
Intratumoral Canine Distemper Virus Infection Inhibits Tumor Growth by Modulation of the Tumor Microenvironment in a Murine Xenograft Model of Canine Histiocytic Sarcoma

 ,
,
Abstract
:1. Introduction
2. Results
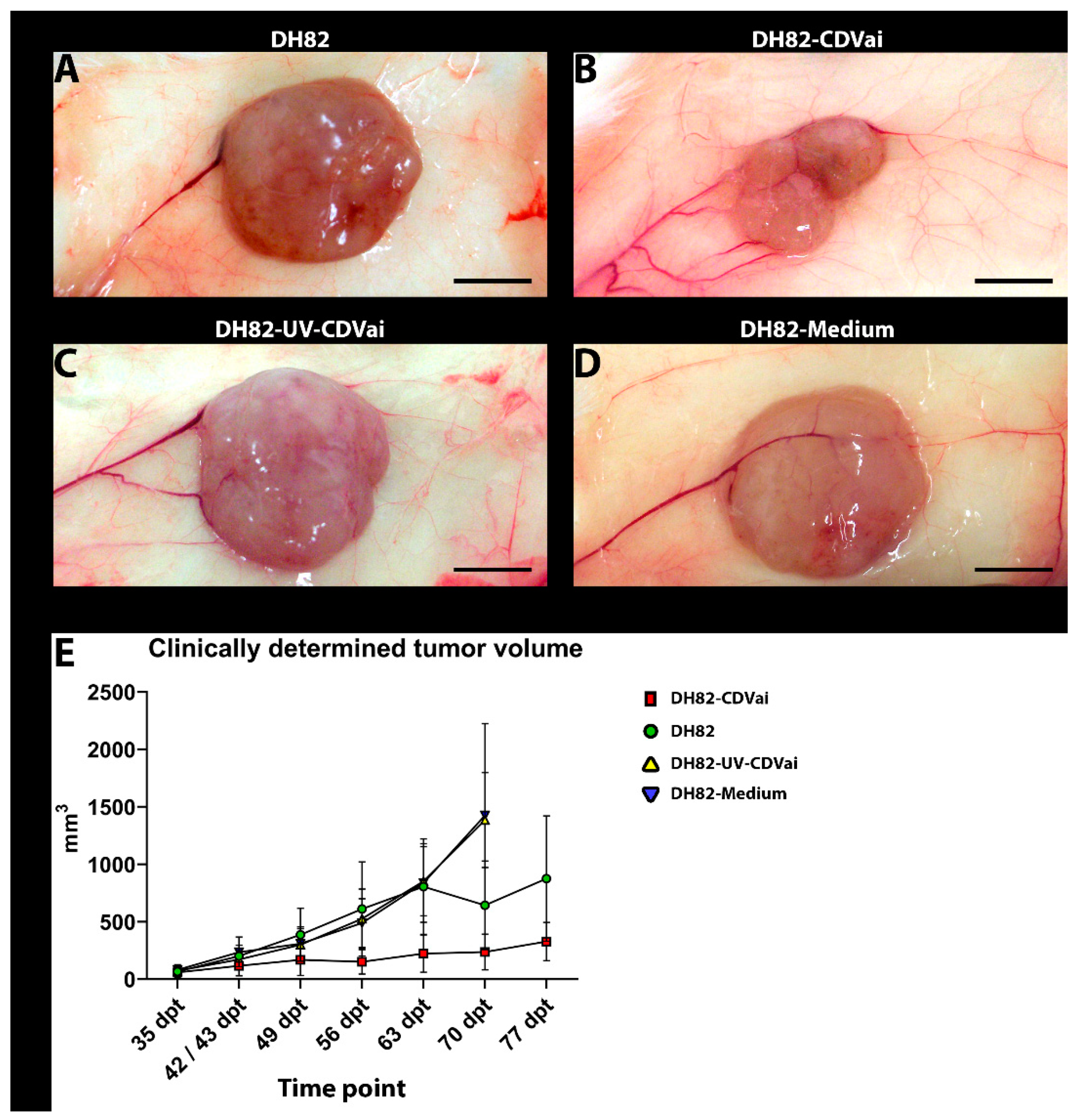
2.1. A Tenfold Intratumoral Infection Leads to a Significantly Reduced Tumor Growth Compared to Controls
2.2. Murine Xenotransplantation Model of Canine Histiocytic Sarcoma Was Successfully Confirmed
2.3. Acute CDV-Infection Leads to a Growth Retardation of Xenotransplants Accompanied by Necrosis in a Murine Model of Canine Histiocytic Sarcoma
2.4. CDV-Nucleoprotein Was Detected in Intratumorally Treated DH82 Cell Xenografts
2.5. Acute CDV-Infection Leads to an Enhanced Apoptosis in DH82 Cell Xenografts at Early Time Points
2.6. Acute Intratumoral CDV-Infection Is Associated with Increased Numbers of Tumor-Associated Macrophages in DH82 Cell Xenografts
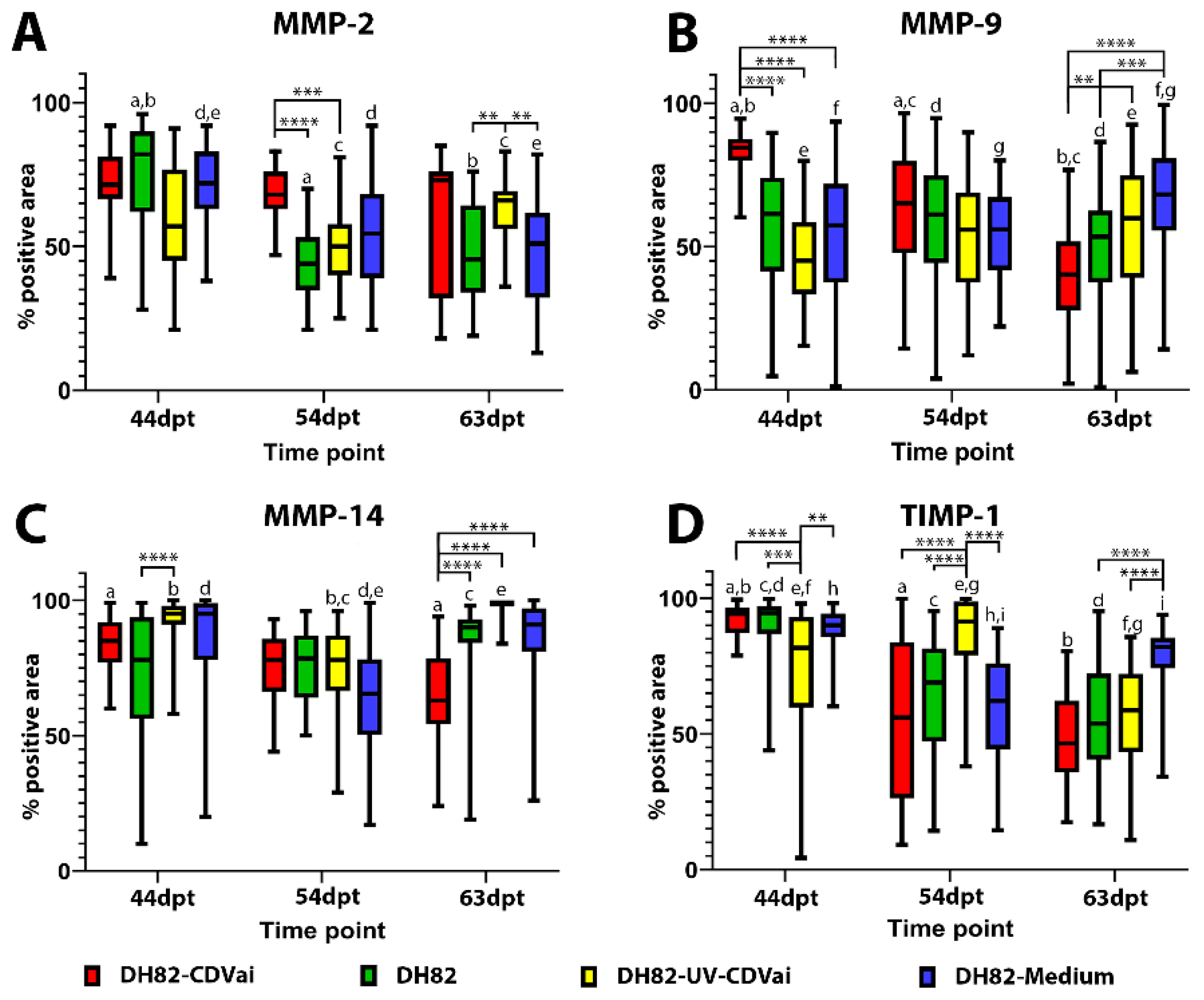
2.7. Acute CDV-Infection Leads to a Modulation of the MMPs and TIMP-1 Expression in DH82 Cell Xenografts
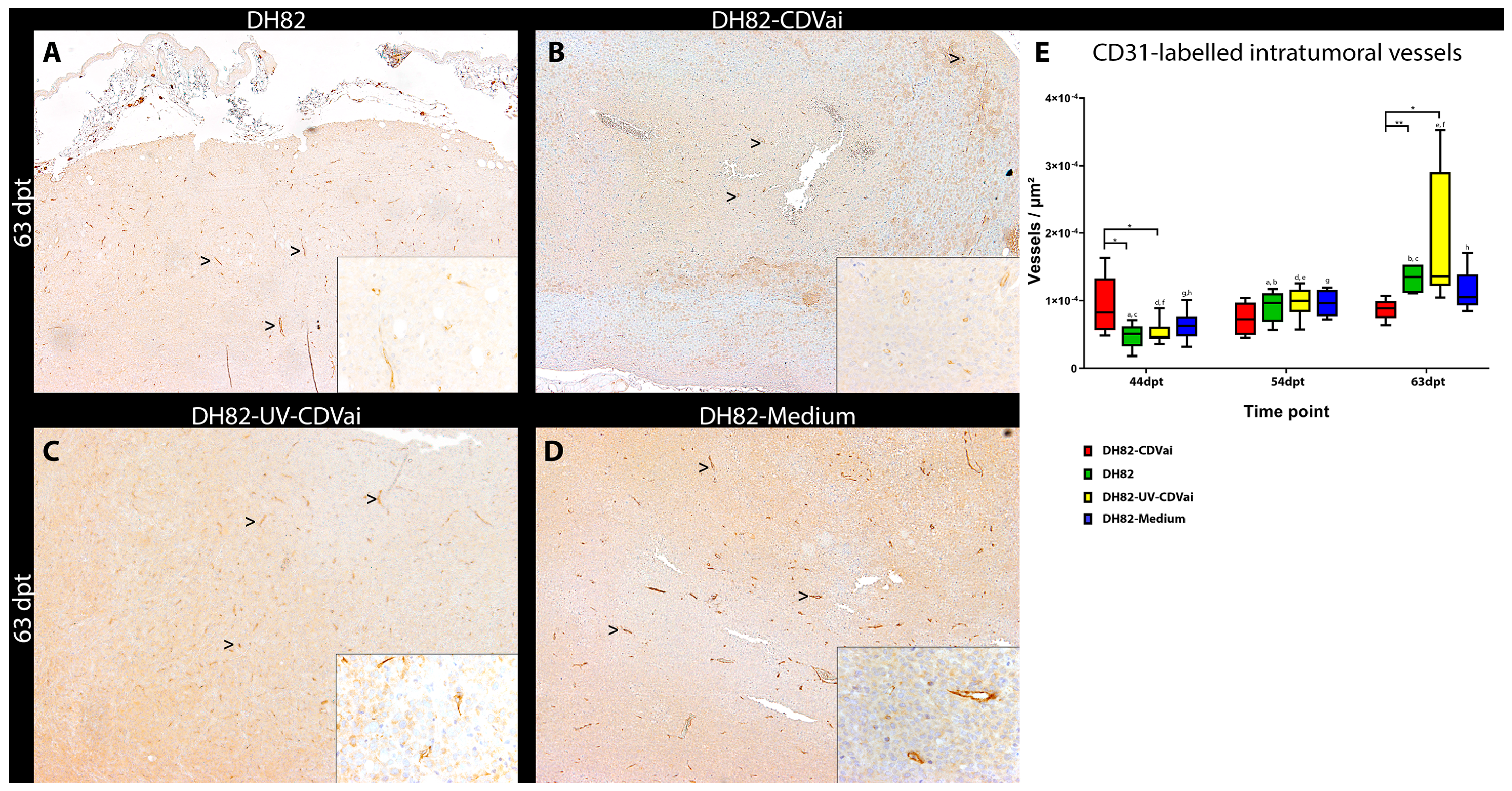
2.8. Acute CDV-Infection Influences Microvascular Density in DH82 Xenografts
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Establishment of Murine Xenografts
4.3. Intratumoral Treatment of Xenotransplanted Histiocytic Sarcomas
4.4. Histology and Immunohistochemistry
4.5. Histological and Immunohistochemical Analysis, Including Morphometric Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, D.G.; Church, D.B.; McGreevy, P.D.; Thomson, P.C.; Brodbelt, D.C. Longevity and mortality of owned dogs in England. Vet. J. 2013, 198, 638–643. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Lewis, T.W.; Wiles, B.M.; Llewellyn-Zaidi, A.M.; Evans, K.M.; O’Neill, D.G. Longevity and mortality in Kennel Club registered dog breeds in the UK in 2014. Canine Genet. Epidemiol. 2018, 5, 10. [Google Scholar] [CrossRef]
- Bilici, A. Prognostic factors related with survival in patients with pancreatic adenocarcinoma. World J. Gastroenterol. 2014, 20, 10802–10812. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, C.A.; Gardner, H.L.; Mathie, T.; Stingle, N.; Portela, R.; Pennell, M.L.; Clifford, C.A.; Rosenberg, M.P.; Vail, D.M.; Williams, L.E.; et al. Impact of Toceranib/Piroxicam/Cyclophosphamide maintenance therapy on outcome of dogs with appendicular osteosarcoma following amputation and Carboplatin chemotherapy: A multi-institutional study. PLoS ONE 2015, 10, e0124889. [Google Scholar] [CrossRef] [PubMed]
