
Xanthine Oxidoreductase-Mediated Superoxide Production Is Not Involved in the Age-Related Pathologies in Sod1-Deficient Mice


Abstract
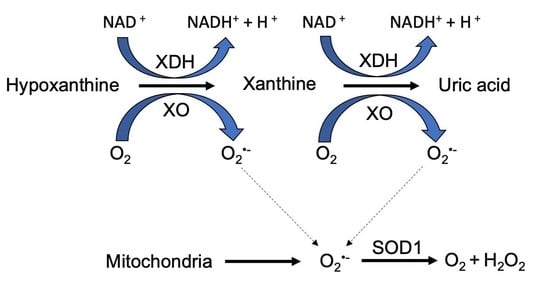
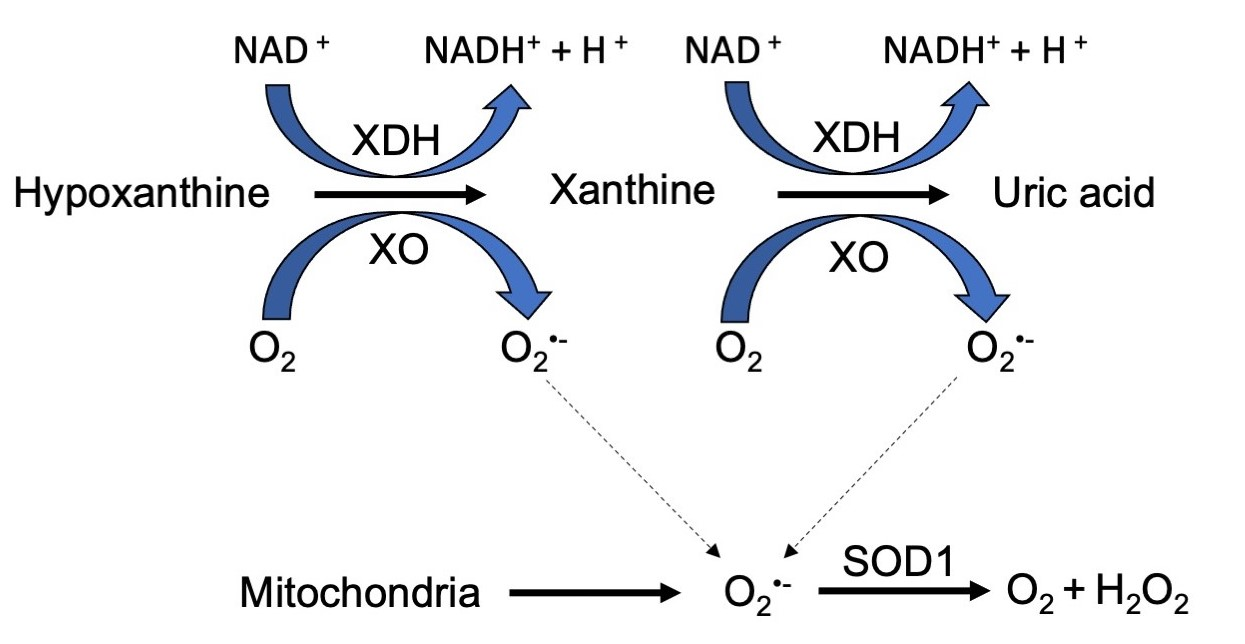
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
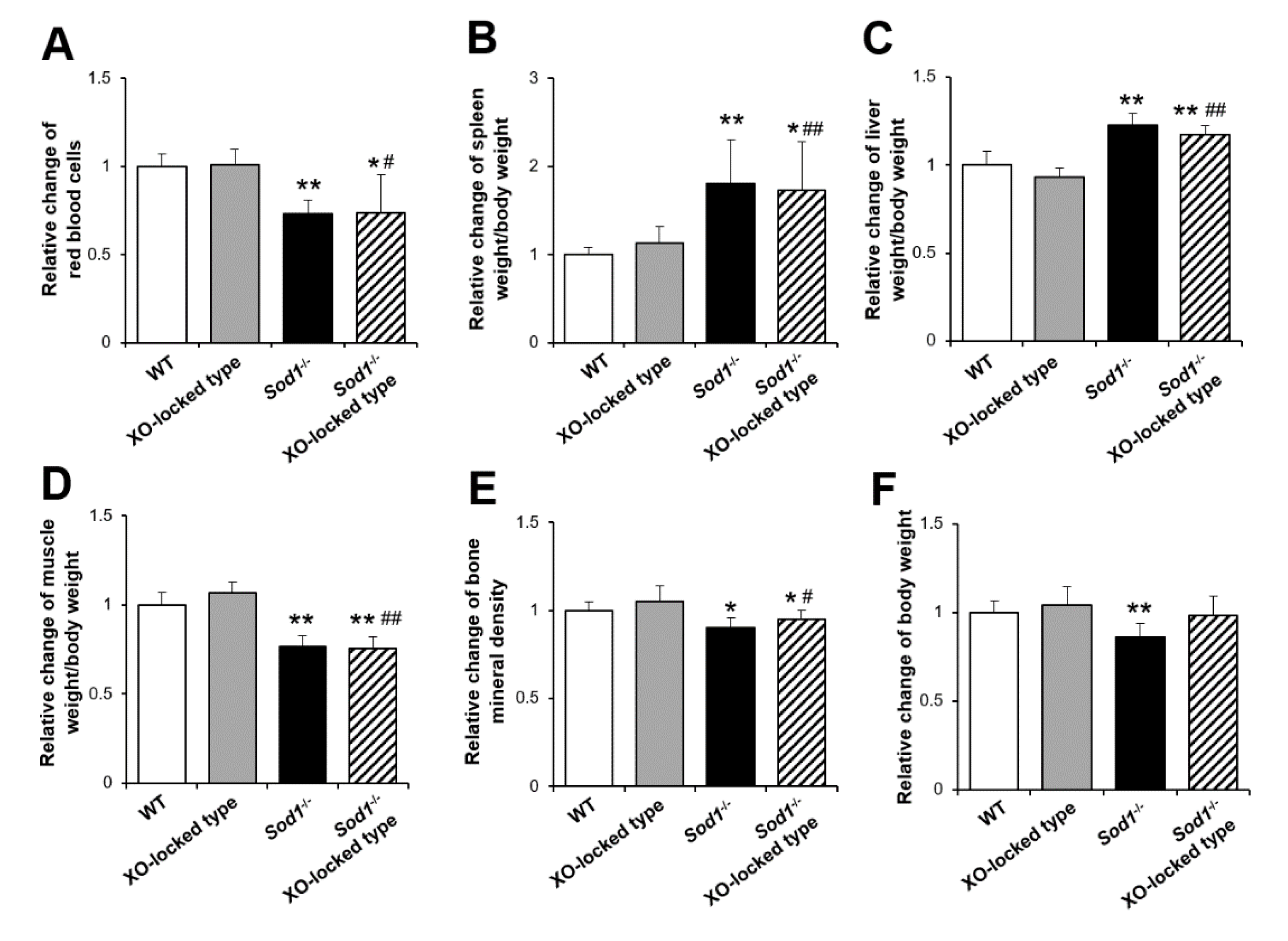
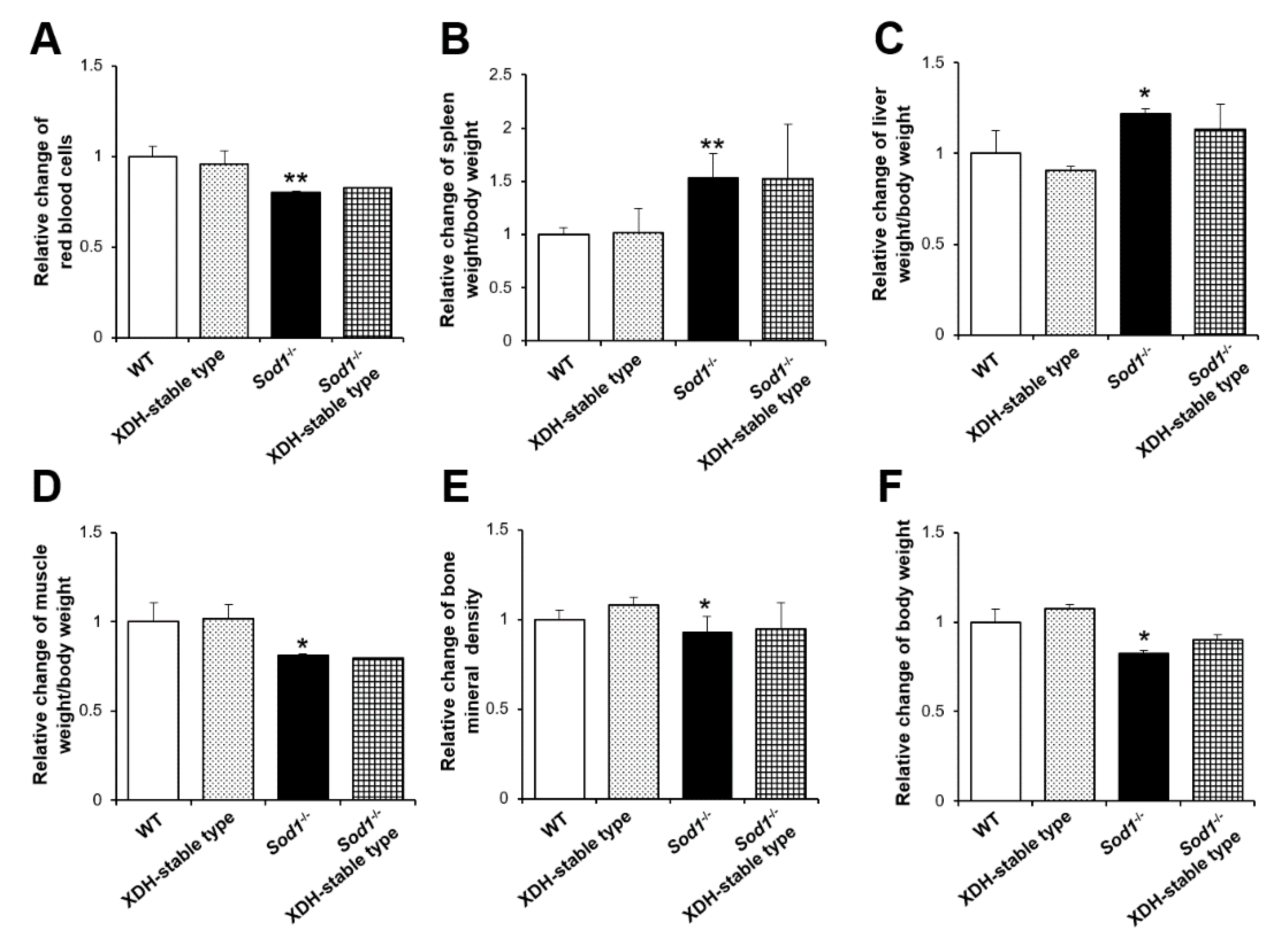
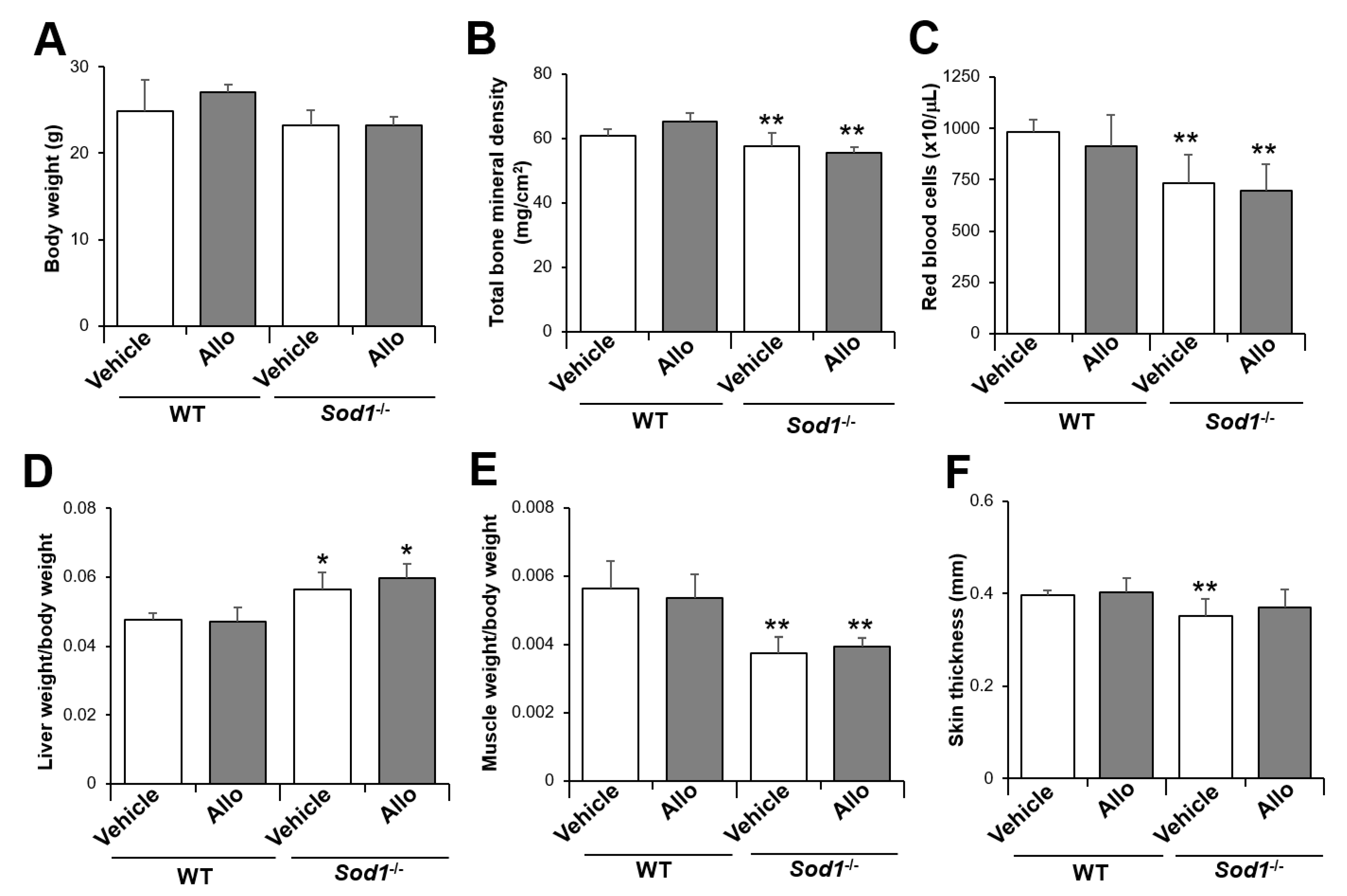
2.1. XO-Locked or XDH-Stable Types Failed to Improve the Aging-Like Phenotypes of Sod1−/− Mice
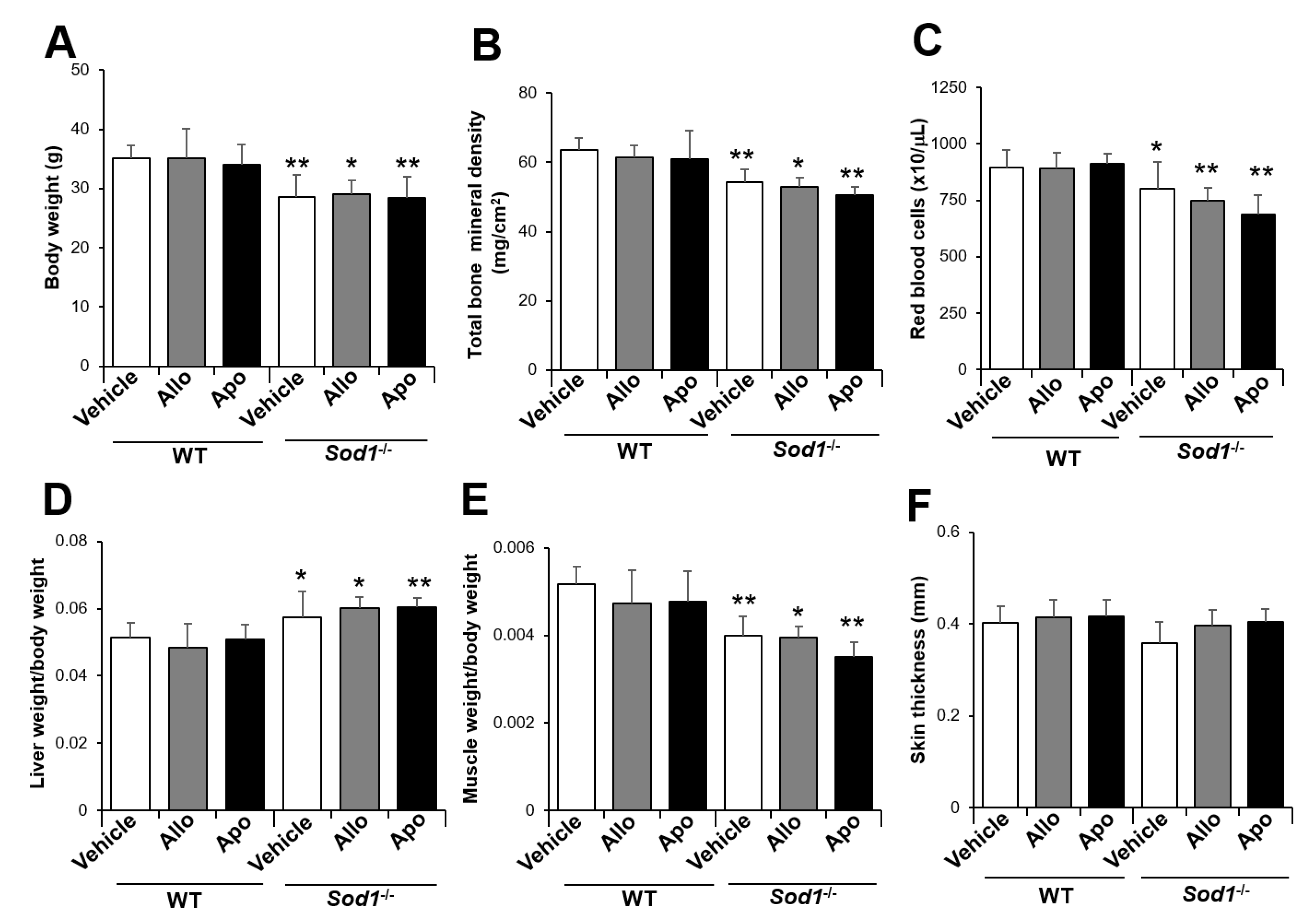
2.2. XO Inhibitor Fails to Improve the Aging-Like Pathologies in Sod1−/− Mice
3. Discussion
3.1. Contribution of XO-Derived O2•− to Tissue Pathology
3.2. Contribution of XO-Derived O2− to Tissue Pathology
3.3. Pathological Effect of XO on Aging and Tumorigenesis
4. Materials and Methods
4.1. Animals and Genotyping
4.2. Administration of Allopurinol and Apocynin
4.3. Analysis of Aging-Like Pathologies
4.4. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | Reactive oxygen species |
| O2•− | Superoxide |
| XOR | Xanthine oxidoreductase |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NOX | NADPH oxidase |
| NO | Nitric oxide |
| SOD | Superoxide dismutase |
| Sod1−/− | Sod1-deficient |
| XO | Xanthin oxidase |
| XDH | Xanthin dehydrogenase |
| KI | Knock-in |
| WT | Wild type |
References
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Shah, A.M. Endothelial cell superoxide generation: Regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, 1014–1030. [Google Scholar] [CrossRef] [PubMed]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, S.; Shimizu, T.; Shirasawa, T. CuZn-SOD deficiency causes ApoB degradation and induces hepatic lipid accumulation by impaired lipoprotein secretion in mice. J. Biol. Chem. 2006, 281, 31713–31719. [Google Scholar] [CrossRef]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef] [Green Version]
