstrated by numerous studies, determines the need for a better study of the mechanisms of participation of ABCA1 and reverse cholesterol transport.
In the modern scientific literature there are quite a lot of data describing the biological functions of ABCA1. It belongs to a large family of membrane proteins that transport chemically diverse substrates through the lipid bilayer of cell membranes, accompanied by ATP hydrolysis. In clinical practice mutations in the ABCA1 gene are known as the cause of a rare genetic disease-Tangier disease. The disease is characterized by a significant decrease in HDL level and a high incidence of cardiovascular diseases [26,27,28].
ABCA1 is localized on the plasma membrane of cells and is expressed in many organs and tissues. It participates in the reverse transport of cholesterol, exporting cholesterol and phospholipids from cells to extracellular acceptors [29,30,31]. In addition to the plasma membrane, ABCA1 is also found in the Golgi complex and in lysosomes, which confirms the information about the mobility of the transporter, which can move between the plasma membrane, the Golgi complex, and lysosomes, ensuring the functioning of the lipid transport route [32,33,34,35,36].
ABCA1 expression has complex regulatory pathways that are carried out both at the transcriptional and post-transcriptional levels [37,38,39]. Excess of cholesterol in macrophages leads to the formation of oxysterols that stimulate ABCA1 expression via LXR (liver X receptor) [40,41,42,43]. LXR forms a heterodimer with RXR (retinoid X receptor), and they form a transcription factor together, connecting with specific sites on the ABCA1 gene promoter for ABCA1 expression increase [44,45,46,47].
The participation of the ABCA1 transporter in the formation of HDL has determined its leading role in the pathogenesis of atherosclerosis, which is the subject of researchers’ close attention [48]. However, in recent years there is more and more evidence that the transporter is also involved in the regulation of inflammation [27], which is carried out through various mechanisms, including participation in the immune response and phagocytosis, which increases the interest of clinicians in ABCA1 as the pathogenesis link of other diseases in addition to atherosclerosis [49,50].
The important role of both the transporter itself and lipid homeostasis in lung function in general is indicated by the fact that the expression of ABCA1 in lung tissues takes the second place after the liver [26]. This observation is extremely important, taking into account the great clinical significance of lipids for respiratory function and the influence of oxidative processes to which lipids are exposed during smoking. To the greatest extent, ABCA1 is present in alveolar macrophages, alveolar pneumocytes of types I and II [51,52], which characterizes its involvement in various biological processes [53]. Indeed, the function of the protein is not limited to the simple exchange of cholesterol and understanding all the mechanisms in which the transporter is involved requires serious research.
3. Participation of ABCA1 in the Regulation of Inflammation in COPD
It is believed that the ABCA1 transporter can participate in the regulation of inflammation in the lungs. The mechanisms of this involvement are diverse and in many respects are not studied completely. It is assumed that the intracellular accumulation of cholesterol in macrophages acts as a trigger of the cellular inflammatory response [54,55]. Consequently, the ABCA1 transport activity can be anti-inflammatory, through the removal of cholesterol excess [26,56,57,58,59].
There is evidence that cigarette smoke modulates signaling pathways that regulate ABCA1 expression in macrophages [60,61]. The reduced ABCA1 transport activity due to smoking leads to intracellular accumulation of cholesterol or even the formation of so-called “foam cells” [62,63,64]. Research data have shown that ABCA1 expression changes in COPD [60,65]. It was found that in patients with moderate and severe COPD ABCA1 expression in lung tissues was for certain lower than in healthy subjects [60].
The role of ABCA1 in pulmonary inflammation is well demonstrated in mouse models. In ABCA1 knockout mice, compared to the wild type a violation of lipid metabolism is observed, including a significant decrease in plasma the level of cholesterol and phospholipids (by about 70%) due to almost undetectable levels of HDL and ApoA-I [66]. Analyzing these data, it is necessary to note that mice differ from humans in a number of features of HDL metabolism (in mice, unlike humans, protein CETP (cholesterol ester transfer protein) in the blood plasma is absent, it transfers cholesterol esters from HDL to LDL and VLDL containing AрoВ, as a result of which they have naturally low LDL and high HDL levels) [67,68].
In the ABCA1 knockout mice, pulmonary focal lesions were found, including thickening of the interalveolar septa, foam alveolar macrophages and hyperplasia of type II alveolar pneumocytes, the surfactant was characterized by alveolar proteinosis [69,70,71]. With age, the alveolar architecture of these mice was destroyed, and the remaining alveoli were epithelized due to severe hypertrophy and hyperplasia of type II pneumocytes [69,70]. The importance of ABCA1 in normal lung physiology is confirmed by the fact that in mice with ApoA-I knockout increased infiltration by inflammatory cells (especially neutrophils), collagen deposition and airway hyperreactivity, as well as impaired lung vasodilation were observed [69,70,71]. These observations demonstrate the role of ABCA1-mediated reverse lipid transport in inflammatory lung diseases.
3.1. Participation of ABCA1 in the Regulation of Inflammation with the Participation of TLR4
The lungs contact constantly with a huge number of pollutants and pathogenic microorganisms through the inhaled air, and therefore they are populated with immune cells densely. As the first line of defense for the lungs, the innate immune system is thought to rely on a large family of PRR (pattern-recognition receptors) for detection of standard molecular structures (patterns) specific to large groups of pathogens, including viruses, bacteria, fungi, parasites, and protozoa. An important role in the initiation of inflammation in COPD is played by Toll-like receptors (TLR) of macrophages, which are a family of transmembrane receptors that are expressed by many cell types, including epithelial cells, endothelial cells, monocytes, macrophages, dendritic cells, and T- and B-lymphocytes [72,73].
The most studied Toll-like receptor TLR4 recognizes lipopolysaccharides of the cell wall of Gram-negative bacteria (LPS) and is localized both on the plasma membrane and in endosomes. When LPS is being recognized, conformational changes in TLR4 receptors lead to the recruitment of its intracellular toll-interleukin 1 receptor (TIR) domain (TIR-domains) containing molecules-adapters, realizing MyD88-dependent and MyD88-independent pathways.
Signaling via TLR4 is of importance in COPD [74,75,76]. In addition to inflammation, it is involved in other processes, such as angiogenesis [77]. TLR4 in the lungs can be activated by either LPS or exogenous oxidants [78] and, consequently, modulate inflammatory responses. It is known that the components of cigarette smoke are also able to activate TLR4, which is important, taking into account its role in the etiology of COPD [79,80].
The evidence that saturated fatty acids can also activate TLR4 is of considerable interest [81,82]. Moreover, unlike saturated fatty acids, unsaturated ones do not have such an effect [82]. These data support clinical observations in which overweight and obese people showed increased TLR4 expression on peripheral blood mononuclear cells and in adipose tissue compared to low body weight, and TLR4 expression levels increased significantly with increase of body mass index [83,84]. Interestingly, that polyunsaturated fatty acids can destabilize ABCA1, disrupt the reverse transport of cholesterol and HDL formation [85,86,87,88,89]. This information is relevant when analyzing the comorbid relationship between COPD and atherosclerosis. There is an assumption that the prevalence of cardiovascular diseases (in particular, coronary artery disease (CAD) and peripheral artery disease (PAD)) may be higher in individuals with a higher body mass index (BMI) and, as it has been noted previously, predominantly bronchitic form of COPD [23,24,90]. At the same time, a low BMI increases the risk of mortality in patients with COPD [91,92], and overweight and obesity are positive predictors of long-term survival [93,94]. This phenomenon got the name “the obesity paradox” [95,96,97]. Its mechanisms are not completely clear, but participation of cytokines, contributing to the development of cachexia, such as TNF (Tumor Necrosis Factor) and IL-6 (Interleukin-6), is assumed [98,99]. TNF is a proinflammatory cytokine that is highly expressed in COPD patients [100]. It is known that TNF activates a number of signaling pathways that induce changes in ABCA1 expression [100], including nuclear factor-kB (NF-kB), sterol regulatory element binding protein 2 (SREBP-2), and janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) [101,102,103]. However, currently there is contradictory information about the TNF role in the expression of ABCA1 in macrophages and reverse cholesterol transport. In foamy cells derived from THP-1 macrophages, TNF suppresses ABCA1 expression via the NF-kB-dependent pathway [104], while in mouse peritoneal macrophages it induces ABCA1 expression [105].
The results of investigations show that the ABCA1 expression in macrophages can play a physiologically significant role, also through the removal of LPS from macrophages, which helps to restore a normal immune response.
Although TLRs have developed evolutionarily for detection of exogenous pathogens, providing innate immune responses, it is becoming apparent that TLR4 activation is also modulated by endogenous molecules, including lipids.
Recent data suggest that TLR 4, when activated, is localized in the so-called “lipid rafts” of plasma membranes, which can regulate its activity [106]. The plasma membrane is heterogeneous in its structure and functions. The structure of the plasma membrane that separates cells from their environment is the subject of numerous studies. According to current data, the plasma membrane is represented by a lipid bilayer containing various types of lipids distributed asymmetrically between two bilayer sheets in which proteins are embedded. In addition, plasma membranes are assumed to contain so-called lipid rafts-dynamic signaling platforms, which are areas of the plasma membrane enriched with glycosphingolipids, sphingomyelin, cholesterol, glycophosphatidylinositol anchor proteins and signaling proteins [104,107,108,109].
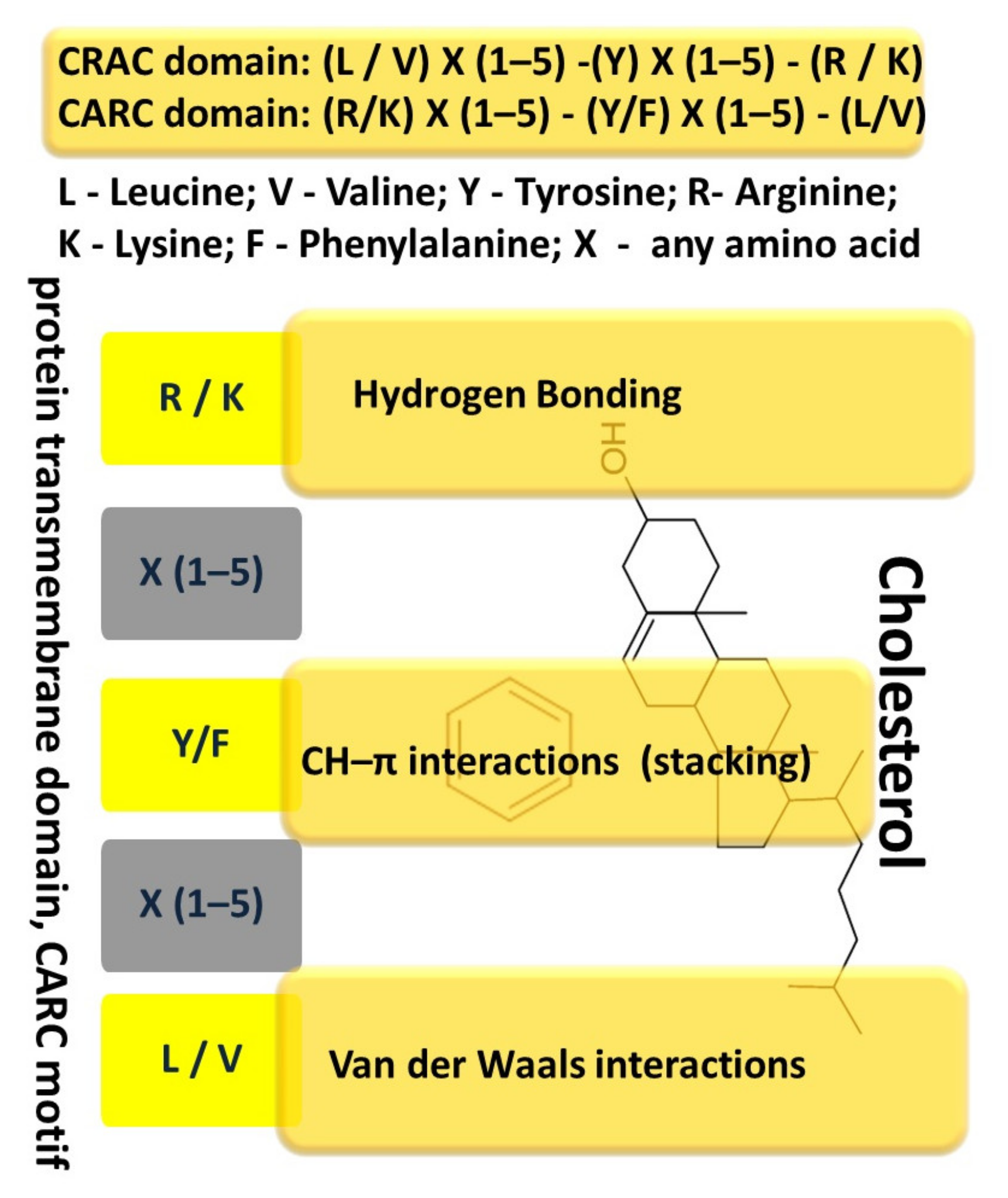
Cholesterol is the main component of lipid rafts, accounting for about 50% of the lipids present in these domains. Cholesterol has many functions, including participation in maintaining the spatial structure of the plasma membrane. In this case, most cholesterol molecules are located by their hydroxyl groups close to the glycerol region of the framework of the lipid bilayer, and their hydrophobic rings are located in the hydrophobic core of the bilayer. Changes in the cholesterol content in the plasma membrane affect its structure and function. Due to the influence on the biophysical properties of the membrane, as well as through the direct interaction of the sterol with specific protein sites, cholesterol can participate in the regulation of the function of transmembrane proteins [110]. It has been shown that proteins that interact with cholesterol or bind it may contain characteristic amino acid sequences that play a definite role in this interaction [106].
