
Glucose Metabolism in Osteoblasts in Healthy and Pathophysiological Conditions

 ,
,
Abstract
:1. Introduction
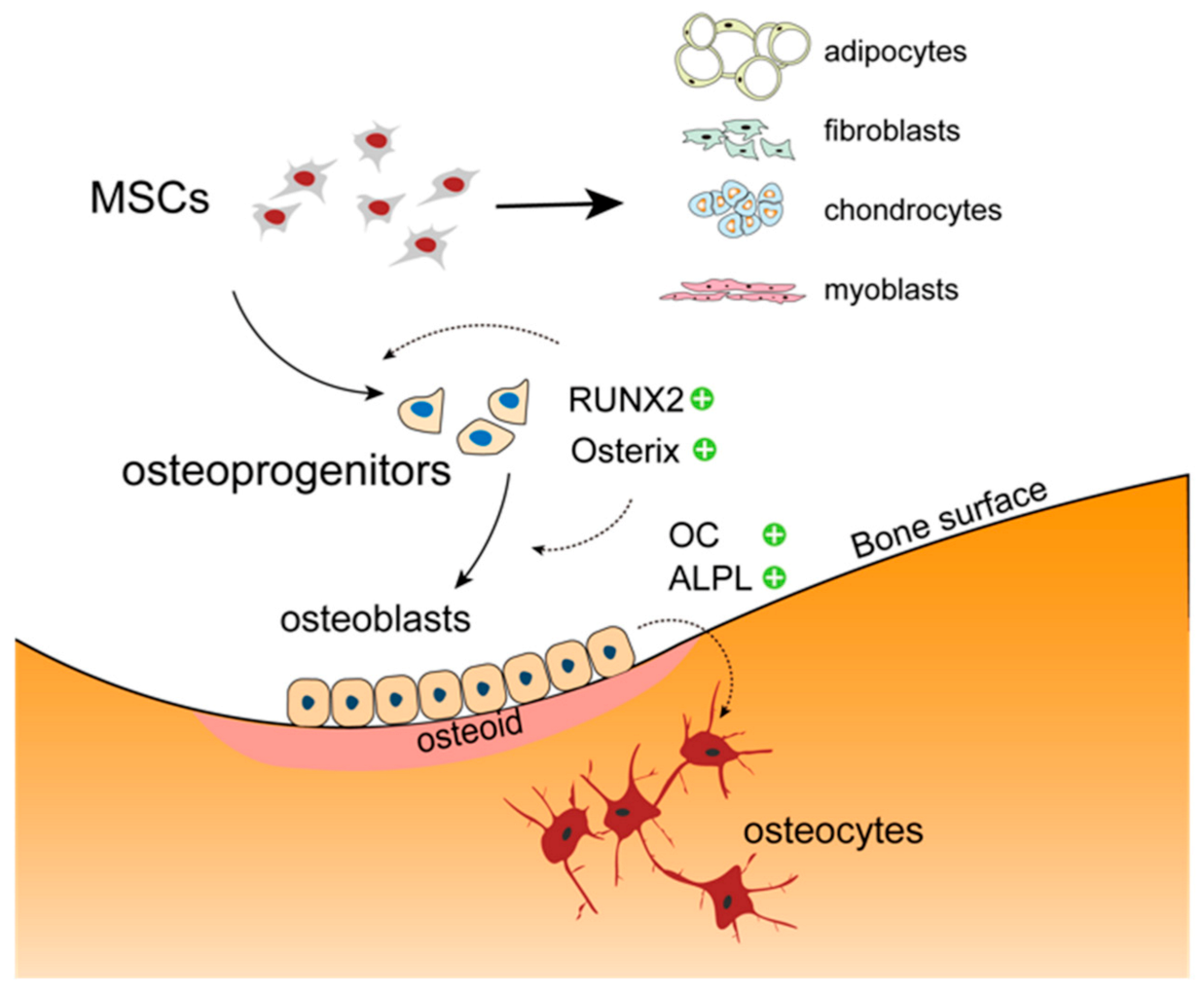
2. The Osteoblast in Bone Physiology
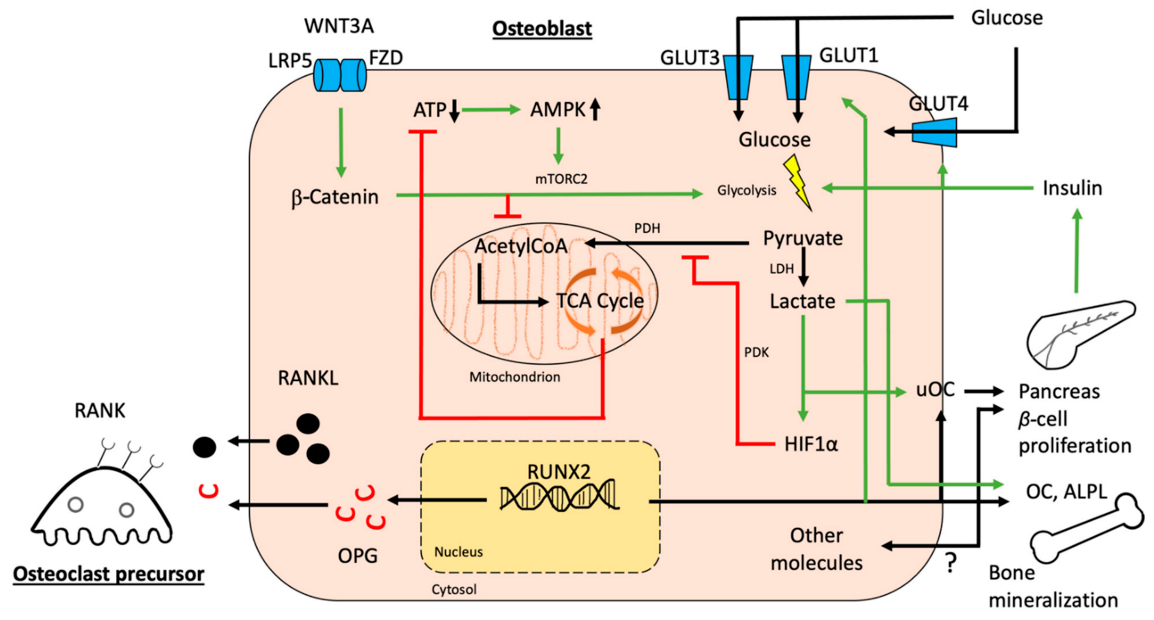
3. Interactions between Glucose Homeostasis and Osteoblasts
3.1. Osteoblastic Glucose Receptors and Glucose Utilization
3.2. Osteoblastic Effects on Overall Glucose Homeostasis
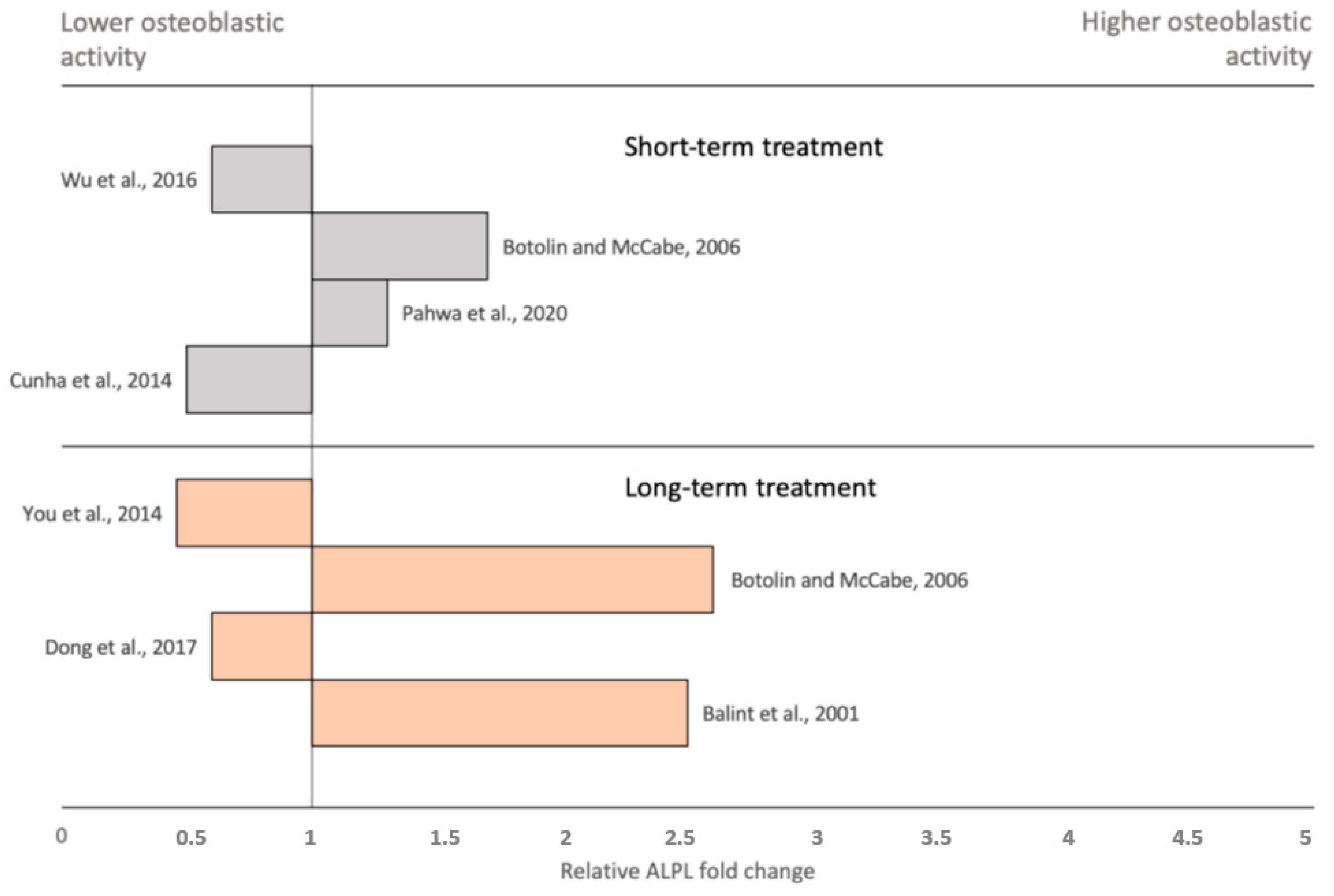
4. Disruption of Osteoblast Function in Anorexia Nervosa and Diabetes
5. Implications for Anti-Diabetic Drugs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lee, N.K.; Sowa, H.; Hinoi, E.; Ferron, M.; Ahn, J.D.; Confavreux, C.; Dacquin, R.; Mee, P.J.; McKee, M.D.; Jung, D.Y.; et al. Endocrine regulation of energy metabolism by the skeleton. Cell 2007, 130, 456–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferron, M.; Wei, J.; Yoshizawa, T.; Del Fattore, A.; DePinho, R.A.; Teti, A.; Ducy, P.; Karsenty, G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 2010, 142, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levinger, I.; Seeman, E.; Jerums, G.; McConell, G.K.; Rybchyn, M.S.; Cassar, S.; Byrnes, E.; Selig, S.; Mason, R.S.; Ebeling, P.R.; et al. Glucose-loading reduces bone remodeling in women and osteoblast function in vitro. Physiol. Rep. 2016, 4, e12700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, J.S.; Ferreira, V.M.; Maquigussa, E.; Naves, M.A.; Boim, M.A. Effects of high glucose and high insulin concentrations on osteoblast function in vitro. Cell Tissue Res. 2014, 358, 249–256. [Google Scholar] [CrossRef]
- Legroux-Gérot, I.; Vignau, J.; D’Herbomez, M.; Collier, F.; Marchandise, X.; Duquesnoy, B.; Cortet, B. Evaluation of bone loss and its mechanisms in anorexia nervosa. Calcif. Tissue Int. 2007, 81, 174–182. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 2019, 568, 541–545. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Mundlos, S.; Otto, F.; Mundlos, C.; Mulliken, J.B.; Aylsworth, A.S.; Albright, S.; Lindhout, D.; Cole, W.G.; Henn, W.; Knoll, J.H.; et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997, 89, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.; Beddington, R.S.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Isefuku, S.; Joyner, C.J.; Reed, A.A.; Simpson, A.H. Distraction osteogenesis in the Cbfa-1+/- mouse. J. Orthop. Res. 2004, 22, 1276–1282. [Google Scholar] [CrossRef]
- Liu, W.; Toyosawa, S.; Furuichi, T.; Kanatani, N.; Yoshida, C.; Liu, Y.; Himeno, M.; Narai, S.; Yamaguchi, A.; Komori, T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J. Cell Biol. 2001, 155, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Tarkkonen, K.; Hieta, R.; Kytölä, V.; Nykter, M.; Kiviranta, R. Comparative analysis of osteoblast gene expression profiles and Runx2 genomic occupancy of mouse and human osteoblasts in vitro. Gene 2017, 626, 119–131. [Google Scholar] [CrossRef]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Renn, J.; Winkler, C. Osterix-mCherry transgenic medaka for in vivo imaging of bone formation. Dev. Dyn. 2009, 238, 241–248. [Google Scholar] [CrossRef]