- Murakami, K.; Murata, N.; Noda, Y.; Irie, K.; Shirasawa, T.; Shimizu, T. Stimulation of the amyloidogenic pathway by cytoplasmic superoxide radicals in an Alzheimer’s disease mouse model. Biosci. Biotechnol. Biochem. 2012, 76, 1098–1103. [Google Scholar] [CrossRef]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, K.; Hirasawa, M.; Imamura, Y.; Noda, S.; Shimizu, T.; Shinoda, K.; Kurihara, T.; Noda, K.; Ozawa, Y.; Ishida, S.; et al. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am. J. Pathol. 2008, 172, 1325–1331. [Google Scholar] [CrossRef] [Green Version]
- Kojima, T.; Wakamatsu, T.H.; Dogru, M.; Ogawa, Y.; Igarashi, A.; Ibrahim, O.M.; Inaba, T.; Shimizu, T.; Noda, S.; Obata, H.; et al. Age-related dysfunction of the lacrimal gland and oxidative stress: Evidence from the Cu,Zn-superoxide dismutase-1 (Sod1) knockout mice. Am. J. Pathol. 2012, 180, 1879–1896. [Google Scholar] [CrossRef]
- Ibrahim, O.M.; Dogru, M.; Matsumoto, Y.; Igarashi, A.; Kojima, T.; Wakamatsu, T.H.; Inaba, T.; Shimizu, T.; Shimazaki, J.; Tsubota, K. Oxidative stress induced age dependent meibomian gland dysfunction in cu, zn-superoxide dismutase-1 (sod1) knockout mice. PLoS ONE 2014, 9, e99328. [Google Scholar] [CrossRef] [Green Version]
- Iuchi, Y.; Okada, F.; Onuma, K.; Onoda, T.; Asao, H.; Kobayashi, M.; Fujii, J. Elevated oxidative stress in erythrocytes due to a SOD1 deficiency causes anaemia and triggers autoantibody production. Biochem. J. 2007, 402, 219–227. [Google Scholar] [CrossRef]
- Nojiri, H.; Saita, Y.; Morikawa, D.; Kobayashi, K.; Tsuda, C.; Miyazaki, T.; Saito, M.; Marumo, K.; Yonezawa, I.; Kaneko, K.; et al. Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J. Bone Miner. Res. 2011, 26, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, D.; Nojiri, H.; Saita, Y.; Kobayashi, K.; Watanabe, K.; Ozawa, Y.; Koike, M.; Asou, Y.; Takaku, T.; Kaneko, K.; et al. Cytoplasmic reactive oxygen species and SOD1 regulate bone mass during mechanical unloading. J. Bone Miner. Res. 2013, 28, 2368–2380. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Inagaki, J.; Saito, M.; Ikeda, Y.; Tsuda, C.; Noda, Y.; Kawakami, S.; Shirasawa, T.; Shimizu, T. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochem. Biophys. Res. Commun. 2009, 382, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, S.; Ozawa, Y.; Toda, T.; Watanabe, K.; Tometsuka, C.; Ogura, T.; Koyama, Y.; Shimizu, T. Collagen peptide and vitamin C additively attenuate age-related skin atrophy in Sod1-deficient mice. Biosci. Biotechnol. Biochem. 2014, 78, 1212–1220. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.T.; Epstein, C.J.; Roberts, L.J., 2nd; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef]
- Noda, Y.; Ota, K.; Shirasawa, T.; Shimizu, T. Copper/zinc superoxide dismutase insufficiency impairs progesterone secretion and fertility in female mice. Biol. Reprod. 2012, 86, 1–8. [Google Scholar] [CrossRef]
- Sagi, H.; Shibuya, S.; Kato, T.; Nakanishi, Y.; Tsuboi, A.; Moriya, S.; Ohno, H.; Miyamoto, H.; Kodama, H.; Shimizu, T. SOD1 deficiency alters gastrointestinal microbiota and metabolites in mice. Exp. Gerontol. 2020, 130, 110795. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Shibuya, S.; Ozawa, Y.; Nojiri, H.; Izuo, N.; Yokote, K.; Shimizu, T. Superoxide dismutase 1 loss disturbs intracellular redox signaling, resulting in global age-related pathological changes. BioMed Res. Int. 2014, 2014, 140165. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kusano, T.; Nishino, T. Chemical nature and reaction mechanisms of the molybdenum cofactor of xanthine oxidoreductase. Curr. Pharm. Des. 2013, 19, 2606–2614. [Google Scholar] [CrossRef] [Green Version]
- Kusano, T.; Ehirchiou, D.; Matsumura, T.; Chobaz, V.; Nasi, S.; Castelblanco, M.; So, A.; Lavanchy, C.; Acha-Orbea, H.; Nishino, T.; et al. Targeted knock-in mice expressing the oxidase-fixed form of xanthine oxidoreductase favor tumor growth. Nat. Commun. 2019, 10, 4904. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Terada, L.S.; Grosso, M.A.; Whitmann, G.J.; Velasco, S.E.; Patt, A.; Harken, A.H.; Repine, J.E. Xanthine oxidase produces hydrogen peroxide which contributes to reperfusion injury of ischemic, isolated, perfused rat hearts. J. Clin. Investig. 1988, 81, 1297–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terada, L.S.; Rubinstein, J.D.; Lesnefsky, E.J.; Horwitz, L.D.; Leff, J.A.; Repine, J.E. Existence and participation of xanthine oxidase in reperfusion injury of ischemic rabbit myocardium. Am. J. Physiol. 1991, 260, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yan, D.; Li, S.; Liu, S.; Zeng, F.; Cheung, C.W.; Liu, H.; Irwin, M.G.; Huang, H.; Xia, Z. Allopurinol reduces oxidative stress and activates Nrf2/p62 to attenuate diabetic cardiomyopathy in rats. J. Cell. Mol. Med. 2020, 24, 1760–1773. [Google Scholar] [CrossRef] [Green Version]
- Baloglu, M.; Gokalp Ozkorkmaz, E. Neuroprotective effects of allopurinol on spinal cord injury in rats: A biochemical and immunohistochemical study. Folia Morphol. 2019, 78, 676–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorbach, C.; Scriven, A.; Capecchi, M.R. The housekeeping gene xanthine oxidoreductase is necessary for milk fat droplet enveloping and secretion: Gene sharing in the lactating mammary gland. Genes Dev. 2002, 16, 3223–3235. [Google Scholar] [CrossRef] [Green Version]
- Nasi, S.; Castelblanco, M.; Chobaz, V.; Ehirchios, D.; So, A.; Bernabei, I.; Kusano, T.; Nishino, T.; Okamoto, K.; Busso, N. Xanthine oxidoreductase Is involved in chondrocyte mineralization and expressed in osteoarthritic damaged cartilage. Front. Cell. Dev. Biol. 2021, 9, 612440. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, T.; Nakayama, T.; Heinig, M.; Zhang, L.; Yuzawa, Y.; Sanchez-Lozada, L.G.; Roncal, C.; Johnson, R.J.; Nakagawa, T. Effect of lowering uric acid on renal disease in the type 2 diabetic db/db mice. Am. J. Physiol. Renal. Physiol. 