
Genome-Wide Methylation Mapping Using Nanopore Sequencing Technology Identifies Novel Tumor Suppressor Genes in Hepatocellular Carcinoma

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
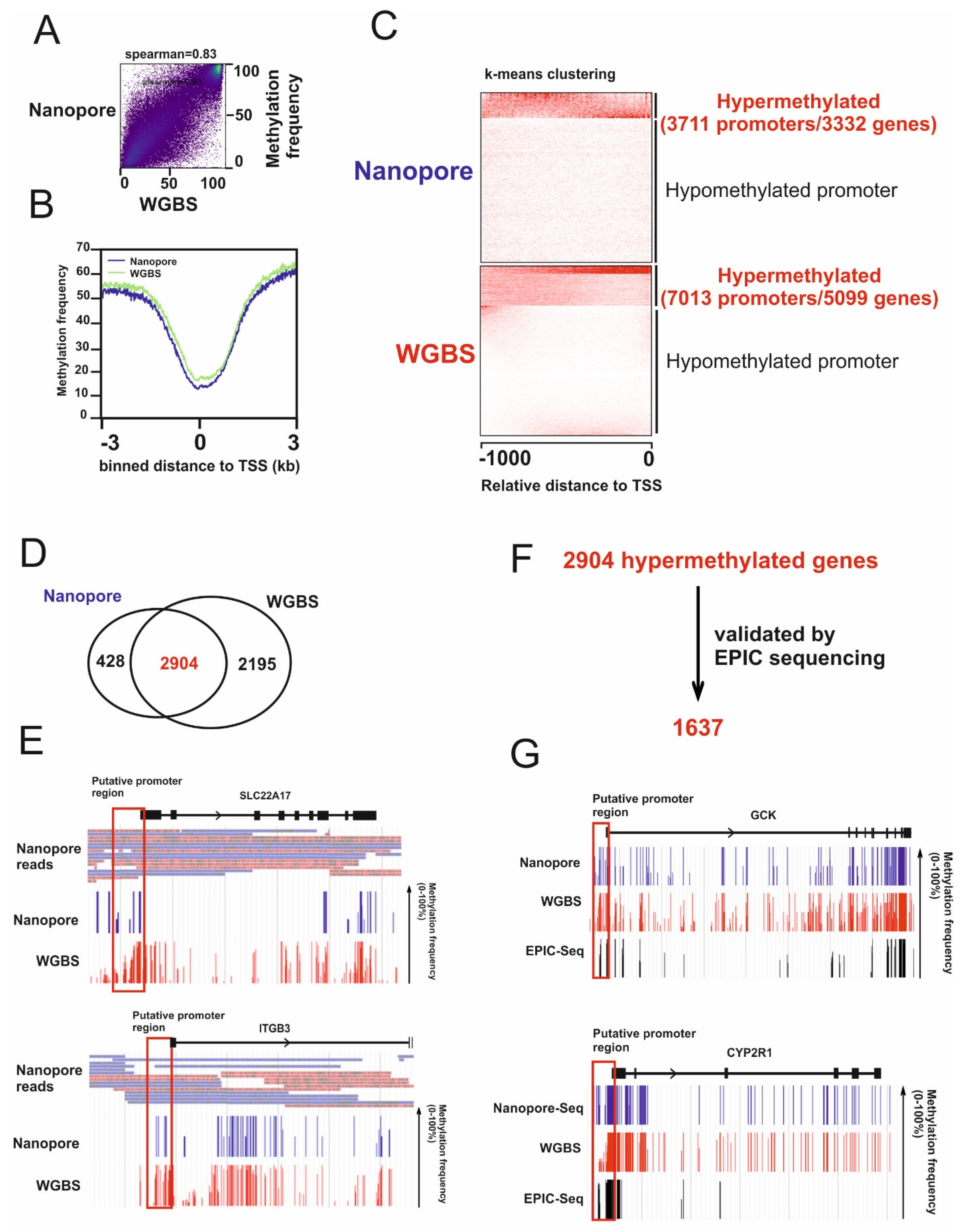
2.1. Identification of Hypermethylated Genes in HCC Cells
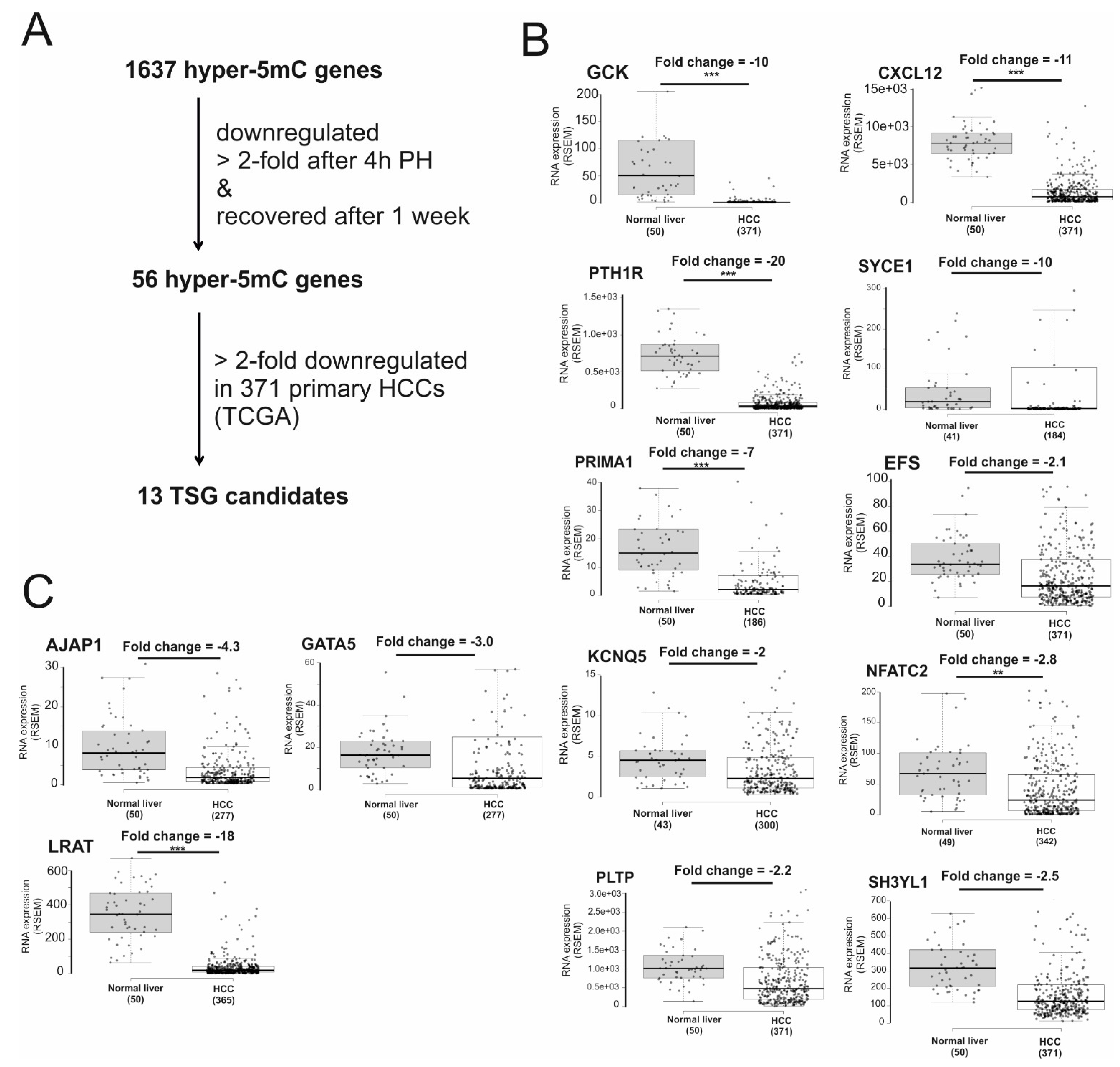
2.2. Identification of Candidates for Novel TSGs in HCC
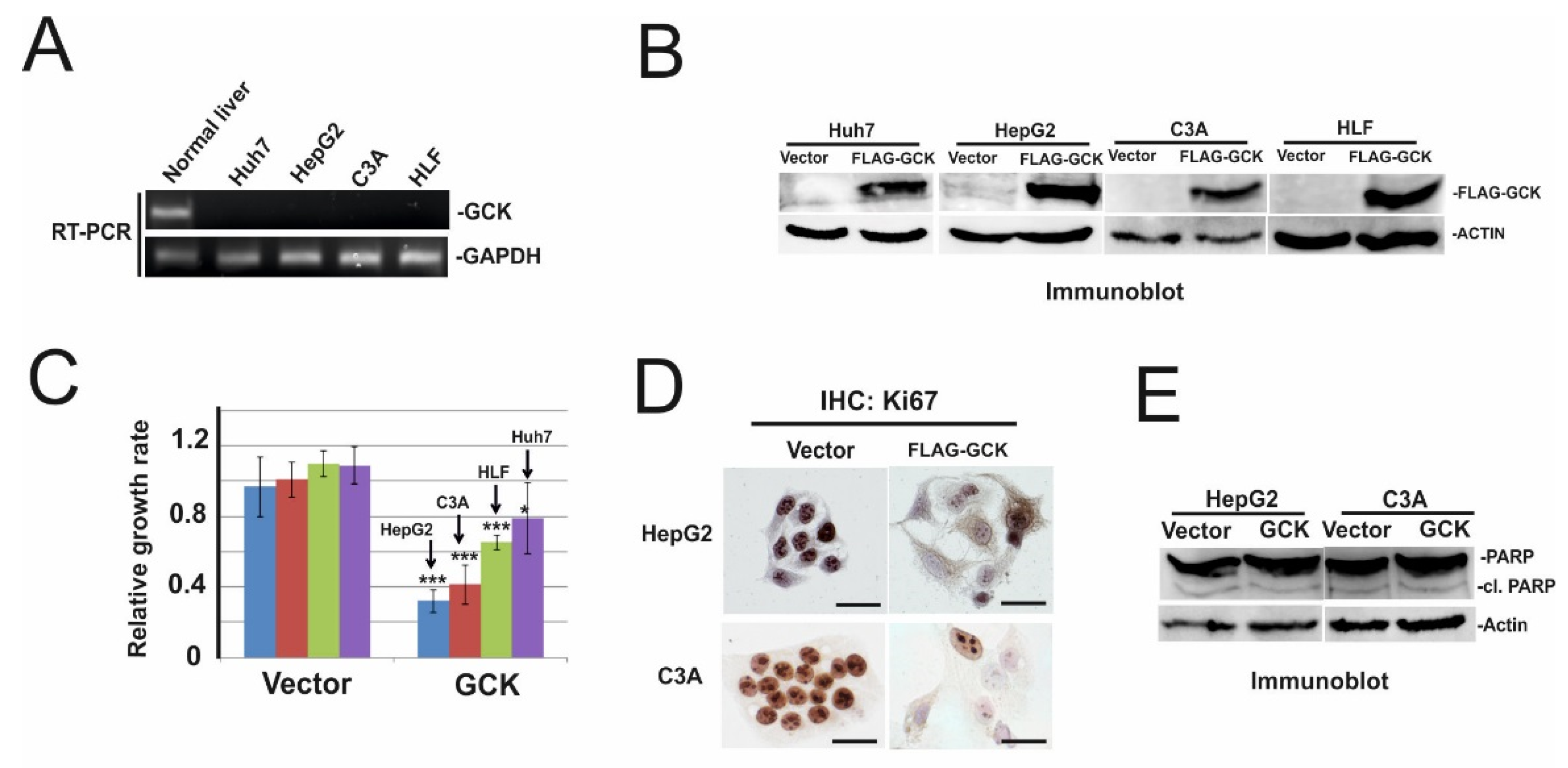
2.3. Ectopic Expression of GCK Inhibits Proliferation of HCC Cells
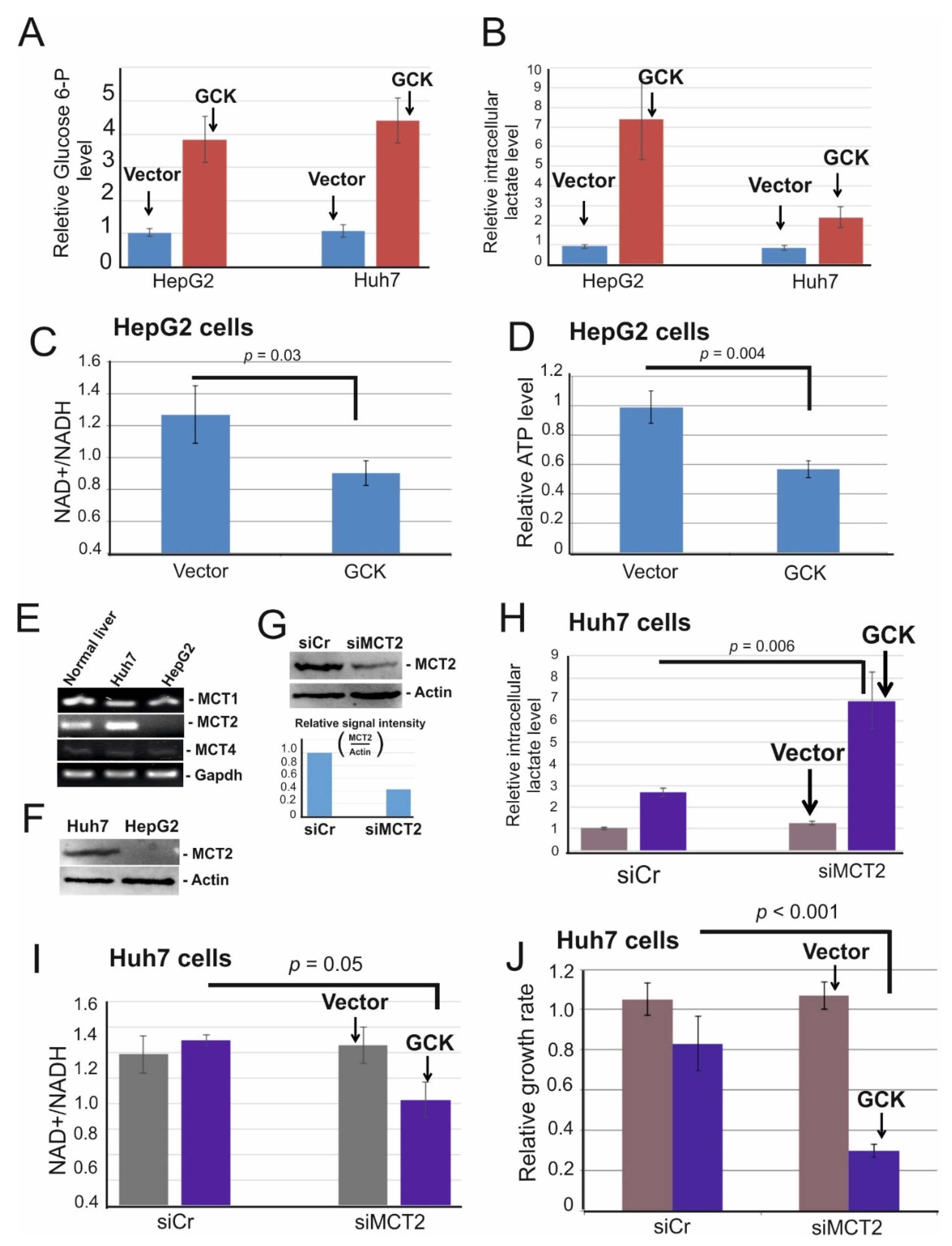
2.4. Ectopic Expression of Glucokinase Caused Intracellular Lactate Accumulation and Induced Energy Crisis Due to NAD+ Depletion
2.5. MCT2 Is Required to Reduce Intracellular Lactate Accumulation in GCK Overexpressing Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture, siRNA, and Transfection
4.2. Immunohistochemistry (IHC)
4.3. Immunoblotting Procedures
4.4. DNA and RNA Extraction
4.5. Nanopore Sequencing
4.6. Methylation Calling for Nanopore Reads
4.7. EPIC Sequencing
4.7.1. Library Generation, Quality Control, and Quantification
4.7.2. Library Denaturation and Sequencing Run
4.7.3. Illumina Sequence Data Analysis
4.8. Semi-Quantitative RT-PCR and qRT-PCR Analysis
4.9. Glucose-6-Phosphate (Glucose 6-P) Measurement
4.10. Lactate Measurement
4.11. NAD+/NADH Measurement
4.12. ATP Measurement
4.13. Crystal Violet Assay
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA A Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [PubMed]
- Allaire, M.; Nault, J.C. Type 2 diabetes-associated hepatocellular carcinoma: A molecular profile. Clin. Liver Dis. 2016, 8, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Krohler, T.; Kessler, S.M.; Hosseini, K.; List, M.; Barghash, A.; Patial, S.; Laggai, S.; Gemperlein, K.; Haybaeck, J.; Muller, R.; et al. The mRNA-binding Protein TTP/ZFP36 in Hepatocarcinogenesis and Hepatocellular Carcinoma. Cancers 2019, 11, 1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Reizel, T.; Wang, Y.J.; Lapiro, J.L.; Kren, B.T.; Schug, J.; Rao, S.; Morgan, A.; Herman, A.; Shekels, L.L.; et al. A negative reciprocal regulatory axis between cyclin D1 and HNF4alpha modulates cell cycle progression and metabolism in the