- Kaback, L.A.; Soung, D.Y.; Naik, A.; Smith, N.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Osterix/Sp7 regulates mesenchymal stem cell mediated endochondral ossification. J. Cell Physiol. 2008, 214, 173–182. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, Z.; Feng, J.Q.; Dusevich, V.M.; Sinha, K.; Zhang, H.; Darnay, B.G.; de Crombrugghe, B. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc. Natl. Acad. Sci. USA 2010, 107, 12919–12924. [Google Scholar] [CrossRef] [Green Version]
- Baek, W.Y.; Lee, M.A.; Jung, J.W.; Kim, S.Y.; Akiyama, H.; de Crombrugghe, B.; Kim, J.E. Positive regulation of adult bone formation by osteoblast-specific transcription factor osterix. J. Bone Miner. Res. 2009, 24, 1055–1065. [Google Scholar] [CrossRef]
- Weng, J.J.; Su, Y. Nuclear matrix-targeting of the osteogenic factor Runx2 is essential for its recognition and activation of the alkaline phosphatase gene. Biochim. Biophys. Acta 2013, 1830, 2839–2852. [Google Scholar] [CrossRef]
- Quarles, L.D.; Yohay, D.A.; Lever, L.W.; Caton, R.; Wenstrup, R.J. Distinct proliferative and differentiated stages of murine MC3T3-E1 cells in culture: An in vitro model of osteoblast development. J. Bone Miner. Res. 1992, 7, 683–692. [Google Scholar] [CrossRef]
- Wennberg, C.; Hessle, L.; Lundberg, P.; Mauro, S.; Narisawa, S.; Lerner, U.H.; Millán, J.L. Functional characterization of osteoblasts and osteoclasts from alkaline phosphatase knockout mice. J. Bone Miner. Res. 2000, 15, 1879–1888. [Google Scholar] [CrossRef]
- Bouillon, R.; Bex, M.; Van Herck, E.; Laureys, J.; Dooms, L.; Lesaffre, E.; Ravussin, E. Influence of age, sex, and insulin on osteoblast function: Osteoblast dysfunction in diabetes mellitus. J. Clin. Endocrinol. Metab. 1995, 80, 1194–1202. [Google Scholar] [CrossRef]
- Lumachi, F.; Camozzi, V.; Tombolan, V.; Luisetto, G. Bone mineral density, osteocalcin, and bone-specific alkaline phosphatase in patients with insulin-dependent diabetes mellitus. Ann. N. Y. Acad. Sci. 2009, 1173 (Suppl. 1), E64–E67. [Google Scholar] [CrossRef]
- Weiss, M.J.; Cole, D.E.; Ray, K.; Whyte, M.P.; Lafferty, M.A.; Mulivor, R.A.; Harris, H. A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proc. Natl. Acad. Sci. USA 1988, 85, 7666–7669. [Google Scholar] [CrossRef] [Green Version]
- Whyte, M.P.; Zhang, F.; Wenkert, D.; McAlister, W.H.; Mack, K.E.; Benigno, M.C.; Coburn, S.P.; Wagy, S.; Griffin, D.M.; Ericson, K.L.; et al. Hypophosphatasia: Validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone 2015, 75, 229–239. [Google Scholar] [CrossRef]
- Bouillon, R.; Marcocci, C.; Carmeliet, G.; Bikle, D.; White, J.H.; Dawson-Hughes, B.; Lips, P.; Munns, C.F.; Lazaretti-Castro, M.; Giustina, A.; et al. Skeletal and Extraskeletal Actions of Vitamin D: Current Evidence and Outstanding Questions. Endocr. Rev. 2019, 40, 1109–1151. [Google Scholar] [CrossRef] [Green Version]
- Henthorn, P.S.; Raducha, M.; Fedde, K.N.; Lafferty, M.A.; Whyte, M.P. Different missense mutations at the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited forms of mild and severe hypophosphatasia. Proc. Natl. Acad. Sci. USA 1992, 89, 9924–9928. [Google Scholar] [CrossRef] [Green Version]
- Jandl, N.M.; Schmidt, T.; Rolvien, T.; Stürznickel, J.; Chrysostomou, K.; von Vopelius, E.; Volk, A.E.; Schinke, T.; Kubisch, C.; Amling, M.; et al. Genotype-Phenotype Associations in 72 Adults with Suspected ALPL-Associated Hypophosphatasia. Calcif. Tissue Int. 2021, 108, 288–301. [Google Scholar] [CrossRef]