One such known sequence, the amino acid cholesterol-binding domain (CRAC, Cholesterol Recognition/interaction Amino acid Consensus sequence), has been identified in proteins that interact with cholesterol or are regulated by it [102,111,112,113]. The amino acid sequence, referred to as CRAC, is defined by the following set of amino acids: (L/V)–X(1–5)–(Y)–X(1–5)–(R/K) [112,112], the CARC motive, has similar properties in binding to transmembrane proteins and has an inverse sequence of amino acids: (R/K)–X(1–5)−(Y/F)–X(1-5)−(L/V) (with X = any amino acid), and tyrosine can be replaced by phenylalanine [112] (Figure 1).
Taking into account, that the probability of the presence of such a domain is very high for many proteins, it is believed that the very presence of CRAC does not indicate the occurrence of specific cholesterol–protein interactions [112,114] and experimental data are necessary for their confirmation [115]. It is also assumed that the CRAC sequence contributes to the localization of membrane proteins in lipid rafts to a certain extent [116]. The presence of several CRAC or CARC sequences, in the transmembrane region or near it, may indicate a possible participation of cholesterol in the accomplishment of protein function.
Analysis of the TLR4 receptor structure allows to reveal the presence of both CRAC and CARC sequences near the transmembrane domain, which can provide a link between cholesterol and the regulation of signal transduction of the receptor [106]. Interestingly, that the CARC-CRAC-CARC domains in TLR4 are located close to the membrane, in front of the TIR domain. This may indicate that this intracellular region of TLR4 binds cholesterol specifically [106].
The interaction of transmembrane proteins, including TLR4, with cholesterol is possible due to its structure. Cholesterol is a polycyclic amphipathic molecule derived from sterane. The cholesterol molecule has a polar and apolar parts. The polar part is represented by a hydroxyl group, which allows to establish hydrogen bonds. The apolar part has an asymmetric structure, including a flat α surface and a β surface with aliphatic groups (two methyl groups and a terminal isooctyl chain). Sphingolipids usually interact with the α-surface of cholesterol, and transmembrane domains of proteins interact with the β-face [112,117]. It is believed that the side chains of branched amino acids, such as valine or leucine, can “permeate” these aliphatic groups and therefore they are particularly suitable for association with the β surface of cholesterol [112] through numerous van der Waals contacts between these residues and cholesterol. The interaction between the aromatic amino acid and cholesterol occurs in the apolar region of the membrane, far from the hydroxyl group of cholesterol, and the interaction with cholesterol is mediated almost exclusively by the CH-π-stacking binding between the aromatic ring of the amino acid (either tyrosine or phenylalanine) and one of the sterane rings of cholesterol [110,117] (Figure 1).
Thus, ABCA1, by changing the cholesterol content in the plasma membranes of macrophages and ensuring the stability of lipid rafts, can regulate the activity of TLR4 [60], which can occur in COPD. In its turn, TLR4 activation inhibits ABCA1 expression, which reduces the outflow of cholesterol from macrophages [118,119]. The activity of IRAK1 (IL-1R-associated kinase 1) plays a key role in this process [120].
Studies of the biological role of ABCA1 have determined that in addition to lipid export, it can mediate intracellular cholesterol transport and allow the movement of lipids between the inner and outer sheets of the plasma membrane [28,121]. These findings have improved our understanding of the role of the ABCA1 transporter in inflammation through the regulation of phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] in plasma membranes. PI(4,5)P2 is the main cellular type of PIP and it is mainly localized in the inner sheet of the plasma membrane, where it is involved in many cellular processes, such as endocytosis, exocytosis, protein transport, and receptor-mediated signal transduction [122,123,124,125,126]. In the plasma membrane, it is concentrated mainly in lipid rafts. It has been found out that ABCA1 participates in the exchange of PI (4,5)P2, redistributing it from the inner to the outer sheet of the plasma membrane [123,125]. It is believed that PI (4,5) P2, moved to the outer side of the plasma membrane, ensures the binding of apoA-I and the outflow of lipids during the formation of HDL. In addition to it, ABCA1 exports PI (4,5)P2 to nascent HDL, reducing its amount in the plasma membrane [122]. Accordingly, a decrease in the functional activity of ABCA1 may increase the amount of PI (4,5)P2 in the inner sheet of the plasma membrane.
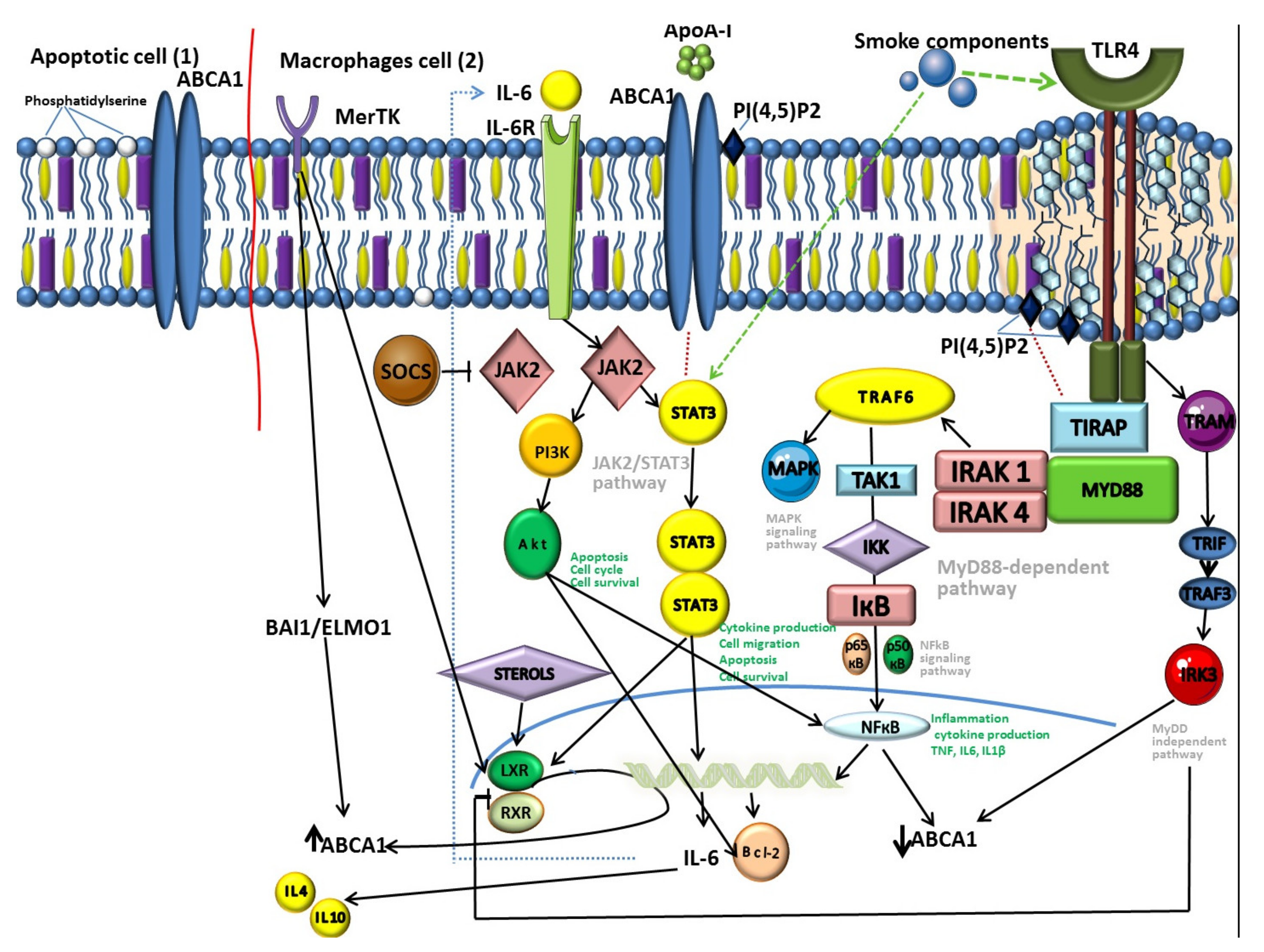
Recent data have shown that PI (4,5)P2, localized in the outer sheet of the plasma membrane, participates in the regulation of cell adhesion and cell motility [126]. One of the most important biological functions of PI(4,5)P2 is related to the fact that it is necessary for the functioning of the sorting adapter TIRAP, a member of the MyD88-dependent TLR4 pathway (Figure 2) [127,128,129]. The MyD88-dependent pathway is regulated by two adapter-associated proteins: Myeloid differentiation primary response gene (88) (MyD88) and toll-interleukin 1 receptor (TIR) domain containing adapter protein (TIRAP) [130].
The MyD88 protein is a key link in the inflammatory signaling pathways of Toll-like receptors (TLR) and interleukin-1 (IL-1) receptors [131]. MyD88 forms a protein complex with kinases of the interleukin-1 receptor-associated kinase (IRAK) family, called the Middosome. MyD88 has a limited ability to interact directly with TLR. This requires an intermediate protein that binds activated TLRs to MyD88. Most plasma membrane-localized TLRs, including TLR4, use the TIRAP sorting adapter to recruit MyD88. The ability of TIRAP to function as a sorting adapter depends on its N-terminal motive rich in positively charged lysine residues [106,128], which interacts with PI (4,5) P2 and other lipids (phosphatidylserine (PS)) localized on the plasma membrane.
3.2. ABCA1 Cross-Links and JAK2/STAT3 Pathways
It is interesting to learn about another mechanism associated with inflammation, in which the transporter is involved, and which is implemented in parallel with the reverse transport of cholesterol. It has been found out that the interaction of ABCA1 and ApoA-I increases phosphorylation and activates JAK2, which, in its turn, increases the binding activity of ApoA-I and ABCA1, which is responsible for the export of lipids [57,134,135]. At the same time, JAK2 increases the transport activity of ABCA1 [136,137], which is known to have an anti-inflammatory effect.
JAK2, activated by the interaction of ABCA1 and ApoA-I, activates STAT3 additionally [57,138], which is independent of the lipid transport function of ABCA1 [37]. ABCA1 contains two potential docking units with STAT3, necessary for phosphorylation of the latter by ApoA-I/ABCA1/JAK2 [136]. It is considered that the transcription factor STAT3 mediates IL-6 signaling pathways [57,138] and performs an anti-inflammatory function in macrophages [138,139]. This fact allows to suggest the functioning of ABCA1 as a direct anti-inflammatory receptor due to the activation of JAK2/STAT3 [37,138].
The JAK2/STAT3 pathway can also exhibit a proinflammatory effect [57,138,139,140,141]. Such multidirectional participation highlights the complexity of parallel processes and their insufficient investigation. STAT3 regulates a number of fundamental cellular processes, including inflammation, proliferation, differentiation, and cell migration [142]. STAT3 can also regulate apoptosis by inducing the expression of the apoptosis inhibitor Bcl-2 (B-cell lymphoma 2) [143,144].