- Schwens, C.; Thom, N.; Moritz, A. Reactive and neoplastic histiocytic diseases in the dog. Tierärztl. Prax. Ausg. K Kleintiere Heimtiere 2011, 39, 176–190. [Google Scholar] [PubMed]
- Takahashi, E.; Nakamura, S. Histiocytic sarcoma: An updated literature review based on the 2008 WHO classification. J. Clin. Exp. Hematop. 2013, 53, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Erich, S.A.; Rutteman, G.R.; Teske, E. Causes of death and the impact of histiocytic sarcoma on the life expectancy of the Dutch population of Bernese mountain dogs and Flat-coated retrievers. Vet. J. 2013, 198, 678–683. [Google Scholar] [CrossRef]
- Gounder, M.; Desai, V.; Kuk, D.; Agaram, N.; Arcila, M.; Durham, B.; Keohan, M.L.; Dickson, M.A.; D’Angelo, S.P.; Shukla, N.; et al. Impact of surgery, radiation and systemic therapy on the outcomes of patients with dendritic cell and histiocytic sarcomas. Eur. J. Cancer 2015, 51, 2413–2422. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Tomiyasu, H.; Hotta, E.; Asada, H.; Fukushima, K.; Kanemoto, H.; Fujino, Y.; Ohno, K.; Uchida, K.; Nakayama, H.; et al. Clinical characteristics and prognostic factors in dogs with histiocytic sarcomas in Japan. J. Vet. Med. Sci. 2014, 76, 661–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, P.F. A review of histiocytic diseases of dogs and cats. Vet. Pathol. 2014, 51, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Skorupski, K.A.; Clifford, C.A.; Paoloni, M.C.; Lara-Garcia, A.; Barber, L.; Kent, M.S.; LeBlanc, A.K.; Sabhlok, A.; Mauldin, E.A.; Shofer, F.S.; et al. CCNU for the treatment of dogs with histiocytic sarcoma. J. Vet. Intern. Med. 2007, 21, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Sinkovics, J.G.; Horvath, J.C. Natural and genetically engineered viral agents for oncolysis and gene therapy of human cancers. Arch Immunol. Ther. Exp. (Warsz) 2008, 56 (Suppl. 1), 3s–59s. [Google Scholar] [CrossRef]
- Kuruppu, D.; Tanabe, K.K. Viral oncolysis by herpes simplex virus and other viruses. Cancer Biol. Ther. 2005, 4, 524–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesonen, S.; Kangasniemi, L.; Hemminki, A. Oncolytic adenoviruses for the treatment of human cancer: Focus on translational and clinical data. Mol. Pharm. 2011, 8, 12–28. [Google Scholar] [CrossRef]