- López-Delgado, L.; Riancho-Zarrabeitia, L.; García-Unzueta, M.T.; Tenorio, J.A.; García-Hoyos, M.; Lapunzina, P.; Valero, C.; Riancho, J.A. Abnormal bone turnover in individuals with low serum alkaline phosphatase. Osteoporos. Int. 2018, 29, 2147–2150. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.; Catala-Lehnen, P.; Huebner, A.K.; Jeschke, A.; Heckt, T.; Lueth, A.; Krause, M.; Koehne, T.; Albers, J.; Schulze, J.; et al. Calcitonin controls bone formation by inhibiting the release of sphingosine 1-phosphate from osteoclasts. Nat. Commun. 2014, 5, 5215. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, K.; Halladay, D.L.; Miles, R.R.; Yang, X.; Galvin, R.J.; Chandrasekhar, S.; Martin, T.J.; Onyia, J.E. The osteoblast-specific transcription factor Cbfa1 contributes to the expression of osteoprotegerin, a potent inhibitor of osteoclast differentiation and function. J. Biol. Chem. 2000, 275, 25163–25172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiotani, A.; Takami, M.; Itoh, K.; Shibasaki, Y.; Sasaki, T. Regulation of osteoclast differentiation and function by receptor activator of NFkB ligand and osteoprotegerin. Anat. Rec. 2002, 268, 137–146. [Google Scholar] [CrossRef]
- Shimizu-Ishiura, M.; Kawana, F.; Sasaki, T. Osteoprotegerin administration reduces femural bone loss in ovariectomized mice via impairment of osteoclast structure and function. J. Electron. Microsc. Tokyo 2002, 51, 315–325. [Google Scholar] [CrossRef]
- Gori, F.; Hofbauer, L.C.; Dunstan, C.R.; Spelsberg, T.C.; Khosla, S.; Riggs, B.L. The expression of osteoprotegerin and RANK ligand and the support of osteoclast formation by stromal-osteoblast lineage cells is developmentally regulated. Endocrinology 2000, 141, 4768–4776. [Google Scholar] [CrossRef]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Uda, Y.; Azab, E.; Sun, N.; Shi, C.; Pajevic, P.D. Osteocyte Mechanobiology. Curr. Osteoporos. Rep. 2017, 15, 318–325. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, F.; Zhang, J.; Shao, D.; Wang, Y.; Li, S.; Lv, S.; Chi, G.; Zhang, M.; Chen, L.; et al. Bioactive carbon dots direct the osteogenic differentiation of human bone marrow mesenchymal stem cells. Colloids Surf. B Biointerfaces 2019, 179, 1–8. [Google Scholar] [CrossRef]
- Watanuki, S.; Kobayashi, H.; Sorimachi, Y.; Yamamoto, M.; Okamoto, S.; Takubo, K. ATP turnover and glucose dependency in hematopoietic stem/progenitor cells are increased by proliferation and differentiation. Biochem. Biophys. Res. Commun. 2019, 514, 287–294. [Google Scholar] [CrossRef]
- Wei, J.; Shimazu, J.; Makinistoglu, M.P.; Maurizi, A.; Kajimura, D.; Zong, H.; Takarada, T.; Lezaki, T.; Pessin, J.E.; Hinoi, E.; et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 2015, 161, 1576–1591. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Frey, J.L.; Wong, G.W.; Faugere, M.C.; Wolfgang, M.J.; Kim, J.K.; Riddle, R.C.; Clemens, T.L. Glucose Transporter-4 Facilitates Insulin-Stimulated Glucose Uptake in Osteoblasts. Endocrinology 2016, 157, 4094–4103. [Google Scholar] [CrossRef]
- Heilig, C.W.; Saunders, T.; Brosius, F.C., 3rd; Moley, K.; Heilig, K.; Baggs, R.; Guo, L.; Conner, D. Glucose transporter-1-deficient mice exhibit impaired development and deformities that are similar to diabetic embryopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 15613–15618. [Google Scholar] [CrossRef] [Green Version]