The JAK2/STAT3 pathway is involved in the regulation of airway inflammation in COPD (Figure 2) [144]. This agrees well with the fact that cigarette smoke, the main etiological factor of COPD, can activate STAT3 in the lungs [44,144,145]. In patients with COPD the STAT3 expression in the lung tissue is increased significantly [146,147]. Moreover, activation levels correlate with the degree of bronchial inflammation, but not with air flow obstruction [148]. This may be explained by the function of STAT3 in the regulation of inflammation, protease production and apoptosis [149,150,151], underlying the pathogenesis of COPD [152,153].
Taking into account the fact that many of the cytokines considered to be the key participants of the persistent inflammation observed in COPD implement their action through JAK/STAT or are produced as a result of its activation [153,154], the interest of researchers in this pathway has increased significantly in recent years. It is noteworthy that ABCA1 mutations, violating the ABCA1/STAT-3 complex did not affect the lipid outflow of ABCA1, but blocked the ability of ABCA1 to suppress cytokine secretion in response to LPS [155].
The data, received as a result of experiments, indicate that the cholesterol load of macrophages associated with ABCA1 inhibition leads to an increase in IL-6 production [118]. In this regard, the increased levels of IL-6 observed in the induced sputum and lung tissue of patients with COPD [156,157] are in good agreement with the data on the reduction of the ABCA1 transport function in macrophages during smoking, as well as with the fact that IL-6 is known to activate STAT3 [158].
IL-6 is well known as a participant in the pathogenesis of many diseases [159], and is often considered as one of the factors of systemic generalization of inflammation and comorbidity of COPD. It is known that IL-6 contributes to the development of emphysema due to activation of STAT3-independent apoptosis [148]. In addition, a correlation between high IL-6 level and mortality of COPD patients has been demonstrated previously [160,161]. At the same time, it should be noted that the function of IL-6 is complex and not unambiguous. It is believed that IL-6 can control inflammatory processes associated with the involvement of adaptive and innate immunity. For example, it can attract myeloid cells to areas of inflammation [162,163]. The experimental results indicate that IL-6 can reduce the proinflammatory response of human macrophages due to induction of anti-inflammatory IL-4 and IL-10 and secretion decrease of the proinflammatory cytokine Il-1β [118]. IL-10 induced by IL-6 may be involved in activation support of STAT3 in macrophages with the help of the specific receptor IL-10R [118,164].
It is also interesting that IL-6 induces ABCA1 expression and enhances the transporter-mediated outflow of cholesterol from human macrophages to apoA-I with the participation of the JAK-2/STAT3 pathway [118,165]. Thus, lipid-loaded macrophages, producing a significant amount of IL-6 promote the induction of ABCA1 gene expression, which leads to an increase in ABCA1-mediated cholesterol outflow through activation of the Jak-2/STAT3 pathway, thereby reducing the formation of foam cells and the accumulation of free cholesterol, respectively.
It has been found out that not only IL-6, but also a number of other cytokines can affect the expression of ABCA1. They can inhibit it as, for example, interferon (IFN) -γ, IL-1β or Platelet-derived growth factor (PDGF), or enhance it as anti-inflammatory cytokines such as IL-10 and TGF-β1 [37].
These and other data suggest that in cholesterol-loaded macrophages Jak2/STAT3 may represent a key signaling pathway for weakening both the accumulation of cellular lipids and the proinflammatory response [57,118,166]. Such anti-inflammatory effect corresponds to the previously described mechanism, in which the interaction of apoAI with ABCA1 activates the Jak-2/STAT3 pathway and participates in the establishment of an anti-inflammatory response in human macrophages [138,166].
The presented data correspond to the concept of macrophage heterogeneity in COPD, according to which proinflammatory M1 and “alternatively activated” (anti-inflammatory, reparative) M2 macrophages producing proinflammatory (including TNF, IL-1ß, IL-6) and anti-inflammatory (e.g., IL-10) cytokines, respectively, are found in the focus of persistent inflammation at the same time [6,167].
However, in COPD, there are obviously violations of the described anti-inflammatory mechanisms, and the implementation of the JAK-2/STAT3 pathway may be one of the links in a complex chain of mechanisms of COPD pathogenesis [144].
3.3. Other Mechanisms of ABCA1 Participation in Inflammation
There are other known mechanisms of inflammatory activation of macrophages associated with intracellular cholesterol accumulation. Intracellular cholesterol in crystalline form can participate in the activation of inflammation via Nod-like receptors (NLR) acting as intracellular observation molecules [168,169,170,171]. The ability of various crystalline substances to activate NLRP3 inflammation is well known for many both exogenous substances and endogenous molecules, for example, silicon dioxide and monosodium sulfate, and it has also been described for cholesterol crystals [168,169,170,171]. It should be noted that nowadays the possibilities of activation of NLRP3 inflammation in COPD due to the accumulation of crystalline forms of cholesterol in myeloid cells are not clear, but this is well known by the example of atherosclerosis [168,169,170,171,172]. At the same time, recent studies have shown that NLRP3 is highly expressed in the lungs, which is due to the large number of immune cells characteristic of this organ, and emerging scientific evidence suggests that the activation of NLRP3 inflammasome may be involved in the onset of COPD pathogenesis [173,174,175,176].
Infectious exacerbations of COPD are an important characteristic of the disease and largely determine the rates of progression and prognosis. It has been found out that bacterial colonization of the bronchi makes a contribution to the progression of the disease. Bacterial colonization of the bronchi caused by a defect in phagocytosis in COPD [177]. Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis and Pseudomonas aeruginosa are most often found in the lower respiratory tract in COPD [178,179]. Bacterial colonization leads to dysregulation of the immune response, also with the participation of TLR4.
Information about the ability of some bacteria to influence ABCA1-mediated lipid transport in lung epithelial cells is interesting. In particular, it has been shown that Ps. aeruginosa can impair lung function through the induction of ABCA1-mediated export of the surfactant phosphatidylcholine (PtdCho) in the alveolar epithelium of mice. The regulation of the transporter expression is probably carried out via the PPARa (peroxisome-proliferator-activated receptor-a)/RXR pathway [135]. Previously it has been found out that the effect of LPS on macrophages leads to a rapid dose-dependent increase in the expression of ABCA1 mRNA [180]. It has also been shown that infection Chlamydia pneumoniae infection reduces ABCA1 expression in A549 lung epithelial carcinoma cells [66].
In general, violations of ABCA1-mediated lipid transport due to bacterial colonization of the bronchi are of considerable clinical and research interest for assessment its role in infectious exacerbations of COPD.
4. Participation of ABCA1 in Phagocytosis and Apoptosis
Due to chronic inflammation of the respiratory tract in COPD there is a significant increase in the number of cells undergoing apoptosis [181]. Apoptosis is the most important mechanism for ensuring of cell self-renewal and plays an important role in responses to damage or infection by controlling the number of cells involved in the process of inflammation [182,183]. The removal of apoptotic cells is mainly carried out by macrophages in the process of efferocytosis [181].
It is known that efferocytosis is impaired in patients with COPD, but the mechanisms of these disorders are not clear nowadays [184,185]. An accumulation of apoptotic epithelial, endothelial, and immune cells in the lungs is noted in these patients [186,187,188]. In addition, the induction of structural apoptosis of airway cells may be the cause of the development of emphysematous changes [189].
Effective removal of cells that have undergone apoptosis from tissues requires their specific recognition either by neighboring cells or by specialized phagocytes [182,190]. Although cells undergoing apoptosis retain the integrity of the plasma membrane, the resulting changes in the composition of membrane lipids, carbohydrates and proteins provide the necessary molecular signals, marking them for recognition by other cells. One of the signs of apoptosis is the translocation of phosphatidylserine (PtdSer) from the inner sheet of the plasma membrane to the outer sheet [182,183]. It has been shown that this process is connected with the ABCA1 transporter function [191,192]. The involvement of the transporter in phagocytosis is well known from the animal models, that we have analyzed before [193].
It is believed that the localization of phosphatidylserine in the outer sheet of the plasma membrane of apoptotic cells is an almost universal signal for recognition by phagocytes [182,194,195]. This process occurs due to binding to various receptors on the cell surface of phagocytes, for example, merthyrosine kinase (MerTK) [44,196,197]. An increased expression of MerTK on the macrophages of the respiratory tract is detected in smokers [198].
Studies have shown that during MerTK-regulated efferocytosis in lung tissue, the LXR pathway is activated, resulting in increased expression of ABCA1 (Figure 2) [181,192,199]. The independent of LXR pathway of ABCA1 activation in phagocytic macrophages, such as BAI1/ELMO1/Rac, is known [200]. This pathway includes receptor brain-specific angiogenesis inhibitor 1 (BAI1), which recognizes phosphatidylserine on apoptotic cells. BAI1 is also identified as a pattern recognition receptor (PRR), which detects LPS and mediates phagocytosis of Gram-negative bacteria by macrophages [201,202]. Despite this, the role of BAI1 in COPD has not been known yet.
The increase of ABCA1-mediated reverse transport of cholesterol from macrophages during phagocytosis is physiologically explicable. As phagocytes absorb objects, rich in cholesterol, ABCA1 provides protection of cells from its overload [104]. Although ABCA1 does not participate directly in the phagocytosis of apoptotic cells, it can help phagocytes recover [104,191]. In addition, ABCA1 induction provides protection of macrophages from oxidative stress caused by the uptake of oxidized lipids [104].
5. Participation of ABCA1 in the Development of COPD Phenotypes
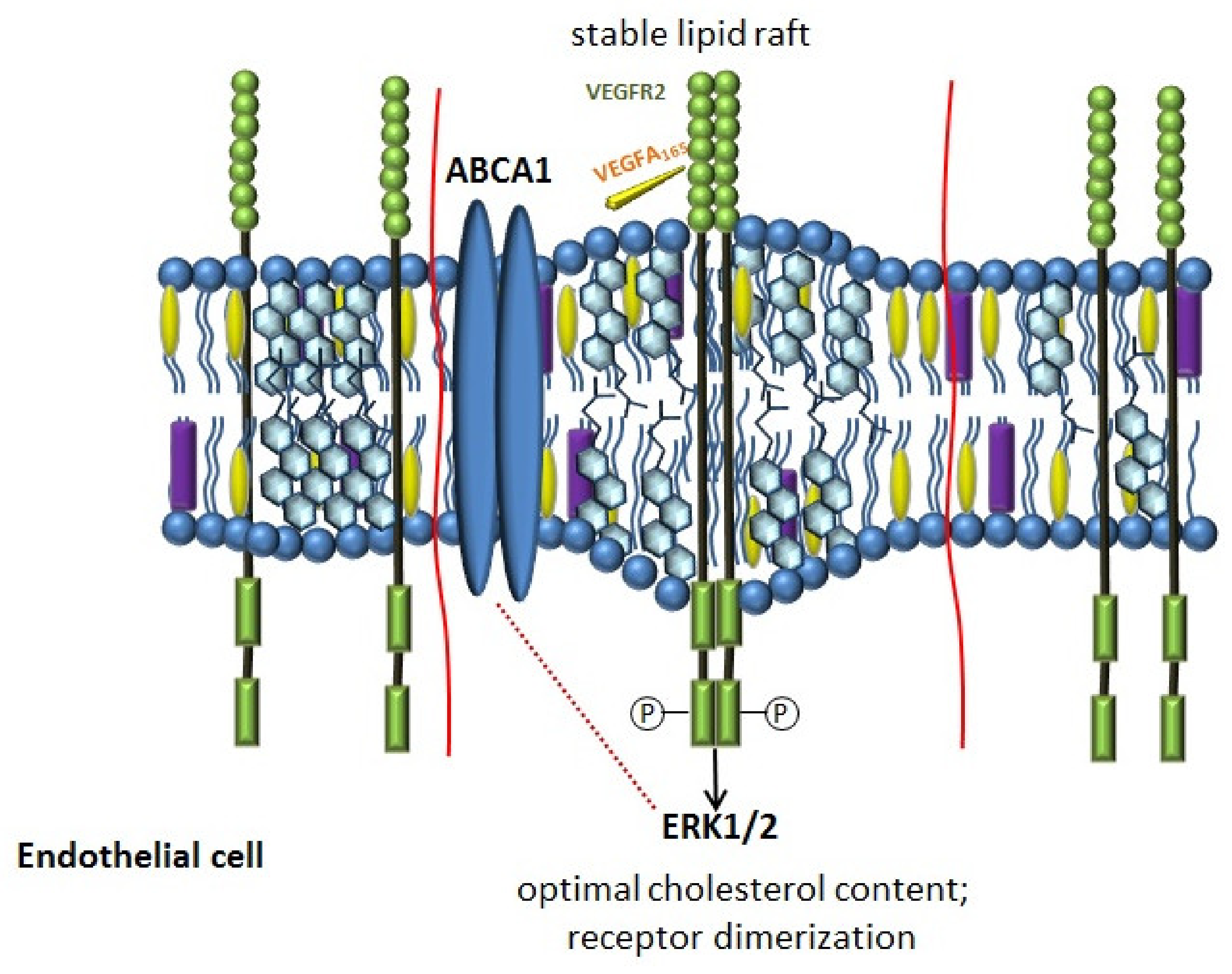
The clinical heterogeneity of COPD is an important characteristic of the disease. The mechanism explaining why different patients with the same risk factor have an emphysematous or bronchitic phenotype is largely unknown. In accordance with the vascular hypothesis, it is assumed that the development of emphysema in COPD may include the progressive loss of endothelial and epithelial cells in the process of apoptosis [203,204,205,206,207]. The results of the studies indicate that vascular endothelial growth factor VEGFA is involved in this process [208,209]. Its role in the pathogenesis of COPD is diverse. The activation of the VEGFA pathway explains the hyperproduction of mucus in the bronchitis phenotype, since VEGFA was originally described as a factor, increasing vascular permeability [210]. Inhibition of the VEGFA pathway, on the contrary, contributes to the disruption of endothelial cell renewal, avascularization of the alveolar septa and their subsequent destruction [209,211]. Experiments on animals have confirmed that blockade of the VEGFA signaling pathway with a VEGFR inhibitor led to apoptosis of endothelial cells in the lungs and morphological changes characteristic of emphysema [212].