- Maitra, R.; Ghalib, M.H.; Goel, S. Reovirus: A targeted therapeutic—Progress and potential. Mol. Cancer Res. 2012, 10, 1514–1525. [Google Scholar] [CrossRef] [Green Version]
- Lapp, S.; Pfankuche, V.M.; Baumgärtner, W.; Puff, C. Viral oncolysis—Can insights from measles be transferred to canine distemper virus? Viruses 2014, 6, 2340–2375. [Google Scholar] [CrossRef] [Green Version]
- Gentschev, I.; Adelfinger, M.; Josupeit, R.; Rudolph, S.; Ehrig, K.; Donat, U.; Weibel, S.; Chen, N.G.; Yu, Y.A.; Zhang, Q.; et al. Preclinical evaluation of oncolytic vaccinia virus for therapy of canine soft tissue sarcoma. PLoS ONE 2012, 7, e37239. [Google Scholar] [CrossRef]
- Patil, S.S.; Gentschev, I.; Nolte, I.; Ogilvie, G.; Szalay, A.A. Oncolytic virotherapy in veterinary medicine: Current status and future prospects for canine patients. J. Transl. Med. 2012, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K.; Doley, J.; Kumar, G.R.; Sahoo, A.P.; Tiwari, A.K. Oncolytic viruses & their specific targeting to tumour cells. Indian J. Med. Res. 2012, 136, 571–584. [Google Scholar]
- Tollefson, A.E.; Ryerse, J.S.; Scaria, A.; Hermiston, T.W.; Wold, W.S. The E3-11.6-kDa adenovirus death protein (ADP) is required for efficient cell death: Characterization of cells infected with adp mutants. Virology 1996, 220, 152–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtrichman, R.; Kleinberger, T. Adenovirus type 5 E4 open reading frame 4 protein induces apoptosis in transformed cells. J. Virol. 1998, 72, 2975–2982. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit. Rev. Oncol. Hematol. 2007, 62, 179–213. [Google Scholar] [CrossRef]
- De Silva, N.; Atkins, H.; Kirn, D.H.; Bell, J.C.; Breitbach, C.J. Double trouble for tumours: Exploiting the tumour microenvironment to enhance anticancer effect of oncolytic viruses. Cytokine Growth Factor Rev. 2010, 21, 135–141. [Google Scholar] [CrossRef]
- Angarita, F.A.; Acuna, S.A.; Ottolino-Perry, K.; Zerhouni, S.; McCart, J.A. Mounting a strategic offense: Fighting tumor vasculature with oncolytic viruses. Trends Mol. Med. 2013, 19, 378–392. [Google Scholar] [CrossRef]
- Overall, C.M.; López-Otín, C. Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef]