- Katz, E.B.; Stenbit, A.E.; Hatton, K.; DePinho, R.; Charron, M.J. Cardiac and adipose tissue abnormalities but not diabetes in mice deficient in GLUT4. Nature 1995, 377, 151–155. [Google Scholar] [CrossRef]
- Guntur, A.R.; Gerencser, A.A.; Le, P.T.; DeMambro, V.E.; Bornstein, S.A.; Mookerjee, S.A.; Maridas, D.E.; Clemmons, D.E.; Brand, M.D.; Rosen, C.J. Osteoblast-like MC3T3-E1 Cells Prefer Glycolysis for ATP Production but Adipocyte-like 3T3-L1 Cells Prefer Oxidative Phosphorylation. J. Bone Miner. Res. 2018, 33, 1052–1065. [Google Scholar] [CrossRef]
- Lee, W.C.; Ji, X.; Nissim, I.; Long, F. Malic Enzyme Couples Mitochondria with Aerobic Glycolysis in Osteoblasts. Cell Rep. 2020, 32, 108108. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Lyons, D.G.; Parpaleix, A.; Roche, M.; Charpak, S. Mapping oxygen concentration in the awake mouse brain. Elife 2016, 5. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, M.; Feng, H.; Peng, Y.; Sun, J.; Qu, X.; Li, C. Lactate induces osteoblast differentiation by stabilization of HIF1α. Mol. Cell Endocrinol. 2017, 452, 84–92. [Google Scholar] [CrossRef]
- Dirckx, N.; Tower, R.J.; Mercken, E.M.; Vangoitsenhoven, R.; Moreau-Triby, C.; Breugelmans, T.; Nefyodova, E.; Cardoen, R.; Mathieu, C.; Van der Schueren, B.; et al. Vhl deletion in osteoblasts boosts cellular glycolysis and improves global glucose metabolism. J. Clin. Investig. 2018, 128, 1087–1105. [Google Scholar] [CrossRef]
- Regan, J.N.; Lim, J.; Shi, Y.; Joeng, K.S.; Arbeit, J.M.; Shohet, R.V.; Long, F. Up-regulation of glycolytic metabolism is required for HIF1α-driven bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 8673–8678. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, N.; Kim, J.; Liu, Y.; Logan, T.M.; Ma, T. Gas chromatography-mass spectrometry analysis of human mesenchymal stem cell metabolism during proliferation and osteogenic differentiation under different oxygen tensions. J. Biotechnol. 2014, 169, 95–102. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, Q.; Chen, Q.; Zhou, Q.; Zhang, Y.; Shi, Z.; Nong, H.; Liu, M.; Zeng, G.; Zong, S. Conditional knockout of the PDK-1 gene in osteoblasts affects osteoblast differentiation and bone formation. J. Cell Physiol. 2021, 236, 5432–5445. [Google Scholar] [CrossRef]
- Li, S.; Xu, W.; Xing, Z.; Qian, J.; Chen, L.; Gu, R.; Guo, W.; Lai, X.; Zhao, W.; Li, S.; et al. A Conditional Knockout Mouse Model Reveals a Critical Role of PKD1 in Osteoblast Differentiation and Bone Development. Sci. Rep. 2017, 7, 40505. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Kim, M.J.; Lee, S.J.; Kim, B.G.; Choi, J.Y.; Lee, S.M.; Ham, H.J.; Koh, J.M.; Jeon, J.H.; Lee, I.K. PDK2 Deficiency Prevents Ovariectomy-Induced Bone Loss in Mice by Regulating the RANKL-NFATc1 Pathway During Osteoclastogenesis. J. Bone Miner. Res. 2021, 36, 553–566. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, W.; Masuyama, R.; Fukuyama, R.; Ito, M.; Zhang, Q.; Komori, H.; Murakami, T.; Moriishi, T.; Miyazaki, T.; et al. Pyruvate dehydrogenase kinase 4 induces bone loss at unloading by promoting osteoclastogenesis. Bone 2012, 50, 409–419. [Google Scholar] [CrossRef]