liver. Proc. Natl. Acad. Sci. USA 2020, 117, 17177–17186. [Google Scholar] [CrossRef] [PubMed]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics of hepatocellular carcinoma. Clin. Transl. Med. 2019, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.D.H.; Koch, A.; Allister, A.; Saran, S.; Ewald, F.; Koch, M.; Nashan, B.; Tamura, T. Treatment with MAPKAP2 (MK2) inhibitor and DNA methylation inhibitor, 5-aza dC, synergistically triggers apoptosis in hepatocellular carcinoma (HCC) via tristetraprolin (TTP). Cell. Signal. 2016, 28, 1872–1880. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression--belts, braces, and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Skvortsova, K.; Zotenko, E.; Luu, P.L.; Gould, C.M.; Nair, S.S.; Clark, S.J.; Stirzaker, C. Comprehensive evaluation of genome-wide 5-hydroxymethylcytosine profiling approaches in human DNA. Epigenetics Chromatin 2017, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M.; et al. High-fidelity CRISPR/Cas9-based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 3509. [Google Scholar] [CrossRef]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE 2010, 5, e8888. [Google Scholar] [CrossRef] [Green Version]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef]
- Schatz, M.C. Nanopore sequencing meets epigenetics. Nat. Methods 2017, 14, 347–348. [Google Scholar] [CrossRef]
- Schreiber, J.; Wescoe, Z.L.; Abu-Shumays, R.; Vivian, J.T.; Baatar, B.; Karplus, K.; Akeson, M. Error rates for nanopore discrimination among cytosine, methylcytosine, and hydroxymethylcytosine along individual DNA strands. Proc. Natl. Acad. Sci. USA 2013, 110, 18910–18915. [Google Scholar] [CrossRef] [Green Version]
- Dreos, R.; Ambrosini, G.; Groux, R.; Cavin Perier, R.; Bucher, P. The eukaryotic promoter database in its 30th year: Focus on non-vertebrate organisms. Nucleic Acids Res. 2017, 45, D51–D55. [Google Scholar] [CrossRef] [Green Version]
- Dreos, R.; Ambrosini, G.; Cavin Perier, R.; Bucher, P. EPD and EPDnew, high-quality promoter resources in the next-generation sequencing era. Nucleic Acids Res. 2013, 41, D157–164. [Google Scholar] [CrossRef]