It is believed that a decrease, as well as an increase in the amount of cholesterol in the plasma membrane, affects the structure of lipid rafts and disrupts the signaling of VEGFR2, the main angiogenic receptor on the cell membrane [213]. The restoration of the normal cholesterol content in the lipid rafts of endothelial cells stabilizes the dimeric state of VEGFR2 and angiogenesis [213]. Thus, ABCA1, providing the stability of lipid rafts through the regulation of cholesterol content in the plasma membrane, can participate in the signal transduction of VEGFR2 (Figure 3) [214]. The results of the studies confirmed that the activation of LXR can disrupt angiogenesis, which is associated with their effect on the homeostasis of endothelial cholesterol [214,215,216].
VEGFA, binding to VEGFR2 triggers its autophosphorylation, activating various intracellular pathways, including ERK1/2 [216]. ERK1/2 are involved in many cellular processes, such as embryogenesis, differentiation, proliferation, and cell death [217,218]. It has been shown that inhibition of ERK1/2 affects both the expression and the activity of ABCA1 [219,220,221]. In macrophages inhibition of ERK1/2 induces expression of ABCA1 [219] via LXR, which significantly increases cholesterol export. In general, the available literature data allow to suggest that ERK1/2 activity may play an important role in the cholesterol metabolism of macrophages.
It has been shown that HDL can also affect angiogenesis, demonstrating both pro- and antiangiogenic actions, which are implemented in several ways, including through VEGFA and sphingosine-1-phosphate (S1P) [213,222,223,224,225]. Moreover, ABCA1 may be involved in the regulation of these pathways [226]. In vitro, under hypoxic conditions, reconstituted HDL (rHDL) enhanced VEGFR2 activation and enhanced phosphorylation of the downstream angiogenesis signaling proteins ERK1/2 and p38 MAPK [222]. Thus, promoting angiogenesis, induced by ischemia, HDL can suppress angiogenesis induced by inflammation [227,228,229]. The antiangiogenic action can be realized by suppression of NF-kB and activation of macrophages [228].
Thus, the ABCA1 transporter may be involved in the pathogenesis of COPD phenotypes. The development of emphysema involves several mechanisms, many of which have not been identified yet. ABCA1 is involved in angiogenesis, apoptosis, and inflammation, which indicates its significant role not only as a simple lipid transporter, but as a regulator of many important biological processes.
6. Conclusions
Thus, the mechanisms of COPD initiation, development and progression are complex and involve many different pathways. At the same time, the importance of lipid metabolism disorders in these processes does not raise any doubts.
The performed analysis of the data showed that ABCA1 can take part in various processes that are disrupted in COPD. These conclusions are supported by the fact that tobacco smoke, the main etiological factor of COPD, can disrupt the expression and function of ABCA1.
ABCA1 in COPD, due to a violation of its lipid transport function, does not provide adequate reverse transport of cholesterol in macrophages, which may cause inflammatory activation of these cells. The mechanisms through which the transporter is involved in the activation of inflammation are different. The analysis of the data from available studies has shown that one of these key pathways is the participation of the transporter in the activation of the TLR4 receptor signaling pathway, which is well known in the pathogenesis of COPD. Activation of both TLR4 itself and its signaling pathway links is possible through participation in the stabilization of lipid rafts, the accumulation of excess cholesterol in cells, and participation in ensuring the functioning of the descending links of the signaling pathway.
It is interesting to learn about the presence of crosstalk between ABCA1 and the JAK2/STAT3 pathway, which takes an active part in the pathogenesis of COPD, demonstrating both anti- and proinflammatory properties, which shows the complexity of simultaneous processes. The associations of ABCA1 with cytokines such as IL-6, IL-1β, which are involved actively in the pathogenesis of COPD, in addition enhance the significance of the transporter and disorders of its functioning.
Taking into account the fact that part of the pathogenesis of COPD is an increase in bacterial colonization of the bronchi, as well as the presence of infectious exacerbations of the disease, the possible participation of bacteria in the regulation of ABCA1 activity opens up new prospects for studying these relationships. The crosstalk of ABCA1 and TLR4, which is also involved in bacterial colonization of the bronchi, reinforces this interest.
Since disorders of phagocytosis and apoptosis are an important link in the development of COPD, data on the participation of ABCA1 in these processes are significant. The function of the protein in this process is related to the fact that the reverse transport of cholesterol, mediated by ABCA1, provides protection of cells from cholesterol overload during phagocytosis. In addition, ABCA1 participates in the exposure of phospholipid ligands on the surface of apoptotic cells.
It is known that COPD is a clinically heterogeneous disease. This heterogeneity includes both pulmonary and extrapulmonary components, such as cardiovascular comorbidity. The involvement of ABCA1 in the pathogenesis of atherosclerosis, through the function of reverse cholesterol transport, as well as participation in the development of lung emphysema through the regulation of angiogenesis, indicate an important contribution of the transporter to various mechanisms of COPD development.
Thus, the closely intertwined disorders of ABCA1-mediated cellular lipid export, homeostasis of membrane lipid rafts and inflammatory activation of macrophages make a significant contribution to the pathogenesis of COPD [230].
The performed analysis showed that ABCA1 and reverse cholesterol transport are involved in many links of the pathogenesis of COPD and, accordingly, take part in determination of the character of the natural course of the disease, including a decrease in lung function, infectious exacerbations, pulmonary and extrapulmonary clinical heterogeneity.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- May, S.M.; Li, J.T. Burden of chronic obstructive pulmonary disease: Healthcare costs and beyond. Allergy Asthma Proc. 2015, 36, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Quaderi, S.A.; Hurst, J.R. The unmet global burden of COPD. Glob. Health Epidemiol. Genom. 2018, 3, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, J.B.; Zielinski, J.; Price, D. Screening for and early detection of chronic obstructive pulmonary disease. Lancet 2009, 374, 721–732. [Google Scholar] [CrossRef]
- Barnes, P.J. COPD 2020: New directions needed. Am. J. Physiol. Cell Mol. Physiol. 2020, 319, L884–L886. [Google Scholar] [CrossRef] [PubMed]
- Vogelmeier, C.F.; Criner, G.J.; Martinez, F.J.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Chen, R.; Decramer, M.; Fabbri, L.M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report: GOLD Executive Summary. Arch. Bronconeumol. 2017, 53, 128–149. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Eeden, S.F.V. Lung Macrophage Phenotypes and Functional Responses: Role in the Pathogenesis of COPD. Int. J. Mol. Sci. 2018, 19, 582. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.E.; Culpitt, S.V.; DeMatos, C.; Donnelly, L.; Smith, M.; Wiggins, J.; Barnes, P.J. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2002, 26, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I.; Bornfeldt, K.E. Intracellular and Intercellular Aspects of Macrophage Immunometabolism in Atherosclerosis. Circ. Res. 2020, 126, 1209–1227. [Google Scholar] [CrossRef]
- Agustí, A.; Celli, B. Natural history of COPD: Gaps and opportunities. ERJ Open Res. 2017, 3, 117–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oelsner, E.; Balte, P.; Schwartz, J.E.; Burkart, K.M.; Cassano, P.; Jacobs, D.R.; Kalhan, R.; Kronmal, R.; Loehr, L.R.; O’Connor, G.T.; et al. Late-Breaking Abstract: High density lipoprotein cholesterol (HDL-C) and longitudinal lung function in six United States (US) cohorts. Eur. Respir. J. 2016, 48, 2001. [Google Scholar] [CrossRef]
- Burkart, K.M.; Ahmed, F.S.; Watson, K.; Hoffman, E.A.; Burke, G.L.; Barr, R.G. Association Between High Density Lipoproteins (HDL) Cholesterol And CT Percent Emphysema. The MESA Lung Study In B37. Chronic Obstructive Pulmonary Disease Pathogenesis I. Am. J. Crit. Care Med. 2010, 181, A2878. [Google Scholar]
- Park, J.H.; Mun, S.; Choi, D.P.; Lee, J.Y.; Kim, H.C. Association between high-density lipoprotein cholesterol level and pulmonary function in healthy Korean adolescents: The JS high school study. BMC Pulm. Med. 2017, 17, 190. [Google Scholar] [CrossRef] [Green Version]
- Burkart, K.M.; Manichaikul, A.; Wilk, J.B.; Ahmed, F.S.; Burke, G.L.; Enright, P.; Hansel, N.N.; Haynes, D.; Heckbert, S.R.; Hoffman, E.A.; et al. APOM and high-density lipoprotein cholesterol are associated with lung function and per cent emphysema. Eur. Respir. J. 2014, 43, 1003–1017. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, Y.; Wang, L.; Shen, T.; Du, W.; Liu, Z.; Chen, R.; Hu, M. High apolipoprotein M serum levels correlate with chronic obstructive pulmonary disease. Lipids Health Dis. 2016, 15, 59. [Google Scholar] [CrossRef] [Green Version]
- Can, U.; Yerlikaya, F.H.; Yosunkaya, S. Role of oxidative stress and serum lipid levels in stable chronic obstructive pulmonary disease. J. Chin. Med. Assoc. 2015, 78, 702–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, L.; Han, F.; Gong, L.; Lv, Y.; Wan, Z.; Liu, H.; Zhang, D.; Jia, Y.; Yang, S.; Ren, L.; et al. Association between chronic obstructive pulmonary disease and serum lipid levels: A meta-analysis. Lipids Health Dis. 2018, 17, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafirova-Ivanovska, B.; Stojkovikj, J.; Dokikj, D.; Anastasova, S.; Debresliovska, A.; Zejnel, S.; Stojkovikj, D. The Level of Cholesterol in COPD Patients with Severe and Very Severe Stage of the Disease. Open Access Maced J. Med. Sci. 2016, 4, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Jiang, B.; Miao, X.; Ma, J.; Wang, J.; Ding, K.; Chen, X.; Hu, Q.; Fu, F.; Zeng, T.; et al. The Relationship of Lymphocyte to High-Density Lipoprotein Ratio with Pulmonary Function in COPD. Int. J. Chron Obstruct Pulmon Dis. 2020, 15, 3159–3169. [Google Scholar] [CrossRef] [PubMed]
- Reed, R.M.; Hashmi, S.; Eberlein, M.; Iacono, A.; Netzer, G.; DeFilippis, A.; Girgis, R.E.; Toth, P.P.; Scharf, S.; Jones, S. Impact of lung transplantation on serum lipids in COPD. Respir. Med. 2011, 105, 1961–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria-Florido, M.T.; Schroder, H.; Grau, M.; Fito, M.; Lassale, C. High density lipoprotein functionality and cardiovascular events and mortality: A systematic review and meta-analysis. Atherosclerosis 2020, 302, 36–42. [Google Scholar] [CrossRef]
- Bandeali, S.; Farmer, J. High-density lipoprotein and atherosclerosis: The role of antioxidant activity. Curr. Atheroscler. Rep. 2012, 14, 101–107. [Google Scholar] [CrossRef]