- Suter, S.E.; Chein, M.B.; von Messling, V.; Yip, B.; Cattaneo, R.; Vernau, W.; Madewell, B.R.; London, C.A. In vitro canine distemper virus infection of canine lymphoid cells: A prelude to oncolytic therapy for lymphoma. Clin. Cancer Res. 2005, 11, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Beineke, A.; Puff, C.; Seehusen, F.; Baumgärtner, W. Pathogenesis and immunopathology of systemic and nervous canine distemper. Vet. Immunol. Immunopathol. 2009, 127, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Kojimoto, A.; Sakai, H.; Uchida, K.; Sugano, S.; Tateyama, S. Growth characteristics of canine distemper virus in a new cell line CCT cells originated from canine malignant histiocytosis. J. Vet. Med. Sci. 2005, 67, 203–206. [Google Scholar] [CrossRef] [Green Version]
- Puff, C.; Krudewig, C.; Imbschweiler, I.; Baumgärtner, W.; Alldinger, S. Influence of persistent canine distemper virus infection on expression of RECK, matrix-metalloproteinases and their inhibitors in a canine macrophage/monocytic tumour cell line (DH82). Vet. J. 2009, 182, 100–107. [Google Scholar] [CrossRef]
- Fayyad, A.; Lapp, S.; Risha, E.; Pfankuche, V.M.; Rohn, K.; Barthel, Y.; Schaudien, D.; Baumgärtner, W.; Puff, C. Matrix metalloproteinases expression in spontaneous canine histiocytic sarcomas and its xenograft model. Vet. Immunol. Immunopathol. 2018, 198, 54–64. [Google Scholar] [CrossRef]
- Pfankuche, V.M.; Sayed-Ahmed, M.; Contioso, V.B.; Spitzbarth, I.; Rohn, K.; Ulrich, R.; Deschl, U.; Kalkuhl, A.; Baumgärtner, W.; Puff, C. Persistent Morbillivirus infection leads to altered cortactin distribution in histiocytic sarcoma cells with decreased cellular migration capacity. PLoS ONE 2016, 11, e0167517. [Google Scholar] [CrossRef]
- Pfankuche, V.M.; Spitzbarth, I.; Lapp, S.; Ulrich, R.; Deschl, U.; Kalkuhl, A.; Baumgärtner, W.; Puff, C. Reduced angiogenic gene expression in morbillivirus-triggered oncolysis in a translational model for histiocytic sarcoma. J. Cell. Mol. Med. 2017, 21, 816–830. [Google Scholar] [CrossRef]
- Armando, F.; Ferrari, L.; Arcari, M.L.; Azzali, G.; Dallatana, D.; Ferrari, M.; Lombardi, G.; Zanfabro, M.; Di Lecce, R.; Lunghi, P.; et al. Endocanalicular transendothelial crossing (ETC): A novel intravasation mode used by HEK-EBNA293-VEGF-D cells during the metastatic process in a xenograft model. PLoS ONE 2020, 15, e0239932. [Google Scholar] [CrossRef]
- Puff, C.; Risha, E.; Baumgärtner, W. Regression of canine cutaneous histiocytoma is associated with an orchestrated expression of matrix metalloproteinases. J. Comp. Pathol. 2013, 149, 208–215. [Google Scholar] [CrossRef]