2009, 297, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Derbre, F.; Ferrando, B.; Gomez-Cabrera, M.C.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Olaso-Gonzalez, G.; Diaz, A.; Gratas-Delamarche, A.; Cerda, M.; Vina, J. Inhibition of xanthine oxidase by allopurinol prevents skeletal muscle atrophy: Role of p38 MAPKinase and E3 ubiquitin ligases. PLoS ONE 2012, 7, e46668. [Google Scholar] [CrossRef]
- Kato, S.; Shirakami, Y.; Yamaguchi, K.; Mizutani, T.; Ideta, T.; Nakamura, H.; Ninomiya, S.; Kubota, M.; Sakai, H.; Ibuka, T.; et al. Allopurinol suppresses azoxymethane-induced colorectal tumorigenesis in C57BL/KsJ-db/db mice. Gastrointest. Disord. 2020, 2, 35. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [Green Version]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturtz, L.A.; Diekert, K.; Jensen, L.T.; Lill, R.; Culotta, V.C. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 2001, 276, 38084–38089. [Google Scholar] [CrossRef]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocheme, H.M.; Murphy, M.P. Complex I is the major site of mitochondrial superoxide production by paraquat. J. Biol. Chem. 2008, 283, 1786–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.S.; Gargano, M.; Cao, J.; Bronson, R.T.; Heimler, I.; Hutz, R.J. Reduced fertility in female mice lacking copper-zinc superoxide dismutase. J. Biol. Chem. 1998, 273, 7765–7769. [Google Scholar] [CrossRef] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Shibuya, S.; Ozawa, Y.; Watanabe, K.; Izuo, N.; Toda, T.; Yokote, K.; Shimizu, T. Palladium and platinum nanoparticles attenuate aging-like skin atrophy via antioxidant activity in mice. PLoS ONE 2014, 9, e109288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qaisar, R.; Bhaskaran, S.; Ranjit, R.; Sataranatarajan, K.; Premkumar, P.; Huseman, K.; Van Remmen, H. Restoration of SERCA ATPase prevents oxidative stress-related muscle atrophy and weakness. Redox Biol. 2019, 20, 68–74. [Google Scholar] [CrossRef]