- Nejak-Bowen, K.N.; Thompson, M.D.; Singh, S.; Bowen, W.C., Jr.; Dar, M.J.; Khillan, J.; Dai, C.; Monga, S.P. Accelerated liver regeneration and hepatocarcinogenesis in mice overexpressing serine-45 mutant beta-catenin. Hepatology 2010, 51, 1603–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marhenke, S.; Buitrago-Molina, L.E.; Endig, J.; Orlik, J.; Schweitzer, N.; Klett, S.; Longerich, T.; Geffers, R.; Sanchez Munoz, A.; Dorrell, C.; et al. p21 promotes sustained liver regeneration and hepatocarcinogenesis in chronic cholestatic liver injury. Gut 2014, 63, 1501–1512. [Google Scholar] [CrossRef]
- Ang, C.H.; Hsu, S.H.; Guo, F.; Tan, C.T.; Yu, V.C.; Visvader, J.E.; Chow, P.K.H.; Fu, N.Y. Lgr5(+) pericentral hepatocytes are self-maintained in normal liver regeneration and susceptible to hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 19530–19540. [Google Scholar] [CrossRef] [Green Version]
- Rib, L.; Villeneuve, D.; Minocha, S.; Praz, V.; Hernandez, N.; Guex, N.; Herr, W.; Cycli, X.C. Cycles of gene expression and genome response during mammalian tissue regeneration. Epigenetics Chromatin 2018, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Zhang, K.L.; Zhang, J.X.; Zeng, L.; Di, C.H.; Fee, B.E.; Rivas, M.; Bao, Z.S.; Jiang, T.; Bigner, D.; et al. AJAP1 is dysregulated at an early stage of gliomagenesis and suppresses invasion through cytoskeleton reorganization. Cns Neurosci. Ther. 2014, 20, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Qu, W.; Wen, X.; Su, K.; Gou, W. MiR-552 promotes the proliferation, migration and EMT of hepatocellular carcinoma cells by inhibiting AJAP1 expression. J. Cell. Mol. Med. 2019, 23, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Gong, Y.; Zhang, A.; Cai, S.; Zeng, Q. Loss of GATA5 expression due to gene promoter methylation induces growth and colony formation of hepatocellular carcinoma cells. Oncol. Lett. 2016, 11, 861–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, R.; Blobel, G.A. GATA Transcription Factors and Cancer. Genes Cancer 2010, 1, 1178–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Zhu, M.; Zhang, R.; Wang, Q.; Li, W.; Dong, X.; Chen, Y.; Lu, Y.; Liu, K.; Lin, B.; et al. GATA5 inhibits hepatocellular carcinoma cells malignant behaviours by blocking expression of reprogramming genes. J. Cell. Mol. Med. 2019, 23, 2536–2548. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Zhou, T.F.; Qiu, B.A.; Yang, Y.X.; Zhu, Y.J.; An, Y.; Zhao, W.C.; Wu, Y.T.; Ma, P.F.; Li, J.B.; et al. Methylation-Mediated Silencing of GATA5 Gene Suppresses Cholangiocarcinoma Cell Proliferation and Metastasis. Transl. Oncol. 2018, 11, 585–592. [Google Scholar] [CrossRef]
- Revill, K.; Wang, T.; Lachenmayer, A.; Kojima, K.; Harrington, A.; Li, J.; Hoshida, Y.; Llovet, J.M.; Powers, S. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology 2013, 145, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Shirakami, Y.; Gottesman, M.E.; Blaner, W.S. Diethylnitrosamine-induced hepatocarcinogenesis is suppressed in lecithin:retinol acyltransferase-deficient mice primarily through retinoid actions immediately after carcinogen administration. Carcinogenesis 2012, 33, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Mortezaee, K. CXCL12/CXCR4 axis in the microenvironment of solid tumors: A critical mediator of metastasis. Life Sci. 2020, 249, 117534. [Google Scholar] [CrossRef]
- Deneka, A.; Korobeynikov, V.; Golemis, E.A. Embryonal Fyn-associated substrate (EFS) and CASS4: The lesser-known CAS protein family members. Gene 2015, 570, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Sternisha, S.M.; Miller, B.G. Molecular and cellular regulation of human glucokinase. Arch. Biochem. Biophys. 2019, 663, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Abbott, G.W. KCNQs: Ligand- and Voltage-Gated Potassium Channels. Front. Physiol. 2020, 11, 583. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, G.R.; Olson, E.N. NFAT signaling: Choreographing the social lives of cells. Cell 2002, 109, S67–S79. [Google Scholar] [CrossRef] [Green Version]
- Wolfbauer, G.; Albers, J.J.; Oram, J.F. Phospholipid transfer protein enhances removal of cellular cholesterol and phospholipids by high-density lipoprotein apolipoproteins. Biochim. Biophys. Acta 1999, 1439, 65–76. [Google Scholar] [CrossRef]