- Gauvrit, A.; Brandler, S.; Sapede-Peroz, C.; Boisgerault, N.; Tangy, F.; Gregoire, M. Measles virus induces oncolysis of mesothelioma cells and allows dendritic cells to cross-prime tumor-specific CD8 response. Cancer Res. 2008, 68, 4882–4892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, S.; Weibel, S.; Donat, U.; Zhang, Q.; Aguilar, R.J.; Chen, N.G.; Szalay, A.A. Vaccinia virus-mediated intra-tumoral expression of matrix metalloproteinase 9 enhances oncolysis of PC-3 xenograft tumors. BMC Cancer 2012, 12, 366. [Google Scholar] [CrossRef] [Green Version]
- Heinzerling, L.; Künzi, V.; Oberholzer, P.A.; Kündig, T.; Naim, H.; Dummer, R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood 2005, 106, 2287–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Cheng, P. Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy. Mol. Cancer 2020, 19, 158. [Google Scholar] [CrossRef] [PubMed]
- Boisgerault, N.; Guillerme, J.B.; Pouliquen, D.; Mesel-Lemoine, M.; Achard, C.; Combredet, C.; Fonteneau, J.F.; Tangy, F.; Grégoire, M. Natural oncolytic activity of live-attenuated measles virus against human lung and colorectal adenocarcinomas. BioMed Res. Int. 2013, 2013, 387362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Chen, P.; Yang, H.; Wu, Y.; Zeng, X.; Zhao, Y.; Wen, Y.; Zhao, X.; Liu, X.; Wei, Y.; et al. Live attenuated measles virus vaccine induces apoptosis and promotes tumor regression in lung cancer. Oncol. Rep. 2013, 29, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbach, C.J.; Paterson, J.M.; Lemay, C.G.; Falls, T.J.; McGuire, A.; Parato, K.A.; Stojdl, D.F.; Daneshmand, M.; Speth, K.; Kirn, D.; et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol. Ther. 2007, 15, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Gentschev, I.; Patil, S.S.; Petrov, I.; Cappello, J.; Adelfinger, M.; Szalay, A.A. Oncolytic virotherapy of canine and feline cancer. Viruses 2014, 6, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Armando, F.; Gambini, M.; Corradi, A.; Becker, K.; Marek, K.; Pfankuche, V.M.; Mergani, A.E.; Brogden, G.; de Buhr, N.; von Köckritz-Blickwede, M.; et al. Mesenchymal to epithelial transition driven by canine distemper virus infection of canine histiocytic sarcoma cells contributes to a reduced cell motility in vitro. J. Cell. Mol. Med. 2020, 24, 9332–9348. [Google Scholar] [CrossRef]