- Morgan, A.D.; Zakeri, R.; Quint, J.K. Defining the relationship between COPD and CVD: What are the implications for clinical practice? Ther. Adv. Respir. Dis. 2018, 12, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Betsuyaku, T. The chronic obstructive pulmonary disease comorbidity spectrum in Japan differs from that in western countries. Respir. Investig. 2015, 53, 259–270. [Google Scholar] [CrossRef]
- Reed, R.M.; Iacono, A.; DeFilippis, A.; Eberlein, M.; Girgis, R.E.; Jones, S. Advanced chronic obstructive pulmonary disease is associated with high levels of high-density lipoprotein cholesterol. J. Heart. Lung Transpl. 2011, 30, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.B.; Ammit, A.J.; Gelissen, I.C. Examining the role of ABC lipid transporters in pulmonary lipid homeostasis and inflammation. Respir. Res. 2017, 18, 41. [Google Scholar] [CrossRef] [Green Version]
- Bochem, A.E.; van der Valk, F.M.; Tolani, S.; Stroes, E.S.; Westerterp, M.; Tall, A.R. Increased Systemic and Plaque Inflammation in ABCA1 Mutation Carriers With Attenuation by Statins. Arter. Thromb. Vasc. Biol. 2015, 35, 1663–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the Human Lipid Exporter ABCA1. Cell 2017, 169, 1228–1239. [Google Scholar] [CrossRef] [Green Version]
- Nagao, K.; Kimura, Y.; Mastuo, M.; Ueda, K. Lipid outward translocation by ABC proteins. FEBS Lett. 2010, 584, 2717–2723. [Google Scholar] [CrossRef] [Green Version]
- Wellington, C.L.; Walker, E.K.; Suarez, A.; Kwok, A.; Bissada, N.; Singaraja, R.; Yang, Y.Z.; Zhang, L.H.; James, E.; Wilson, J.E.; et al. ABCA1 mRNA and protein distribution patterns predict multiple different roles and levels of regulation. Lab. Investig. 2002, 82, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Jessup, W.; Gelissen, I.C.; Gaus, K.; Kritharides, L. Roles of ATP binding cassette transporters A1 and G1, scavenger receptor BI and membrane lipid domains in cholesterol export from macrophages. Curr. Opin. Lipidol. 2006, 17, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Bochem, A.E.; Yvan-Charvet, L.; Murphy, A.J.; Wang, N.; Tall, A.R. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ. Res. 2014, 114, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeld, E.B.; Remaley, A.T.; Demosky, S.J.; Stonik, J.A.; Cooney, A.M.; Comly, M.; Dwyer, N.K.; Zhang, M.; Blanchette-Mackie, J.; Santamarina-Fojo, S.; et al. Cellular localization and trafficking of the human ABCA1 transporter. J. Biol. Chem. 2001, 276, 27584–27590. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, W.E.; Piehler, A.; Wenzel, J.J. ABC A-subfamily transporters: Structure, function and disease. Biochim. Biophys. Acta 2006, 1762, 510–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orso, E.; Broccardo, C.; Kaminski, W.E.; Bottcher, A.; Liebisch, G.; Drobnik, W.; Gotz, A.; Chambenoit, O.; Diederich, W.; Langmann, T.; et al. Transport of lipids from golgi to plasma membrane is defective in tangier disease patients and Abc1-deficient mice. Nat. Genet. 2000, 24, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.R.; Abe-Dohmae, S.; Ohnishi, T.; Aoki, R.; Morinaga, G.; Okuhira, K.; Ikeda, Y.; Kano, F.; Matsuo, M.; Kioka, N.; et al. Effects of mutations of ABCA1 in the first extracellular domain on subcellular trafficking and ATP binding/hydrolysis. J. Biol. Chem. 2003, 278, 8815–8819. [Google Scholar] [CrossRef]
- Yin, K.; Liao, D.F.; Tang, C.K. ATP-binding membrane cassette transporter A1 (ABCA1): A possible link between inflammation and reverse cholesterol transport. Mol. Med. 2010, 16, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Francone, O.L.; Aiello, R.J. ABCA1: Regulation, function and relationship to atherosclerosis. Curr. Opin. Investig. Drugs 2002, 3, 415–419. [Google Scholar] [PubMed]
- Zanotti, I.; Poti, F.; Pedrelli, M.; Favari, E.; Moleri, E.; Franceschini, G.; Calabresi, L.; Bernini, F. The LXR agonist T0901317 promotes the reverse cholesterol transport from macrophages by increasing plasma efflux potential. J. Lipid. Res. 2008, 49, 954–960. [Google Scholar] [CrossRef] [Green Version]
- Hussein, M.A.; Shrestha, E.; Ouimet, M.; Barrett, T.J.; Leone, S.; Moore, K.J.; Herault, Y.; Fisher, E.A.; Garabedian, M.J. LXR-Mediated ABCA1 Expression and Function Are Modulated by High Glucose and PRMT2. PLoS ONE 2015, 10, e0135218. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A.; Boisvert, W.A.; Lee, C.H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell. 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Pan, H.; Zheng, Y.; Pan, Q.; Chen, H.; Chen, F.; Wu, J.; Di, D. Expression of LXRbeta, ABCA1 and ABCG1 in human triplenegative breast cancer tissues. Oncol. Rep. 2019, 42, 1869–1877. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Pham, T.X.; Lee, J. Lipopolysaccharide represses the expression of ATP-binding cassette transporter G1 and scavenger receptor class B, type I in murine macrophages. Inflamm. Res. 2012, 61, 465–472. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Gelissen, I.C.; Ammit, A.J. Regulation of ATP binding cassette transporter A1 (ABCA1) expression: Cholesterol-dependent and-independent signaling pathways with relevance to inflammatory lung disease. Respir. Res. 2020, 21, 250. [Google Scholar] [CrossRef]
- Wong, J.; Quinn, C.M.; Brown, A.J. SREBP-2 positively regulates transcription of the cholesterol efflux gene, ABCA1, by generating oxysterol ligands for LXR. Biochem. J. 2006, 400, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Tall, A.R. Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arter. Thromb. Vasc. Biol. 2003, 23, 1178–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef] [Green Version]
- Soumian, S.; Albrecht, C.; Davies, A.H.; Gibbs, R.G. ABCA1 and atherosclerosis. Vasc. Med. 2005, 10, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Smith, A.; Gelissen, I.C.; Ammit, A.J. The effect of statins and the synthetic LXR agonist T0901317 on expression of ABCA1 transporter protein in human lung epithelial cell lines in vitro. Pharmacol. Rep. 2019, 71, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Albavera, L.; Dominguez-Perez, M.; Medina-Leyte, D.J.; Gonzalez-Garrido, A.; Villarreal-Molina, T. The Role of the ATP-Binding Cassette A1 (ABCA1) in Human Disease. Int. J. Mol. Sci. 2021, 22, 593. [Google Scholar] [CrossRef] [PubMed]
- Kielar, D.; Dietmaier, W.; Langmann, T.; Aslanidis, C.; Probst, M.; Naruszewicz, M.; Schmitz, G. Rapid quantification of human ABCA1 mRNA in various cell types and tissues by real-time reverse transcription-PCR. Clin. Chem. 2001, 47, 2089–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, S.R.; Tao, J.Q.; Yu, K.J.; Borok, Z.; Crandall, E.D.; Collins, H.L.; Rothblat, G.H. Expression and biological activity of ABCA1 in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vedhachalam, C.; Ghering, A.B.; Davidson, W.S.; Lund-Katz, S.; Rothblat, G.H.; Phillips, M.C. ABCA1-induced cell surface binding sites for ApoA-I. Arter. Thromb. Vasc. Biol. 2007, 27, 1603–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landry, Y.D.; Denis, M.; Nandi, S.; Bell, S.; Vaughan, A.M.; Zha, X. ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions. J. Biol. Chem. 2006, 281, 36091–36101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Birrell, M.A.; Catley, M.C.; Hardaker, E.; Wong, S.; Willson, T.M.; McCluskie, K.; Leonard, T.; Farrow, S.N.; Collins, J.L.; Haj-Yahia, S.; et al. Novel role for the liver X nuclear receptor in the suppression of lung inflammatory responses. J. Biol. Chem. 2007, 282, 31882–31890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Houston, B.A.; Storey, C.; LeBoeuf, R.C. Both STAT3 activation and cholesterol efflux contribute to the anti-inflammatory effect of apoA-I/ABCA1 interaction in macrophages. J. Lipid Res. 2016, 57, 848–857. [Google Scholar] [CrossRef] [Green Version]
- Francone, O.L.; Royer, L.; Boucher, G.; Haghpassand, M.; Freeman, A.; Brees, D.; Aiello, R.J. Increased cholesterol deposition, expression of scavenger receptors, and response to chemotactic factors in Abca1-deficient macrophages. Arter. Thromb. Vasc. Biol. 2005, 25, 1198–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Ishibashi, M.; Seimon, T.; Lee, M.; Sharma, S.M.; Fitzgerald, K.A.; Samokhin, A.O.; Wang, Y.; Sayers, S.; Aikawa, M.; et al. Free cholesterol accumulation in macrophage membranes activates Toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ. Res. 2009, 104, 455–465. [Google Scholar] [CrossRef]
- Zhu, X.; Owen, J.S.; Wilson, M.D.; Li, H.; Griffiths, G.L.; Thomas, M.J.; Hiltbold, E.M.; Fessler, M.B.; Parks, J.S. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J. Lipid Res. 2010, 51, 3196–3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonett, J.; Goldklang, M.; Sklepkiewicz, P.; Gerber, A.; Trischler, J.; Zelonina, T.; Westerterp, M.; Lemaitre, V.; Okada, Y.; D’Armiento, J. A critical role for ABC transporters in persistent lung inflammation in the development of emphysema after smoke exposure. FASEB J. 2018, 13, 81. [Google Scholar] [CrossRef]
- Song, W.; Wang, W.; Dou, L.Y.; Wang, Y.; Xu, Y.; Chen, L.F.; Yan, X.W. The implication of cigarette smoking and cessation on macrophage cholesterol efflux in coronary artery disease patients. J. Lipid Res. 2015, 56, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fessler, M.B. A New Frontier in Immunometabolism. Cholesterol in Lung Health and Disease. Ann. Am. Thorac. Soc. 2017, 14, S399–S405. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.M.; Nair, P.; Hargreave, F.E.; Efthimiadis, A.E.; Anvari, M.; Allen, C.J.; Group, E.R.S. Lipid and smoker’s inclusions in sputum macrophages in patients with airway diseases. Respir. Med. 2011, 105, 1691–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basset-Leobon, C.; Lacoste-Collin, L.; Aziza, J.; Bes, J.C.; Jozan, S.; Courtade-Saidi, M. Cut-off values and significance of Oil Red O-positive cells in bronchoalveolar lavage fluid. Cytopathology 2010, 21, 245–250. [Google Scholar] [CrossRef]
- Korhonen, J.T.; Olkkonen, V.M.; Lahesmaa, R.; Puolakkainen, M. ABC-cassette transporter 1 (ABCA1) expression in epithelial cells in Chlamydia pneumoniae infection. Microb. Pathog. 2013, 61–62, 57–61. [Google Scholar] [CrossRef]
- Dusuel, A.; Deckert, V.; de Barros, J.P.P.; van Dongen, K.; Choubley, H.; Charron, E.; Le Guern, N.; Labbe, J.; Mandard, S.; Grober, J.; et al. Human cholesteryl ester transfer protein lacks lipopolysaccharide transfer activity, but worsens inflammation and sepsis outcomes in mice. J. Lipid Res. 2020, 62, 100011. [Google Scholar] [CrossRef]
- Shrestha, S.; Wu, B.J.; Guiney, L.; Barter, P.J.; Rye, K.A. Cholesteryl ester transfer protein and its inhibitors. J. Lipid Res. 2018, 59, 772–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, S.R.; Tao, J.Q.; Collins, H.L.; Francone, O.L.; Rothblat, G.H. Pulmonary abnormalities due to ABCA1 deficiency in mice. Am. J. Physiol. Cell Mol. Physiol. 2005, 289, L980–L989. [Google Scholar] [CrossRef] [PubMed]