- Chan, W.K.; Chen, V.P.; Luk, W.K.; Choi, R.C.; Tsim, K.W. N-linked glycosylation of proline-rich membrane anchor (PRiMA) is not required for assembly and trafficking of globular tetrameric acetylcholinesterase. Neurosci. Lett. 2012, 523, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Rodriguez-Hernandez, C.J.; Mateo-Lozano, S.; Perez-Jaume, S.; Goncalves-Alves, E.; Lavarino, C.; Mora, J.; de Torres, C. Parathyroid hormone-like hormone plays a dual role in neuroblastoma depending on PTH1R expression. Mol. Oncol. 2019, 13, 1959–1975. [Google Scholar] [CrossRef] [Green Version]
- Blessing, A.M.; Ganesan, S.; Rajapakshe, K.; Ying Sung, Y.; Reddy Bollu, L.; Shi, Y.; Cheung, E.; Coarfa, C.; Chang, J.T.; McDonnell, D.P.; et al. Identification of a Novel Coregulator, SH3YL1, That Interacts With the Androgen Receptor N-Terminus. Mol. Endocrinol. 2015, 29, 1426–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, O.M.; Davies, O.R. Molecular structure of human synaptonemal complex protein SYCE1. Chromosoma 2019, 128, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Valera, A.; Bosch, F. Glucokinase expression in rat hepatoma cells induces glucose uptake and is rate limiting in glucose utilization. Eur. J. Biochem. 1994, 222, 533–539. [Google Scholar] [CrossRef]
- Stambaugh, R.; Post, D. Substrate and product inhibition of rabbit muscle lactic dehydrogenase heart (H4) and muscle (M4) isozymes. J. Biol. Chem. 1966, 241, 1462–1467. [Google Scholar] [CrossRef]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. The SLC16 gene family-structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef]
- Sun, S.; Liu, J.; Zhao, M.; Han, Y.; Chen, P.; Mo, Q.; Wang, B.; Chen, G.; Fang, Y.; Tian, Y.; et al. Loss of the novel mitochondrial protein FAM210B promotes metastasis via PDK4-dependent metabolic reprogramming. Cell Death Dis. 2017, 8, e2870. [Google Scholar] [CrossRef]
- Yang, L.; Hou, Y.; Yuan, J.; Tang, S.; Zhang, H.; Zhu, Q.; Du, Y.E.; Zhou, M.; Wen, S.; Xu, L.; et al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and p53 signaling pathways. Oncotarget 2015, 6, 25755–25769. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Li, J.; Xin, J.; Wu, J.; Guo, J.; Zhang, L.; Jiang, L.; Zhang, W.; Yang, Z.; Li, L. Methylation profile of single hepatocytes derived from hepatitis B virus-related hepatocellular carcinoma. PLoS ONE 2011, 6, e19862. [Google Scholar] [CrossRef]