- Armando, F.; Gambini, M.; Corradi, A.; Giudice, C.; Pfankuche, V.M.; Brogden, G.; Attig, F.; von Köckritz-Blickwede, M.; Baumgärtner, W.; Puff, C. Oxidative stress in canine histiocytic sarcoma cells induced by an infection with canine distemper virus led to a dysregulation of HIF-1α downstream pathway resulting in a reduced expression of VEGF-B in vitro. Viruses 2020, 12, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everts, A.; Bergeman, M.; McFadden, G.; Kemp, V. Simultaneous tumor and stroma targeting by oncolytic viruses. Biomedicines 2020, 8, 474. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Miao, Q.; Baumgärtner, W.; Failing, K.; Alldinger, S. Phase-dependent expression of matrix metalloproteinases and their inhibitors in demyelinating canine distemper encephalitis. Acta Neuropathol. 2003, 106, 486–494. [Google Scholar] [CrossRef]
- Gröters, S.; Alldinger, S.; Baumgärtner, W. Up-regulation of mRNA for matrix metalloproteinases-9 and -14 in advanced lesions of demyelinating canine distemper leukoencephalitis. Acta Neuropathol. 2005, 110, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Bregano, L.C.; Agostinho, S.D.; Roncatti, F.L.; Pires, M.C.; Riva, H.G.; Luvizotto, M.C.; Cardoso, T.C. Immunohistochemical detection of metalloproteinase-9 (MMP-9), anti-oxidant like 1 protein (AOP-1) and synaptosomal-associated protein (SNAP-25) in the cerebella of dogs naturally infected with spontaneous canine distemper. Folia Histochem. Cytobiol. 2011, 49, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, G.F.; Melo, G.D.; Souza, M.S.; Machado, A.A.; Migliolo, D.S.; Moraes, O.C.; Nunes, C.M.; Ribeiro, E.S. Zymographic patterns of MMP-2 and MMP-9 in the CSF and cerebellum of dogs with subacute distemper leukoencephalitis. Vet. Immunol. Immunopathol. 2013, 154, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Pulz, L.H.; Strefezzi, R.F. Proteases as prognostic markers in human and canine cancers. Vet Comp Oncol 2017, 15, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015, 44–46, 94–112. [Google Scholar] [CrossRef]
- Hornebeck, W.; Lambert, E.; Petitfrère, E.; Bernard, P. Beneficial and detrimental influences of tissue inhibitor of metalloproteinase-1 (TIMP-1) in tumor progression. Biochimie 2005, 87, 377–383. [Google Scholar] [CrossRef]
- Barnes, A.; Bee, A.; Bell, S.; Gilmore, W.; Mee, A.; Morris, R.; Carter, S.D. Immunological and inflammatory characterisation of three canine cell lines: K1, K6 and DH82. Vet. Immunol. Immunopathol. 2000, 75, 9–25. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Fujiwara, Y.; Ohnishi, K.; Takeya, M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv. Drug Deliv. Rev. 2016, 99, 180–185. [Google Scholar] [CrossRef]