- McNeish, J.; Aiello, R.J.; Guyot, D.; Turi, T.; Gabel, C.; Aldinger, C.; Hoppe, K.L.; Roach, M.L.; Royer, L.J.; de Wet, J.; et al. High density lipoprotein deficiency and foam cell accumulation in mice with targeted disruption of ATP-binding cassette transporter-1. Proc. Natl. Acad. Sci. USA 2000, 97, 4245–4250. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, H.; Shi, Y.; Nandedkar, S.; Zhang, H.; Gao, H.; Feroah, T.; Weihrauch, D.; Schulte, M.L.; Jones, D.W.; et al. Genetic deletion of apolipoprotein A-I increases airway hyperresponsiveness, inflammation, and collagen deposition in the lung. J. Lipid Res. 2010, 51, 2560–2570. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.; Ricciardolo, F.L.M.; Caramori, G.; Adcock, I.M.; Chung, K.F.; Barnes, P.J.; Brun, P.; Leonardi, A.; Ando, F.; Vallese, D.; et al. Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur. Respir. J. 2017, 49, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Fenton, M.J. Toll-like receptors: Function and roles in lung disease. Am. J. Physiol. Cell Mol. Physiol. 2004, 286, L887–L892. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Jiang, G.; Cohn, L.; Lee, P.J. Toll-like receptor 4 deficiency causes pulmonary emphysema. J. Clin. Investig. 2006, 116, 3050–3059. [Google Scholar] [CrossRef] [PubMed]
- Haw, T.J.; Starkey, M.R.; Pavlidis, S.; Fricker, M.; Arthurs, A.L.; Nair, P.M.; Liu, G.; Hanish, I.; Kim, R.Y.; Foster, P.S.; et al. Toll-like receptor 2 and 4 have opposing roles in the pathogenesis of cigarette smoke-induced chronic obstructive pulmonary disease. Am. J. Physiol. Cell Mol. Physiol. 2018, 314, L298–L317. [Google Scholar] [CrossRef] [PubMed]
- Bhagwani, A.; Thompson, A.A.R.; Farkas, L. When Innate Immunity Meets Angiogenesis-The Role of Toll-Like Receptors in Endothelial Cells and Pulmonary Hypertension. Front. Med. 2020, 7, 352. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Qureshi, S.; Homer, R.; Medzhitov, R.; Noble, P.W.; Lee, P.J. Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J. Immunol. 2005, 175, 4834–4838. [Google Scholar] [CrossRef]
- Sarir, H.; Mortaz, E.; Karimi, K.; Kraneveld, A.D.; Rahman, I.; Caldenhoven, E.; Nijkamp, F.P.; Folkerts, G. Cigarette smoke regulates the expression of TLR4 and IL-8 production by human macrophages. J. Inflamm. 2009, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Karimi, K.; Sarir, H.; Mortaz, E.; Smit, J.J.; Hosseini, H.; De Kimpe, S.J.; Nijkamp, F.P.; Folkerts, G. Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophages. Respir. Res. 2006, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Kleinridders, A.; Schenten, D.; Konner, A.C.; Belgardt, B.F.; Mauer, J.; Okamura, T.; Wunderlich, F.T.; Medzhitov, R.; Bruning, J.C. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009, 10, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Mauerer, R.; Ebert, S.; Langmann, T. High glucose, unsaturated and saturated fatty acids differentially regulate expression of ATP-binding cassette transporters ABCA1 and ABCG1 in human macrophages. Exp. Mol. Med. 2009, 41, 126–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, F.H.; Grundy, S.M. Comparison of effects of dietary saturated, monounsaturated, and polyunsaturated fatty acids on plasma lipids and lipoproteins in man. J. Lipid Res. 1985, 26, 194–202. [Google Scholar] [CrossRef]
- Hu, Y.W.; Ma, X.; Li, X.X.; Liu, X.H.; Xiao, J.; Mo, Z.C.; Xiang, J.; Liao, D.F.; Tang, C.K. Eicosapentaenoic acid reduces ABCA1 serine phosphorylation and impairs ABCA1-dependent cholesterol efflux through cyclic AMP/protein kinase A signaling pathway in THP-1 macrophage-derived foam cells. Atherosclerosis 2009, 204, e35–e43. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Oram, J.F. Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a phospholipase D2 pathway. J. Biol. Chem. 2005, 280, 35896–35903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.C. Is ABCA1 a lipid transfer protein? J. Lipid Res. 2018, 59, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, E.; Nakano, Y.; Ohara, T.; Muro, S.; Hirai, T.; Sato, S.; Sakai, H.; Tsukino, M.; Kinose, D.; Nishioka, M.; et al. Body mass index in male patients with COPD: Correlation with low attenuation areas on CT. Thorax 2009, 64, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, M.N.; Wouters, E.F.M.; Rutten, E.; Casaburi, R.; Rennard, S.I.; Lomas, D.A.; Bamman, M.; Celli, B.; Agusti, A.; Tal-Singer, R.; et al. It’s more than low BMI: Prevalence of cachexia and associated mortality in COPD. Respir. Res. 2019, 20, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoll, P.; Foerster, S.; Virchow, J.C.; Lommatzsch, M. Overweight is a predictor of long-term survival in hospitalised patients with exacerbations of COPD. Respir. Med. 2016, 116, 59–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Yang, D.; Ge, Z.; Yan, M.; Wu, N.; Liu, Y. Body mass index of patients with chronic obstructive pulmonary disease is associated with pulmonary function and exacerbations: A retrospective real world research. J. Thorac. Dis. 2018, 10, 5086–5099. [Google Scholar] [CrossRef]
- Blum, A.; Simsolo, C.; Sirchan, R.; Haiek, S. "Obesity paradox" in chronic obstructive pulmonary disease. ISR Med. Assoc. J. 2011, 13, 672–675. [Google Scholar]
- Spelta, F.; Pasini, A.M.F.; Cazzoletti, L.; Ferrari, M. Body weight and mortality in COPD: Focus on the obesity paradox. Eat Weight. Disord. 2018, 23, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Ejike, C.O.; Wise, R.A.; McCormack, M.C.; Brigham, E.P. Investigation of the Obesity Paradox in Chronic Obstructive Pulmonary Disease, According to Smoking Status, in the United States. Am. J. Epidemiol. 2019, 188, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Rahman, I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol. Appl. Pharmacol. 2011, 254, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.; Caramori, G.; Oates, T.; Capelli, A.; Lusuardi, M.; Gnemmi, I.; Ioli, F.; Chung, K.F.; Donner, C.F.; Barnes, P.J.; et al. Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. Eur. Respir. J. 2002, 20, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingo, M.R.; Silveira, L.J.; Miller, Y.E.; Friedlander, A.L.; Cosgrove, G.P.; Chan, E.D.; Maier, L.A.; Bowler, R.P. Tumour necrosis factor gene polymorphisms are associated with COPD. Eur. Respir. J. 2008, 31, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Karsan, A. Signaling pathways mediated by tumor necrosis factor alpha. Histol. Histopathol. 2000, 15, 1303–1325. [Google Scholar] [CrossRef] [PubMed]
- Kusnadi, A.; Park, S.H.; Yuan, R.; Pannellini, T.; Giannopoulou, E.; Oliver, D.; Lu, T.; Park-Min, K.H.; Ivashkiv, L.B. The Cytokine TNF Promotes Transcription Factor SREBP Activity and Binding to Inflammatory Genes to Activate Macrophages and Limit Tissue Repair. Immunity 2019, 51, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-kappaB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Gerbod-Giannone, M.C.; Li, Y.; Holleboom, A.; Han, S.; Hsu, L.C.; Tabas, I.; Tall, A.R. TNFalpha induces ABCA1 through NF-kappaB in macrophages and in phagocytes ingesting apoptotic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 3112–3117. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.F.; Yang, X.F.; Cheng, B.; Mei, C.L.; Li, Q.X.; Xiao, H.; Zeng, Q.T.; Liao, Y.H.; Liu, K. Protective effect of Astragalus polysaccharides on ATP binding cassette transporter A1 in THP-1 derived foam cells exposed to tumor necrosis factor-alpha. Phytother. Res. 2010, 24, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Ruysschaert, J.M.; Lonez, C. Role of lipid microdomains in TLR-mediated signalling. Biochim. Biophys. Acta 2015, 1848, 1860–1867. [Google Scholar] [CrossRef]
- Viard, M.; Parolini, I.; Sargiacomo, M.; Fecchi, K.; Ramoni, C.; Ablan, S.; Ruscetti, F.W.; Wang, J.M.; Blumenthal, R. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J. Virol. 2002, 76, 11584–11595. [Google Scholar] [CrossRef] [Green Version]
- Hakomori, S.; Handa, K.; Iwabuchi, K.; Yamamura, S.; Prinetti, A. New insights in glycosphingolipid function: "glycosignaling domain," a cell surface assembly of glycosphingolipids with signal transducer molecules, involved in cell adhesion coupled with signaling. Glycobiology 1998, 8, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Fantini, J.; Epand, R.M.; Barrantes, F.J. Cholesterol-Recognition Motifs in Membrane Proteins. Adv. Exp. Med. Biol. 2019, 1135, 3–25. [Google Scholar] [CrossRef]
- Sharpe, L.J.; Rao, G.; Jones, P.M.; Glancey, E.; Aleidi, S.M.; George, A.M.; Brown, A.J.; Gelissen, I.C. Cholesterol sensing by the ABCG1 lipid transporter: Requirement of a CRAC motif in the final transmembrane domain. Biochim. Biophys. Acta 2015, 1851, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Papadopoulos, V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 1998, 139, 4991–4997. [Google Scholar] [CrossRef]
- Baier, C.J.; Fantini, J.; Barrantes, F.J. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep. 2011, 1, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epand, R.M. Proteins and cholesterol-rich domains. Biochim. Biophys. Acta 2008, 1778, 1576–1582. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Thomas, A.; Brasseur, R.; Epand, R.F. Cholesterol interaction with proteins that partition into membrane domains: An overview. Subcell Biochem. 2010, 51, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Barrantes, F.J. Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim. Biophys. Acta 2009, 1788, 2345–2361. [Google Scholar] [CrossRef] [PubMed]
- Frisdal, E.; Lesnik, P.; Olivier, M.; Robillard, P.; Chapman, M.J.; Huby, T.; Guerin, M.; Le Goff, W. Interleukin-6 protects human macrophages from cellular cholesterol accumulation and attenuates the proinflammatory response. J. Biol. Chem. 2011, 286, 30926–30936. [Google Scholar] [CrossRef] [Green Version]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef]
- Guo, L.; Chen, C.H.; Zhang, L.L.; Cao, X.J.; Ma, Q.L.; Deng, P.; Zhu, G.; Gao, C.Y.; Li, B.H.; Pi, Y.; et al. IRAK1 mediates TLR4-induced ABCA1 downregulation and lipid accumulation in VSMCs. Cell Death Dis. 2015, 6, e1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarling, E.J.; de Vallim, T.Q.A.; Edwards, P.A. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol. Metab. 2013, 24, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Gulshan, K.; Brubaker, G.; Conger, H.; Wang, S.; Zhang, R.; Hazen, S.L.; Smith, J.D. PI(4,5)P2 Is Translocated by ABCA1 to the Cell Surface Where It Mediates Apolipoprotein A1 Binding and Nascent HDL Assembly. Circ. Res. 2016, 119, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doughman, R.L.; Firestone, A.J.; Anderson, R.A. Phosphatidylinositol phosphate kinases put PI4,5P(2) in its place. J. Membr. Biol. 2003, 194, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.L.; Janmey, P.A. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 2003, 65, 761–789. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Thapa, N.; Hedman, A.C.; Anderson, R.A. Phosphatidylinositol 4,5-bisphosphate: Targeted production and signaling. Bioessays 2013, 35, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, A.; Kanemaru, K.; Matsubara, A.; Takai, E.; Shimozawa, M.; Satow, R.; Yamaguchi, H.; Nakamura, Y.; Fukami, K. Phosphatidylinositol 4,5-bisphosphate is localized in the plasma membrane outer leaflet and regulates cell adhesion and motility. Biochem. Biophys. Res. Commun. 2020, 527, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.C.; Choi, S. Insight into Phosphatidylinositol-Dependent Membrane Localization of the Innate Immune Adaptor Protein Toll/Interleukin 1 Receptor Domain-Containing Adaptor Protein. Front. Immunol. 