- Shen, J.; Wang, S.; Zhang, Y.J.; Kappil, M.; Wu, H.C.; Kibriya, M.G.; Wang, Q.; Jasmine, F.; Ahsan, H.; Lee, P.H.; et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology 2012, 55, 1799–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tampe, B.; Tampe, D.; Muller, C.A.; Sugimoto, H.; LeBleu, V.; Xu, X.; Muller, G.A.; Zeisberg, E.M.; Kalluri, R.; Zeisberg, M. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J. Am. Soc. Nephrol. Jan. 2014, 25, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Mouse Genome Sequencing, C.; Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Ezaka, K.; Kanda, M.; Sugimoto, H.; Shimizu, D.; Oya, H.; Nomoto, S.; Sueoka, S.; Tanaka, Y.; Takami, H.; Hashimoto, R.; et al. Reduced Expression of Adherens Junctions Associated Protein 1 Predicts Recurrence of Hepatocellular Carcinoma After Curative Hepatectomy. Ann. Surg. Oncol. 2015, 22, S1499–S1507. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.P.; Choi, R.C.; Chan, W.K.; Leung, K.W.; Guo, A.J.; Chan, G.K.; Luk, W.K.; Tsim, K.W. The assembly of proline-rich membrane anchor (PRiMA)-linked acetylcholinesterase enzyme: Glycosylation is required for enzymatic activity but not for oligomerization. J. Biol. Chem. 2011, 286, 32948–32961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zuo, J.; Ji, G.; Saiyin, H.; Liu, X.; Yin, F.; Cao, N.; Wen, Y.; Li, J.J.; Yu, L. Proapoptotic function of integrin beta(3) in human hepatocellular carcinoma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Tian, F.; Ying, W.; Qian, X. Quantitative proteomics reveal the anti-tumour mechanism of the carbohydrate recognition domain of Galectin-3 in Hepatocellular carcinoma. Sci. Rep. 2017, 7, 5189. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, X.; Wang, T.; Hu, X.; Hui, X.; Yan, M.; Gao, Q.; Chen, T.; Li, J.; Yao, M.; et al. Acetylcholinesterase, a key prognostic predictor for hepatocellular carcinoma, suppresses cell growth and induces chemosensitization. Hepatology 2011, 53, 493–503. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Mathupala, S.; Rempel, A.; Geschwind, J.F.; Ko, Y.H. Mitochondrial bound type II hexokinase: A key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim. Biophys. Acta 2002, 1555, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, E.; Pedersen, P.L. High aerobic glycolysis of rat hepatoma cells in culture: Role of mitochondrial hexokinase. Proc. Natl. Acad. Sci. USA 1977, 74, 3735–3739. [Google Scholar] [CrossRef] [Green Version]