- Lardy, H.A.; Ferguson, S.M. Oxidative phosphorylation in mitochondria. Annu. Rev. Biochem. 1969, 38, 991–1034. [Google Scholar] [CrossRef]
- Jones, S.J.; Gray, C.; Boyde, A.; Burnstock, G. Purinergic transmitters inhibit bone formation by cultured osteoblasts. Bone 1997, 21, 393–399. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.B.; Chellaiah, M.; Zou, J.; Reynolds, M.A.; Costello, L.C. Evidence that Osteoblasts are Specialized Citrate-producing Cells that Provide the Citrate for Incorporation into the Structure of Bone. Open Bone J. 2014, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickens, F. The citric acid content of animal tissues, with reference to its occurrence in bone and tumour. Biochem. J. 1941, 35, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, T.; Bandow, K.; Suzuki, H.; Chiba, N.; Kakimoto, K.; Ohnishi, T.; Kawamoto, S.; Nagaoka, E.; Matsuguchi, T. Osteoblast differentiation is functionally associated with decreased AMP kinase activity. J. Cell Physiol. 2009, 221, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Esen, E.; Chen, J.; Karner, C.M.; Okunade, A.L.; Patterson, B.W.; Long, F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 2013, 17, 745–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karner, C.M.; Esen, E.; Chen, J.; Hsu, F.F.; Turk, J.; Long, F. Wnt Protein Signaling Reduces Nuclear Acetyl-CoA Levels to Suppress Gene Expression during Osteoblast Differentiation. J. Biol. Chem. 2016, 291, 13028–13039. [Google Scholar] [CrossRef] [Green Version]
- Boland, G.M.; Perkins, G.; Hall, D.J.; Tuan, R.S. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J. Cell Biochem. 2004, 93, 1210–1230. [Google Scholar] [CrossRef]
- Xi, G.; Rosen, C.J.; Clemmons, D.R. IGF-I and IGFBP-2 Stimulate AMPK Activation and Autophagy, Which Are Required for Osteoblast Differentiation. Endocrinology 2016, 157, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S. TGF-β regulates β-catenin signaling and osteoblast differentiation in human mesenchymal stem cells. J. Cell Biochem. 2011, 112, 1651–1660. [Google Scholar] [CrossRef] [Green Version]
- Shares, B.H.; Busch, M.; White, N.; Shum, L.; Eliseev, R.A. Active mitochondria support osteogenic differentiation by stimulating β-catenin acetylation. J. Biol. Chem. 2018, 293, 16019–16027. [Google Scholar] [CrossRef] [Green Version]
- Bellido, T.; Ali, A.A.; Plotkin, L.I.; Fu, Q.; Gubrij, I.; Roberson, P.K.; Weinstein, R.S.; O’Brien, C.A.; Manolagas, S.C.; Jilka, R.L. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J. Biol. Chem. 2003, 278, 50259–50272. [Google Scholar] [CrossRef] [Green Version]
- Esen, E.; Lee, S.Y.; Wice, B.M.; Long, F. PTH Promotes Bone Anabolism by Stimulating Aerobic Glycolysis via IGF Signaling. J. Bone Miner. Res. 2015, 30, 1959–1968. [Google Scholar] [CrossRef] [Green Version]
- Wan, M.; Yang, C.; Li, J.; Wu, X.; Yuan, H.; Ma, H.; He, X.; Nie, S.; Chang, C.; Cao, X. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 2008, 22, 2968–2979. [Google Scholar] [CrossRef] [Green Version]