- Jang, Y.C.; Rodriguez, K.; Lustgarten, M.S.; Muller, F.L.; Bhattacharya, A.; Pierce, A.; Choi, J.J.; Lee, N.H.; Chaudhuri, A.; Richardson, A.G.; et al. Superoxide-mediated oxidative stress accelerates skeletal muscle atrophy by synchronous activation of proteolytic systems. Geroscience 2020, 42, 1579–1591. [Google Scholar] [CrossRef]
- Bhaskaran, S.; Pollock, N.; Macpherson, P.C.; Ahn, B.; Piekarz, K.M.; Staunton, C.A.; Brown, J.L.; Qaisar, R.; Vasilaki, A.; Richardson, A.; et al. Neuron-specific deletion of CuZnSOD leads to an advanced sarcopenic phenotype in older mice. Aging Cell. 2020, 19, e13225. [Google Scholar] [CrossRef]
- Kim, J.; Toda, T.; Watanabe, K.; Shibuya, S.; Ozawa, Y.; Izuo, N.; Cho, S.; Seo, D.B.; Yokote, K.; Shimizu, T. Syringaresinol Reverses Age-Related Skin Atrophy by Suppressing FoxO3a-Mediated Matrix Metalloproteinase-2 Activation in Copper/Zinc Superoxide Dismutase-Deficient Mice. J. Investig. Dermatol. 2019, 139, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Sautin, Y.Y.; Johnson, R.J. Uric acid: The oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Ridi, R.; Tallima, H. Physiological functions and pathogenic potential of uric acid: A review. J. Adv. Res. 2017, 8, 487–493. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavaroni, I.; Mazza, S.; Fantuzzi, M.; Dall’Aglio, E.; Bonora, E.; Delsignore, R.; Passeri, M.; Reaven, G.M. Changes in insulin and lipid metabolism in males with asymptomatic hyperuricaemia. J. Intern. Med. 1993, 234, 25–30. [Google Scholar] [CrossRef]
- Amaro, S.; Soy, D.; Obach, V.; Cervera, A.; Planas, A.M.; Chamorro, A. A pilot study of dual treatment with recombinant tissue plasminogen activator and uric acid in acute ischemic stroke. Stroke 2007, 38, 2173–2175. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Ladenheim, B.; Cutler, R.G.; Kruman, I.I.; Cadet, J.L.; Mattson, M.P. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. J. Neurochem. 