- Alves, V.A.; Pinheiro, C.; Morais-Santos, F.; Felipe-Silva, A.; Longatto-Filho, A.; Baltazar, F. Characterization of monocarboxylate transporter activity in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 11780–11787. [Google Scholar] [CrossRef] [Green Version]
- Board, M.; Colquhoun, A.; Newsholme, E.A. High Km glucose-phosphorylating (glucokinase) activities in a range of tumor cell lines and inhibition of rates of tumor growth by the specific enzyme inhibitor mannoheptulose. Cancer Res. 1995, 55, 3278–3285. [Google Scholar]
- Al-Ziaydi, A.G.; Al-Shammari, A.M.; Hamzah, M.I.; Kadhim, H.S.; Jabir, M.S. Hexokinase inhibition using D-Mannoheptulose enhances oncolytic newcastle disease virus-mediated killing of breast cancer cells. Cancer Cell Int. 2020, 20, 420. [Google Scholar] [CrossRef]
- Zhang, Y.; Courtois, P.; Sener, A.; Malaisse, W.J. Dissimilar effects of D-mannoheptulose on the phosphorylation of alpha- versus beta-D-glucose by either hexokinase or glucokinase. Int. J. Mol. Med. 2004, 14, 107–112. [Google Scholar] [PubMed]
- Saran, S.; Tran, D.D.; Ewald, F.; Koch, A.; Hoffmann, A.; Koch, M.; Nashan, B.; Tamura, T. Depletion of three combined THOC5 mRNA export protein target genes synergistically induces human hepatocellular carcinoma cell death. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Description | Potential Function |
|---|---|---|
| CXCL12 | C-X-C motif chemokine ligand 12 | functions as the ligand for the chemokine (C-X-C motif) receptor 4, and plays a role in many diverse cellular functions including cell migration and invasion [36] |
| EFS | embryonal Fyn-associated substrate | associates with FAK and SRC family kinases to activate downstream effectors regulating the actin cytoskeleton [37] |
| GCK | glucokinase | phosphorylates glucose and plays a key regulatory role in glucose uptake [38] |
| KCNQ5 | potassium voltage-gated channel subfamily Q member 5 | voltage-gated potassium (Kv) channel that regulates changes in membrane potential [39] |