2018, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Bonham, K.S.; Orzalli, M.H.; Hayashi, K.; Wolf, A.I.; Glanemann, C.; Weninger, W.; Iwasaki, A.; Knipe, D.M.; Kagan, J.C. A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell 2014, 156, 705–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Varghese, Z.; Moorhead, J.F.; Lee, C.T.; Chen, J.B.; Ruan, X.Z. Cross-talk between TLR4-MyD88-NF-kappaB and SCAP-SREBP2 pathways mediates macrophage foam cell formation. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H874–H884. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, E.; Taboubi, S.; Torres, D.; Delbauve, S.; Hachani, A.; Whitehead, M.A.; Pearce, W.P.; Berenjeno, I.M.; Nock, G.; Filloux, A.; et al. The p110delta isoform of the kinase PI(3)K controls the subcellular compartmentalization of TLR4 signaling and protects from endotoxic shock. Nat. Immunol. 2012, 13, 1045–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plociennikowska, A.; Zdioruk, M.I.; Traczyk, G.; Swiatkowska, A.; Kwiatkowska, K. LPS-induced clustering of CD14 triggers generation of PI(4,5)P2. J. Cell Sci. 2015, 128, 4096–4111. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Vaughan, A.M.; Oram, J.F. Janus kinase 2 modulates the apolipoprotein interactions with ABCA1 required for removing cellular cholesterol. J. Biol. Chem. 2004, 279, 7622–7628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agassandian, M.; Miakotina, O.L.; Andrews, M.; Mathur, S.N.; Mallampalli, R.K. Pseudomonas aeruginosa and sPLA2 IB stimulate ABCA1-mediated phospholipid efflux via ERK-activation of PPARalpha-RXR. Biochem. J. 2007, 403, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.J.; Yin, K.; Fu, Y.C.; Tang, C.K. The interaction of ApoA-I and ABCA1 triggers signal transduction pathways to mediate efflux of cellular lipids. Mol. Med. 2012, 18, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.M.; Tang, C.; Oram, J.F. ABCA1 mutants reveal an interdependency between lipid export function, apoA-I binding activity, and Janus kinase 2 activation. J. Lipid Res. 2009, 50, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Liu, Y.; Kessler, P.S.; Vaughan, A.M.; Oram, J.F. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J. Biol. Chem. 2009, 284, 32336–32343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, L.M.; Sarma, U.; Willets, K.; Smallie, T.; Brennan, F.; Foxwell, B.M. Expression of constitutively active STAT3 can replicate the cytokine-suppressive activity of interleukin-10 in human primary macrophages. J. Biol. Chem. 2007, 282, 6965–6975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 2006, 6, 379–386. [Google Scholar] [CrossRef]
- Hao, X.R.; Cao, D.L.; Hu, Y.W.; Li, X.X.; Liu, X.H.; Xiao, J.; Liao, D.F.; Xiang, J.; Tang, C.K. IFN-gamma down-regulates ABCA1 expression by inhibiting LXRalpha in a JAK/STAT signaling pathway-dependent manner. Atherosclerosis 2009, 203, 417–428. [Google Scholar] [CrossRef]
- Akcora, B.O.; Gabriel, A.V.; Ortiz-Perez, A.; Bansal, R. Pharmacological inhibition of STAT3 pathway ameliorates acute liver injury in vivo via inactivation of inflammatory macrophages and hepatic stellate cells. FASEB Bioadv. 2020, 2, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Ward, P.A. STAT3 and suppressor of cytokine signaling 3: Potential targets in lung inflammatory responses. Expert Opin Ther. Targets 2007, 11, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, P.; Wyman, A.E.; Garcia-Arcos, I.; Dabo, A.J.; Gadhvi, S.; Foronjy, R. STAT3 modulates cigarette smoke-induced inflammation and protease expression. Front. Physiol. 2013, 4, 267. [Google Scholar] [CrossRef] [Green Version]
- Bohadana, A.; Teculescu, D.; Martinet, Y. Mechanisms of chronic airway obstruction in smokers. Respir. Med. 2004, 98, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Carlier, F.; Detry, B.; Sibille, Y.; Pilette, C. STAT-3 is activated in COPD airway epithelial cells. ERJ Open Res. 2019, 5, 214. [Google Scholar] [CrossRef]
- Qu, P.; Roberts, J.; Li, Y.; Albrecht, M.; Cummings, O.W.; Eble, J.N.; Du, H.; Yan, C. Stat3 downstream genes serve as biomarkers in human lung carcinomas and chronic obstructive pulmonary disease. Lung Cancer 2009, 63, 341–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruwanpura, S.M.; McLeod, L.; Miller, A.; Jones, J.; Vlahos, R.; Ramm, G.; Longano, A.; Bardin, P.G.; Bozinovski, S.; Anderson, G.P.; et al. Deregulated Stat3 signaling dissociates pulmonary inflammation from emphysema in gp130 mutant mice. Am. J. Physiol. Cell Mol. Physiol. 2012, 302, L627–L639. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Huang, C.; Huang, K.; Wu, W.; Jiang, T.; Cao, J.; Feng, Z.; Qiu, Z. STAT3 knockdown reduces pancreatic cancer cell invasiveness and matrix metalloproteinase-7 expression in nude mice. PLoS ONE 2011, 6, e25941. [Google Scholar] [CrossRef]
- Camporeale, A.; Poli, V. IL-6, IL-17 and STAT3: A holy trinity in auto-immunity? Front. Biosci. 2012, 17, 2306–2326. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Hong, J.; Wang, Y.C.; Zhang, Y.J.; Wang, P.; Su, W.Y.; Lin, Y.W.; Lu, R.; Zou, W.P.; Xiong, H.; et al. Inhibition of JAK2/STAT3 signalling induces colorectal cancer cell apoptosis via mitochondrial pathway. J. Cell Mol. Med. 2012, 16, 1878–1888. [Google Scholar] [CrossRef]
- Chung, K.F.; Adcock, I.M. Multifaceted mechanisms in COPD: Inflammation, immunity, and tissue repair and destruction. Eur. Respir. J. 2008, 31, 1334–1356. [Google Scholar] [CrossRef] [PubMed]
- Yew-Booth, L.; Birrell, M.A.; Lau, M.S.; Baker, K.; Jones, V.; Kilty, I.; Belvisi, M.G. JAK-STAT pathway activation in COPD. Eur. Respir. J. 2015, 46, 843–845. [Google Scholar] [CrossRef] [Green Version]
- Schindler, C.; Strehlow, I. Cytokines and STAT signaling. Adv. Pharmacol. 2000, 47, 113–174. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Mujawar, Z.; Tamehiro, N. ABC transporters, atherosclerosis and inflammation. Atherosclerosis 2010, 211, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Eickmeier, O.; Huebner, M.; Herrmann, E.; Zissler, U.; Rosewich, M.; Baer, P.C.; Buhl, R.; Schmitt-Grohe, S.; Zielen, S.; Schubert, R. Sputum biomarker profiles in cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD) and association between pulmonary function. Cytokine 2010, 50, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Hlapcic, I.; Belamaric, D.; Bosnar, M.; Kifer, D.; Dugac, A.V.; Rumora, L. Combination of Systemic Inflammatory Biomarkers in Assessment of Chronic Obstructive Pulmonary Disease: Diagnostic Performance and Identification of Networks and Clusters. Diagnostics 2020, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Verma, S.K.; Kumar, S.; Ahmad, M.K.; Nischal, A.; Singh, S.K.; Dixit, R.K. Correlation of severity of chronic obstructive pulmonary disease with potential biomarkers. Immunol. Lett. 2018, 196, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Zhang, S.J.; Shen, H.J.; Guan, Y.; Liu, Q.; Zhao, W.; Jia, Y.L.; Shen, J.; Yan, X.F.; Xie, Q.M. Rac1 signaling regulates cigarette smoke-induced inflammation in the lung via the Erk1/2 MAPK and STAT3 pathways. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Tanni, S.E.; Caram, L.M.; Correa, C.; Correa, C.R.; Godoy, I. Three-year follow-up of Interleukin 6 and C-reactive protein in chronic obstructive pulmonary disease. Respir. Res. 2013, 14, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fielding, C.A.; McLoughlin, R.M.; McLeod, L.; Colmont, C.S.; Najdovska, M.; Grail, D.; Ernst, M.; Jones, S.A.; Topley, N.; Jenkins, B.J. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. 2008, 181, 2189–2195. [Google Scholar] [CrossRef]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8 (Suppl. 2). [Google Scholar] [CrossRef] [Green Version]
- Niemand, C.; Nimmesgern, A.; Haan, S.; Fischer, P.; Schaper, F.; Rossaint, R.; Heinrich, P.C.; Muller-Newen, G. Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J. Immunol. 2003, 170, 3263–3272. [Google Scholar] [CrossRef] [Green Version]
- Perez-Baos, S.; Barrasa, J.I.; Gratal, P.; Larranaga-Vera, A.; Prieto-Potin, I.; Herrero-Beaumont, G.; Largo, R. Tofacitinib restores the inhibition of reverse cholesterol transport induced by inflammation: Understanding the lipid paradox associated with rheumatoid arthritis. Br. J. Pharmacol. 2017, 174, 3018–3031. [Google Scholar] [CrossRef] [Green Version]
- Yin, K.; Deng, X.; Mo, Z.C.; Zhao, G.J.; Jiang, J.; Cui, L.B.; Tan, C.Z.; Wen, G.B.; Fu, Y.; Tang, C.K. Tristetraprolin-dependent post-transcriptional regulation of inflammatory cytokine mRNA expression by apolipoprotein A-I: Role of ATP-binding membrane cassette transporter A1 and signal transducer and activator of transcription 3. J. Biol. Chem. 2011, 286, 13834–13845. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- de la Roche, M.; Hamilton, C.; Mortensen, R.; Jeyaprakash, A.A.; Ghosh, S.; Anand, P.K. Trafficking of cholesterol to the ER is required for NLRP3 inflammasome activation. J. Cell Biol. 2018, 217, 3560–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Rajamaki, K.; Lappalainen, J.; Oorni, K.; Valimaki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [Green Version]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Gemert, S.B.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nardo, D.; De Nardo, C.M.; Latz, E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am. J. Pathol. 2014, 184, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Pinto, A.; Sorrentino, R. Role of the inflammasome in chronic obstructive pulmonary disease (COPD). Oncotarget 2017, 8, 81813–81824. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.-J.; Mao, B.; Wang, S.-J.; Xiao, W.; Li, G.-H.; Wu, L.-H. The activation of NLRP3 inflammasome pathway in COPD is limited in local airways. Eur. Respir. J. 2018, 52, 4981. [Google Scholar] [CrossRef]
- Xiao, W.; Fu, J.-J.; Wang, S.-J.; Wang, Y.; Miao, T.-W.; Mao, B. Andrographolide inhibits the activation of NLRP3 inflammasome in patients with COPD. Eur. Respir. J. 2018, 52, 5246. [Google Scholar] [CrossRef]
- Ghosh, B.; Gaike, A.H.; Pyasi, K.; Brashier, B.; Das, V.V.; Londhe, J.D.; Juvekar, S.; Shouche, Y.S.; Donnelly, L.E.; Salvi, S.S.; et al. Bacterial load and defective monocyte-derived macrophage bacterial phagocytosis in biomass smoke-related COPD. Eur. Respir. J. 2019, 53, 73. [Google Scholar] [CrossRef] [PubMed]
- Sidletskaya, K.; Vitkina, T.; Denisenko, Y. The Role of Toll-Like Receptors 2 and 4 in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Int. J. Chron. Obstruct Pulmon. Dis. 2020, 15, 1481–1493. [Google Scholar] [CrossRef]
- D’Anna, S.E.; Balbi, B.; Cappello, F.; Carone, M.; Di Stefano, A. Bacterial-viral load and the immune response in stable and exacerbated COPD: Significance and therapeutic prospects. Int. J. Chron. Obstruct Pulmon. Dis. 2016, 11, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.; Gan, X.; Menke, J.G.; Wright, S.D.; Cai, T.Q. Bacterial lipopolysaccharide induces expression of ABCA1 but not ABCG1 via an LXR-independent pathway. J. Lipid Res. 2002, 43, 952–959. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Hussell, T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin. Immunopathol. 2016, 38, 409–423. [Google Scholar] [CrossRef] [Green Version]