- Zoidis, E.; Ghirlanda-Keller, C.; Schmid, C. Stimulation of glucose transport in osteoblastic cells by parathyroid hormone and insulin-like growth factor I. Mol. Cell Biochem. 2011, 348, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavassieux, P.M.; Chenu, C.; Valentin-Opran, A.; Merle, B.; Delmas, P.D.; Hartmann, D.J.; Saez, S.; Meunier, P.J. Influence of experimental conditions on osteoblast activity in human primary bone cell cultures. J. Bone Miner. Res. 1990, 5, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Tsao, Y.T.; Huang, Y.J.; Wu, H.H.; Liu, Y.A.; Liu, Y.S.; Lee, O.K. Osteocalcin Mediates Biomineralization during Osteogenic Maturation in Human Mesenchymal Stromal Cells. Int. J. Mol. Sci. 2017, 18, 159. [Google Scholar] [CrossRef]
- Bao, Y.Q.; Zhou, M.; Zhou, J.; Lu, W.; Gao, Y.C.; Pan, X.P.; Tang, J.L.; Lu, H.J.; Jia, W.P. Relationship between serum osteocalcin and glycaemic variability in Type 2 diabetes. Clin. Exp. Pharmacol. Physiol. 2011, 38, 50–54. [Google Scholar] [CrossRef]
- Colleluori, G.; Napoli, N.; Phadnis, U.; Armamento-Villareal, R.; Villareal, D.T. Effect of Weight Loss, Exercise, or Both on Undercarboxylated Osteocalcin and Insulin Secretion in Frail, Obese Older Adults. Oxid. Med. Cell Longev. 2017, 2017, 4807046. [Google Scholar] [CrossRef] [Green Version]
- Takashi, Y.; Ishizu, M.; Mori, H.; Miyashita, K.; Sakamoto, F.; Katakami, N.; Matsuoka, T.A.; Yasuda, T.; Hashida, S.; Matsuhisa, M.; et al. Circulating osteocalcin as a bone-derived hormone is inversely correlated with body fat in patients with type 1 diabetes. PLoS ONE 2019, 14, e0216416. [Google Scholar] [CrossRef]
- Hwang, Y.C.; Jeong, I.K.; Ahn, K.J.; Chung, H.Y. Circulating osteocalcin level is associated with improved glucose tolerance, insulin secretion and sensitivity independent of the plasma adiponectin level. Osteoporos. Int. 2012, 23, 1337–1342. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, J.; Karsenty, G.; Ferron, M. In vivo analysis of the contribution of bone resorption to the control of glucose metabolism in mice. Mol. Metab. 2013, 2, 498–504. [Google Scholar] [CrossRef]
- Ferron, M.; Hinoi, E.; Karsenty, G.; Ducy, P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc. Natl. Acad. Sci. USA 2008, 105, 5266–5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Hanna, T.; Suda, N.; Karsenty, G.; Ducy, P. Osteocalcin promotes β-cell proliferation during development and adulthood through Gprc6a. Diabetes 2014, 63, 1021–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oury, F.; Sumara, G.; Sumara, O.; Ferron, M.; Chang, H.; Smith, C.E.; Hermo, L.; Suarez, S.; Roth, B.L.; Ducy, P.; et al. Endocrine regulation of male fertility by the skeleton. Cell 2011, 144, 796–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otani, T.; Mizokami, A.; Hayashi, Y.; Gao, J.; Mori, Y.; Nakamura, S.; Takeuchi, H.; Hirata, M. Signaling pathway for adiponectin expression in adipocytes by osteocalcin. Cell Signal. 2015, 27, 532–544. [Google Scholar] [CrossRef]
- Pi, M.; Wu, Y.; Quarles, L.D. GPRC6A mediates responses to osteocalcin in β-cells in vitro and pancreas in vivo. J. Bone Miner. Res. 2011, 26, 1680–1683. [Google Scholar] [CrossRef] [Green Version]
- Mizokami, A.; Yasutake, Y.; Higashi, S.; Kawakubo-Yasukochi, T.; Chishaki, S.; Takahashi, I.; Takeuchi, H.; Hirata, M. Oral administration of osteocalcin improves glucose utilization by stimulating glucagon-like peptide-1 secretion. Bone 2014, 69, 68–79. [Google Scholar] [CrossRef]