| NFATC2 | nuclear factor of activated T cells 2 | functions as transcription factor and involves in NFAT signaling [40] |
| PLTP | phospholipid transfer protein | involved in transport of lipids and vitamin E among lipoproteins, and between lipoproteins and cells [41] |
| PRIMA1 | proline-rich membrane anchor 1 | required for the assembly of Acetylcholinesterase, which plays a crucial role in terminating the synaptic transmission in the central nervous system and at the neuromuscular junctions (NMJs) in the peripheral nervous system [42] |
| PTH1R | parathyroid hormone 1 receptor | a receptor for parathyroid hormone (PTH) and for parathyroid hormone-like hormone (PTHLH) and may act as proproliferation factor in neuroblastoma [43] |
| SH3YL1 | SH3 and SYLF domain containing 1 | coregulator of the Androgen Receptor and participates in Androgen-mediated growth of prostate cancer cells [44] |
| SYCE1 | synaptonemal complex central element protein 1 | a member of the synaptonemal complex, which links homologous chromosomes during prophase I of meiosis [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davenport, C.F.; Scheithauer, T.; Dunst, A.; Bahr, F.S.; Dorda, M.; Wiehlmann, L.; Tran, D.D.H. Genome-Wide Methylation Mapping Using Nanopore Sequencing Technology Identifies Novel Tumor Suppressor Genes in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3937. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083937
Davenport CF, Scheithauer T, Dunst A, Bahr FS, Dorda M, Wiehlmann L, Tran DDH. Genome-Wide Methylation Mapping Using Nanopore Sequencing Technology Identifies Novel Tumor Suppressor Genes in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(8):3937. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083937
Chicago/Turabian StyleDavenport, Colin F., Tobias Scheithauer, Alessia Dunst, Frauke Sophie Bahr, Marie Dorda, Lutz Wiehlmann, and Doan Duy Hai Tran. 2021. "Genome-Wide Methylation Mapping Using Nanopore Sequencing Technology Identifies Novel Tumor Suppressor Genes in Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 8: 3937. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083937