- Arienti, S.; Barth, N.D.; Dorward, D.A.; Rossi, A.G.; Dransfield, I. Regulation of Apoptotic Cell Clearance During Resolution of Inflammation. Front. Pharmacol. 2019, 10, 891. [Google Scholar] [CrossRef] [Green Version]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, K.; Janssen, W.J.; Fessler, M.B.; McPhillips, K.A.; Borges, V.M.; Bowler, R.P.; Xiao, Y.Q.; Kench, J.A.; Henson, P.M.; Vandivier, R.W. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J. Immunol. 2006, 176, 7657–7665. [Google Scholar] [CrossRef] [Green Version]
- Mukaro, V.R.; Hodge, S. Airway clearance of apoptotic cells in COPD. Curr. Drug Targets 2011, 12, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am. J. Respir. Crit. Care Med. 2001, 163, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K.; Mercer, B.A.; Schulman, L.L.; Sonett, J.R.; D’Armiento, J.M. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur. Respir. J. 2005, 25, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Makris, D.; Vrekoussis, T.; Izoldi, M.; Alexandra, K.; Katerina, D.; Dimitris, T.; Michalis, A.; Tzortzaki, E.; Siafakas, N.M.; Tzanakis, N. Increased apoptosis of neutrophils in induced sputum of COPD patients. Respir. Med. 2009, 103, 1130–1135. [Google Scholar] [CrossRef] [Green Version]
- Aoshiba, K.; Yokohori, N.; Nagai, A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am. J. Respir. Cell Mol. Biol. 2003, 28, 555–562. [Google Scholar] [CrossRef]
- Fond, A.M.; Ravichandran, K.S. Clearance of Dying Cells by Phagocytes: Mechanisms and Implications for Disease Pathogenesis. Adv. Exp. Med. Biol. 2016, 930, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ. Res. 2010, 106, 1861–1869. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Diaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlyarov, S.N.; Kotlyarova, A.A. Participation of ABCA1 transporter in development of chronic obstructive pulmonary disease. Pavlov. Russ. Med Biol. Her. 2020, 28, 360–370. [Google Scholar] [CrossRef]
- Boada-Romero, E.; Martinez, J.; Heckmann, B.L.; Green, D.R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 2020, 21, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Nagata, S. An Apoptotic ’Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Nishi, C.; Yanagihashi, Y.; Segawa, K.; Nagata, S. MERTK tyrosine kinase receptor together with TIM4 phosphatidylserine receptor mediates distinct signal transduction pathways for efferocytosis and cell proliferation. J. Biol. Chem. 2019, 294, 7221–7230. [Google Scholar] [CrossRef]
- Lemke, G. Phosphatidylserine Is the Signal for TAM Receptors and Their Ligands. Trends Biochem. Sci. 2017, 42, 738–748. [Google Scholar] [CrossRef]
- Kazeros, A.; Harvey, B.G.; Carolan, B.J.; Vanni, H.; Krause, A.; Crystal, R.G. Overexpression of apoptotic cell removal receptor MERTK in alveolar macrophages of cigarette smokers. Am. J. Respir. Cell Mol. Biol. 2008, 39, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, A.N.; Hidalgo, A. Nuclear Receptors and Clearance of Apoptotic Cells: Stimulating the Macrophage’s Appetite. Front. Immunol. 2014, 5, 211. [Google Scholar] [CrossRef] [Green Version]
- Fond, A.M.; Lee, C.S.; Schulman, I.G.; Kiss, R.S.; Ravichandran, K.S. Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J. Clin. Investig. 2015, 125, 2748–2758. [Google Scholar] [CrossRef]
- Das, S.; Owen, K.A.; Ly, K.T.; Park, D.; Black, S.G.; Wilson, J.M.; Sifri, C.D.; Ravichandran, K.S.; Ernst, P.B.; Casanova, J.E. Brain angiogenesis inhibitor 1 (BAI1) is a pattern recognition receptor that mediates macrophage binding and engulfment of Gram-negative bacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 2136–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.Y.; Shin, S.A.; Oh, Y.S.; Park, H.H.; Lee, C.S. Understanding the Role of the BAI Subfamily of Adhesion G Protein-Coupled Receptors (GPCRs) in Pathological and Physiological Conditions. Genes 2018, 9, 597. [Google Scholar] [CrossRef] [Green Version]
- Szucs, B.; Szucs, C.; Petrekanits, M.; Varga, J.T. Molecular Characteristics and Treatment of Endothelial Dysfunction in Patients with COPD: A Review Article. Int. J. Mol. Sci. 2019, 20, 329. [Google Scholar] [CrossRef] [Green Version]
- Liebow, A.A. Pulmonary emphysema with special reference to vascular changes. Am. Rev. Respir. Dis. 1959, 80, 67–93. [Google Scholar] [CrossRef]
- Green, C.E.; Turner, A.M. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Respir. Res. 2017, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- Kanazawa, H.; Yoshikawa, J. Elevated oxidative stress and reciprocal reduction of vascular endothelial growth factor levels with severity of COPD. Chest 2005, 128, 3191–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, S.; Peinado, V.I.; Ramirez, J.; Morales-Blanhir, J.; Bastos, R.; Roca, J.; Rodriguez-Roisin, R.; Barbera, J.A. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Betsuyaku, T.; Nagai, K.; Fuke, S.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Hamamura, I.; Hata, J.; Takahashi, H.; et al. Decreased airway expression of vascular endothelial growth factor in cigarette smoke-induced emphysema in mice and COPD patients. Inhal. Toxicol. 2008, 20, 349–359. [Google Scholar] [CrossRef]
- Bates, D.O. Vascular endothelial growth factors and vascular permeability. Cardiovasc. Res. 2010, 87, 262–271. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Rossiter, H.B.; Wagner, P.D.; Breen, E.C. Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J. Appl. Physiol. 2004, 97, 1559–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath-Morrow, S.A.; Cho, C.; Cho, C.; Zhen, L.; Hicklin, D.J.; Tuder, R.M. Vascular endothelial growth factor receptor 2 blockade disrupts postnatal lung development. Am. J. Respir. Cell Mol. Biol. 2005, 32, 420–427. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Gu, Q.; Fang, L. Cholesterol-mediated regulation of angiogenesis: An emerging paradigm. Cardiol. Plus 2019, 4, 1–9. [Google Scholar] [CrossRef]
- Noghero, A.; Perino, A.; Seano, G.; Saglio, E.; Lo Sasso, G.; Veglio, F.; Primo, L.; Hirsch, E.; Bussolino, F.; Morello, F. Liver X receptor activation reduces angiogenesis by impairing lipid raft localization and signaling of vascular endothelial growth factor receptor-2. Arter. Thromb. Vasc. Biol. 2012, 32, 2280–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrecque, L.; Royal, I.; Surprenant, D.S.; Patterson, C.; Gingras, D.; Beliveau, R. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol. Biol. Cell 2003, 14, 334–347. [Google Scholar] [CrossRef]
- Liao, W.X.; Feng, L.; Zhang, H.; Zheng, J.; Moore, T.R.; Chen, D.B. Compartmentalizing VEGF-induced ERK2/1 signaling in placental artery endothelial cell caveolae: A paradoxical role of caveolin-1 in placental angiogenesis in vitro. Mol. Endocrinol. 2009, 23, 1428–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Lei, T.; Guo, P.; Yu, J.; Xu, Q.; Luo, Y.; Ke, R.; Huang, D. Mechanisms shaping the role of ERK1/2 in cellular senescence (Review). Mol. Med. Rep. 2019, 19, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef]
- Zhou, X.; Yin, Z.; Guo, X.; Hajjar, D.P.; Han, J. Inhibition of ERK1/2 and activation of liver X receptor synergistically induce macrophage ABCA1 expression and cholesterol efflux. J. Biol. Chem. 2010, 285, 6316–6326. [Google Scholar] [CrossRef] [Green Version]
- Mulay, V.; Wood, P.; Manetsch, M.; Darabi, M.; Cairns, R.; Hoque, M.; Chan, K.C.; Reverter, M.; Alvarez-Guaita, A.; Rye, K.A.; et al. Inhibition of mitogen-activated protein kinase Erk1/2 promotes protein degradation of ATP binding cassette transporters A1 and G1 in CHO and HuH7 cells. PLoS ONE 2013, 8, e62667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.H.; Shi, F.F.; Chen, T.; Wei, W.; Zhou, X.M.; Chen, L.D. Inhibition of ERK1/2 improves lipid balance in rat macrophages via ABCA1/G1 and CD36. Mol. Med. Rep. 2016, 13, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Cannizzo, C.M.; Adonopulos, A.A.; Solly, E.L.; Ridiandries, A.; Vanags, L.Z.; Mulangala, J.; Yuen, S.C.G.; Tsatralis, T.; Henriquez, R.; Robertson, S.; et al. VEGFR2 is activated by high-density lipoproteins and plays a key role in the proangiogenic action of HDL in ischemia. FASEB J. 2018, 32, 2911–2922. [Google Scholar] [CrossRef] [Green Version]
- Christoffersen, C.; Nielsen, L.B. Apolipoprotein M: Bridging HDL and endothelial function. Curr. Opin Lipidol. 2013, 24, 295–300. [Google Scholar] [CrossRef]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnstrom, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, M.; Frej, C.; Holmer, A.; Guo, L.J.; Tran, S.; Dahlback, B. High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arter. Thromb. Vasc. Biol. 2017, 37, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, M.; Jentsch, J.A.; Peters, S.; Keul, P.; Weske, S.; Graler, M.H.; Mladenov, E.; Iliakis, G.; Heusch, G.; Levkau, B. Regulation of ABCA1-mediated cholesterol efflux by sphingosine-1-phosphate signaling in macrophages. J. Lipid Res. 2019, 60, 506–515. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Hagemann, N.; Sun, L.; Wu, J.; Doeppner, T.R.; Dai, Y.; Hermann, D.M. High-density lipoprotein (HDL) promotes angiogenesis via S1P3-dependent VEGFR2 activation. Angiogenesis 2018, 21, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Ng, M.K.; Bursill, C.A. The role of high-density lipoproteins in the regulation of angiogenesis. Cardiovasc. Res. 2015, 106, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, H.C.; Tan, J.T.; Dunn, L.L.; Patel, S.; Vanags, L.Z.; Bao, S.; Ng, M.K.; Bursill, C.A. Multifunctional regulation of angiogenesis by high-density lipoproteins. Cardiovasc. Res. 2014, 101, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tang, C. Regulation of ABCA1 functions by signaling pathways. Biochim. Biophys. Acta 2012, 1821, 522–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Cholesterol-binding domain (CRAC and CARC domains).

Figure 2.
Schematic picture of the ATP binding cassette transporter A1 (ABCA1) participation in the mechanisms of inflammation in macrophages in chronic obstructive pulmonary disease (COPD) (1) and apoptosis (2).
Figure 2.
Schematic picture of the ATP binding cassette transporter A1 (ABCA1) participation in the mechanisms of inflammation in macrophages in chronic obstructive pulmonary disease (COPD) (1) and apoptosis (2).

Figure 3.
Schematic picture of the ABCA1 transporter participation in angiogenesis through regulation of the vascular endothelial growth factor (VEGFA) pathway in endothelial cells in COPD.
Figure 3.
Schematic picture of the ABCA1 transporter participation in angiogenesis through regulation of the vascular endothelial growth factor (VEGFA) pathway in endothelial cells in COPD.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kotlyarov, S. Participation of ABCA1 Transporter in Pathogenesis of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 3334. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073334
AMA Style
Kotlyarov S. Participation of ABCA1 Transporter in Pathogenesis of Chronic Obstructive Pulmonary Disease. International Journal of Molecular Sciences. 2021; 22(7):3334. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073334
Chicago/Turabian StyleKotlyarov, Stanislav. 2021. "Participation of ABCA1 Transporter in Pathogenesis of Chronic Obstructive Pulmonary Disease" International Journal of Molecular Sciences 22, no. 7: 3334. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073334
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.