- Pi, M.; Kapoor, K.; Ye, R.; Nishimoto, S.K.; Smith, J.C.; Baudry, J.; Quarles, L.D. Evidence for Osteocalcin Binding and Activation of GPRC6A in β-Cells. Endocrinology 2016, 157, 1866–1880. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, C.V.; Gasparini, S.J.; Tu, J.; Zhou, H.; Seibel, M.J.; Bräuner-Osborne, H. Metabolic and skeletal homeostasis are maintained in full locus GPRC6A knockout mice. Sci. Rep. 2019, 9, 5995. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Kode, A.; Xu, L.; Mosialou, I.; Silva, B.C.; Ferron, M.; Clemens, T.L.; Economides, A.N.; Kousteni, S. Genetic evidence points to an osteocalcin-independent influence of osteoblasts on energy metabolism. J. Bone Miner. Res. 2011, 26, 2012–2025. [Google Scholar] [CrossRef]
- Yao, Q.; Yu, C.; Zhang, X.; Zhang, K.; Guo, J.; Song, L. Wnt/β-catenin signaling in osteoblasts regulates global energy metabolism. Bone 2017, 97, 175–183. [Google Scholar] [CrossRef]
- Diegel, C.R.; Hann, S.; Ayturk, U.M.; Hu, J.C.W.; Lim, K.E.; Droscha, C.J.; Madaj, Z.B.; Foxa, G.E.; Izaguirre, I.; Transgenics Core, V.V.A.; et al. An osteocalcin-deficient mouse strain without endocrine abnormalities. PLoS Genet. 2020, 16, e1008361. [Google Scholar] [CrossRef]
- Moriishi, T.; Ozasa, R.; Ishimoto, T.; Nakano, T.; Hasegawa, T.; Miyazaki, T.; Liu, W.; Fukuyama, R.; Wang, Y.; Komori, H.; et al. Osteocalcin is necessary for the alignment of apatite crystallites, but not glucose metabolism, testosterone synthesis, or muscle mass. PLoS Genet. 2020, 16, e1008586. [Google Scholar] [CrossRef]
- Galusca, B.; Bossu, C.; Germain, N.; Kadem, M.; Frere, D.; Lafage-Proust, M.H.; Lang, F.; Estour, B. Age-related differences in hormonal and nutritional impact on lean anorexia nervosa bone turnover uncoupling. Osteoporos. Int. 2006, 17, 888–896. [Google Scholar] [CrossRef]
- Urano, A.; Hotta, M.; Ohwada, R.; Araki, M. Vitamin K deficiency evaluated by serum levels of undercarboxylated osteocalcin in patients with anorexia nervosa with bone loss. Clin. Nutr. 2015, 34, 443–448. [Google Scholar] [CrossRef]
- Goldstone, A.P.; Howard, J.K.; Lord, G.M.; Ghatei, M.A.; Gardiner, J.V.; Wang, Z.L.; Wang, R.M.; Girgis, S.I.; Bailey, C.J.; Bloom, S.R. Leptin prevents the fall in plasma osteocalcin during starvation in male mice. Biochem. Biophys. Res. Commun. 2002, 295, 475–481. [Google Scholar] [CrossRef]
- Louvet, L.; Leterme, D.; Delplace, S.; Miellot, F.; Marchandise, P.; Gauthier, V.; Hardouin, P.; Chauveau, C.; Ghali Mhenni, O. Sirtuin 1 deficiency decreases bone mass and increases bone marrow adiposity in a mouse model of chronic energy deficiency. Bone 2020, 136, 115361. [Google Scholar] [CrossRef]
- Little, R.R.; Sacks, D.B. HbA1c: How do we measure it and what does it mean? Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 113–118. [Google Scholar] [CrossRef]
- Losada-Grande, E.; Hawley, S.; Soldevila, B.; Martinez-Laguna, D.; Nogues, X.; Diez-Perez, A.; Puig-Domingo, M.; Mauricio, D.; Prieto-Alhambra, D. Insulin use and Excess Fracture Risk in Patients with Type 2 Diabetes: A Propensity-Matched cohort analysis. Sci. Rep. 2017, 7, 3781. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.H.; Sloane, R.; Pieper, C.; Lyles, K.W.; Adler, R.A.; Van Houtven, C.; LaFleur, J.; Colón-Emeric, C. Glycemic Control and Insulin Treatment Alter Fracture Risk in Older Men With Type 2 Diabetes Mellitus. J. Bone Miner. Res. 2019, 34, 2045–2051. [Google Scholar] [CrossRef]