- Grote, D.; Russell, S.J.; Cornu, T.I.; Cattaneo, R.; Vile, R.; Poland, G.A.; Fielding, A.K. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood 2001, 97, 3746–3754. [Google Scholar] [CrossRef] [Green Version]
- Bock, P.; Spitzbarth, I.; Haist, V.; Stein, V.M.; Tipold, A.; Puff, C.; Beineke, A.; Baumgärtner, W. Spatio-temporal development of axonopathy in canine intervertebral disc disease as a translational large animal model for nonexperimental spinal cord injury. Brain Pathol. 2013, 23, 82–99. [Google Scholar] [CrossRef]
- Prajeeth, C.K.; Beineke, A.; Iskandar, C.D.; Gudi, V.; Herder, V.; Gerhauser, I.; Haist, V.; Teich, R.; Huehn, J.; Baumgärtner, W.; et al. Limited role of regulatory T cells during acute Theiler virus-induced encephalitis in resistant C57BL/6 mice. J. Neuroinflamm. 2014, 11, 180. [Google Scholar] [CrossRef] [Green Version]
- Alldinger, S.; Baumgärtner, W.; Kremmer, E.; Fonfara, S. Characterization of a canine CD44 specific monoclonal antibody. Zentralbl. Veterinärmed. A 1999, 46, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Baynosa, R.C.; Browder, L.K.; Jones, S.R.; Oliver, J.A.; Van Der Harten, C.A.; Stephenson, L.L.; Wang, W.Z.; Khiabani, K.T.; Zamboni, W.A. Evaluation of artificial dermis neovascularization in an avascular wound. J. Reconstr. Microsurg. 2009, 25, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, F.; Contioso, V.B.; Stein, V.M.; Carlson, R.; Tipold, A.; Ulrich, R.; Puff, C.; Baumgärtner, W.; Spitzbarth, I. Passage-dependent morphological and phenotypical changes of a canine histiocytic sarcoma cell line (DH82 cells). Vet. Immunol. Immunopathol. 2015, 163, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S.; Hage, E.; Käufer, C.; Gerhauser, I.; Anjum, M.; Li, L.; Baumgärtner, W.; Schulz, T.F.; Löscher, W. Viral mouse models of multiple sclerosis and epilepsy: Marked differences in neuropathogenesis following infection with two naturally occurring variants of Theiler’s virus BeAn strain. Neurobiol. Dis. 2017, 99, 121–132. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Name, Clonality, Source | Demasking Procedure | Dilution of Antibodies | Secondary Antibody | Positive Control |
|---|---|---|---|---|---|
| CD44 | Clone 2D10; monoclonal rat; E. Kremmer, München, Germany | Microwave/CB 20 min | 1:200 | RaR-b | DH82 cell pellet |