2002, 80, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C.; Spitsin, S.; Kean, R.B.; Champion, J.M.; Dickson, G.M.; Chaudhry, I.; Koprowski, H. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA 1998, 95, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Spitsin, S.V.; Scott, G.S.; Mikheeva, T.; Zborek, A.; Kean, R.B.; Brimer, C.M.; Koprowski, H.; Hooper, D.C. Comparison of uric acid and ascorbic acid in protection against EAE. Free Radic. Biol. Med. 2002, 33, 1363–1371. [Google Scholar] [CrossRef]
- Yu, Z.F.; Bruce-Keller, A.J.; Goodman, Y.; Mattson, M.P. Uric acid protects neurons against excitotoxic and metabolic insults in cell culture, and against focal ischemic brain injury in vivo. J. Neurosci. Res. 1998, 53, 613–625. [Google Scholar] [CrossRef]
- Tsukada, K.; Hasegawa, T.; Tsutsumi, S.; Katoh, H.; Kuwano, H.; Miyazaki, T.; Yamamoto, Y. Effect of uric acid on liver injury during hemorrhagic shock. Surgery 2000, 127, 439–446. [Google Scholar] [CrossRef]
- Wang, J.; Fan, Y.; Cai, X.; Gao, Z.; Yu, Z.; Wei, B.; Tang, Y.; Hu, L.; Liu, W.T.; Gu, Y. Uric acid preconditioning alleviated doxorubicin induced JNK activation and Cx43 phosphorylation associated cardiotoxicity via activation of AMPK-SHP2 signaling pathway. Ann. Transl. Med. 2020, 8, 1570. [Google Scholar] [CrossRef]
- Santos, C.X.; Anjos, E.I.; Augusto, O. Uric acid oxidation by peroxynitrite: Multiple reactions, free radical formation, and amplification of lipid oxidation. Arch. Biochem. Biophys. 1999, 372, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Komaki, Y.; Sugiura, H.; Koarai, A.; Tomaki, M.; Ogawa, H.; Akita, T.; Hattori, T.; Ichinose, M. Cytokine-mediated xanthine oxidase upregulation in chronic obstructive pulmonary disease’s airways. Pulm. Pharmacol. Ther. 2005, 18, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Song, S.H.; Kim, H.J.; Ikeno, Y.; Yu, B.P. Modulation of renal xanthine oxidoreductase in aging: Gene expression and reactive oxygen species generation. J. Nutr. Health Aging 1999, 3, 19–23. [Google Scholar] [PubMed]
- Newaz, M.A.; Yousefipour, Z.; Oyekan, A. Oxidative stress-associated vascular aging is xanthine oxidase-dependent but not NAD(P)H oxidase-dependent. J. Cardiovasc. Pharmacol. 2006, 48, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Vida, C.; Rodriguez-Teres, S.; Heras, V.; Corpas, I.; De la Fuente, M.; Gonzalez, E. The aged-related increase in xanthine oxidase expression and activity in several tissues from mice is not shown in long-lived animals. Biogerontology 2011, 12, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Matesanz, N.; Lafuente, N.; Azcutia, V.; Martin, D.; Cuadrado, A.; Nevado, J.; Rodriguez-Manas, L.; Sanchez-Ferrer, C.F.; Peiro, C. Xanthine oxidase-derived extracellular superoxide anions stimulate activator protein 1 activity and hypertrophy in human vascular smooth muscle via c-Jun N-terminal kinase and p38 mitogen-activated protein kinases. J. Hypertens. 2007, 25, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, 453–462. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shibuya, S.; Watanabe, K.; Ozawa, Y.; Shimizu, T. Xanthine Oxidoreductase-Mediated Superoxide Production Is Not Involved in the Age-Related Pathologies in Sod1-Deficient Mice. Int. J. Mol. Sci. 2021, 22, 3542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073542
Shibuya S, Watanabe K, Ozawa Y, Shimizu T. Xanthine Oxidoreductase-Mediated Superoxide Production Is Not Involved in the Age-Related Pathologies in Sod1-Deficient Mice. International Journal of Molecular Sciences. 2021; 22(7):3542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073542
Chicago/Turabian StyleShibuya, Shuichi, Kenji Watanabe, Yusuke Ozawa, and Takahiko Shimizu. 2021. "Xanthine Oxidoreductase-Mediated Superoxide Production Is Not Involved in the Age-Related Pathologies in Sod1-Deficient Mice" International Journal of Molecular Sciences 22, no. 7: 3542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073542