| CDV-NP | Clone D110; monoclonal mouse; A. Zurbriggen, Bern, Switzerland | Microwave/CB 20 min | 1:100 | GaM-b | persistently CDV-Ond-infected DH82 cell pellet |
| Cleaved Caspase 3 (Asp175) | Polyclonal rabbit; Cell Signaling Technology Inc., Danvers, MA, USA | Microwave/CB 20 min | 1:200 | GaR-b | SCID mouse, lymphoid tissue |
| Mac3 (CD107b/LAMP2) | Monoclonal rat; clone: M3/84; AbD Serotec, Oxford, UK | Microwave/CB 20 min | 1:200 | n.a. | SCID mouse, lymphoid tissue |
| CD31 | Polyclonal rabbit; Acris antibodies, Hiddenhausen, Germany | Microwave/CB 20 min | 1:100 | GaR-b | Canine granulation tissue |
| MMP-2 | MAB13405; monoclonal mouse; Millipore, Burlington, MA, USA | None | 1:400 | GaM-b | DH82 cell pellet |
| MMP-9 | RM105-MMP9; polyclonal rabbit; Triple Point Biologics, Forest Grove, OR, USA | None | 1:500 | GaR-b | DH82 cell pellet |
| MMP-14 | RP1-MMP14; polyclonal rabbit; Triple Point Biologics, Forest Grove, OR, USA | None | 1:200 | GaR-b | DH82 cell pellet |
| TIMP-1 | RP3-TIMP1; polyclonal rabbit; Triple Point Biologics, Forest Grove, OR, USA | None | 1:1000 | GaR-b | Canine stillborn puppy, bone marrow |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armando, F.; Fayyad, A.; Arms, S.; Barthel, Y.; Schaudien, D.; Rohn, K.; Gambini, M.; Lombardo, M.S.; Beineke, A.; Baumgärtner, W.; et al. Intratumoral Canine Distemper Virus Infection Inhibits Tumor Growth by Modulation of the Tumor Microenvironment in a Murine Xenograft Model of Canine Histiocytic Sarcoma. Int. J. Mol. Sci. 2021, 22, 3578. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073578
Armando F, Fayyad A, Arms S, Barthel Y, Schaudien D, Rohn K, Gambini M, Lombardo MS, Beineke A, Baumgärtner W, et al. Intratumoral Canine Distemper Virus Infection Inhibits Tumor Growth by Modulation of the Tumor Microenvironment in a Murine Xenograft Model of Canine Histiocytic Sarcoma. International Journal of Molecular Sciences. 2021; 22(7):3578. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073578
Chicago/Turabian StyleArmando, Federico, Adnan Fayyad, Stefanie Arms, Yvonne Barthel, Dirk Schaudien, Karl Rohn, Matteo Gambini, Mara Sophie Lombardo, Andreas Beineke, Wolfgang Baumgärtner, and et al. 2021. "Intratumoral Canine Distemper Virus Infection Inhibits Tumor Growth by Modulation of the Tumor Microenvironment in a Murine Xenograft Model of Canine Histiocytic Sarcoma" International Journal of Molecular Sciences 22, no. 7: 3578. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073578