- Sayinalp, S.; Gedik, O.; Koray, Z. Increasing serum osteocalcin after glycemic control in diabetic men. Calcif. Tissue Int. 1995, 57, 422–425. [Google Scholar] [CrossRef]
- Clowes, J.A.; Hannon, R.A.; Yap, T.S.; Hoyle, N.R.; Blumsohn, A.; Eastell, R. Effect of feeding on bone turnover markers and its impact on biological variability of measurements. Bone 2002, 30, 886–890. [Google Scholar] [CrossRef]
- de Mello-Sampayo, C.; Agripino, A.A.; Stilwell, D.; Vidal, B.; Fernando, A.L.; Silva-Lima, B.; Vaz, M.F.; Canhão, H.; Marques, M.C. Chronic Hyperglycemia Modulates Rat Osteoporotic Cortical Bone Microarchitecture into Less Fragile Structures. Int. J. Endocrinol. 2017, 2017, 4603247. [Google Scholar] [CrossRef] [Green Version]
- Balint, E.; Szabo, P.; Marshall, C.F.; Sprague, S.M. Glucose-induced inhibition of in vitro bone mineralization. Bone 2001, 28, 21–28. [Google Scholar] [CrossRef]
- García-Hernández, A.; Arzate, H.; Gil-Chavarría, I.; Rojo, R.; Moreno-Fierros, L. High glucose concentrations alter the biomineralization process in human osteoblastic cells. Bone 2012, 50, 276–288. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, Z.Y.; Mao, Y.; Xin, H.T.; Ren, G.L.; Bai, X.F. Effects of insulin-like growth factor I on the development of osteoblasts in hyperglycemia. Diabetes Res. Clin. Pract. 2006, 73, 95–97. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Yang, L.; Zhou, J.; Tang, Y.; Zheng, L.; Qin, P. Runx2 alleviates high glucose-suppressed osteogenic differentiation via PI3K/AKT/GSK3β/β-catenin pathway. Cell Biol. Int. 2017, 41, 822–832. [Google Scholar] [CrossRef]
- Gu, H.; Song, M.; Boonanantanasarn, K.; Baek, K.; Woo, K.M.; Ryoo, H.M.; Baek, J.H. Conditions Inducing Excessive O-GlcNAcylation Inhibit BMP2-Induced Osteogenic Differentiation of C2C12 Cells. Int. J. Mol. Sci. 2018, 19, 202. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Ai, W.; Chen, L.; Zhao, S.; Liu, E. Bradykinin receptors and EphB2/EphrinB2 pathway in response to high glucose-induced osteoblast dysfunction and hyperglycemia-induced bone deterioration in mice. Int. J. Mol. Med. 2016, 37, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Pahwa, H.; Khan, M.T.; Sharan, K. Hyperglycemia impairs osteoblast cell migration and chemotaxis due to a decrease in mitochondrial biogenesis. Mol. Cell Biochem. 2020, 469, 109–118. [Google Scholar] [CrossRef]
- Zayzafoon, M.; Stell, C.; Irwin, R.; McCabe, L.R. Extracellular glucose influences osteoblast differentiation and c-Jun expression. J. Cell Biochem. 2000, 79, 301–310. [Google Scholar] [CrossRef]
- Botolin, S.; McCabe, L.R. Chronic hyperglycemia modulates osteoblast gene expression through osmotic and non-osmotic pathways. J. Cell Biochem. 2006, 99, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Hao, P.; Xu, S.; Liu, S.; Zhou, W.; Yue, X.; Rausch-Fan, X.; Liu, Z. Alpha-Lipoic Acid Alleviates High-Glucose Suppressed Osteogenic Differentiation of MC3T3-E1 Cells via Antioxidant Effect and PI3K/Akt Signaling Pathway. Cell Physiol. Biochem. 2017, 42, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Gu, W.; Chen, L.; Pan, L.; Chen, J.; Peng, Y. MiR-378 overexpression attenuates high glucose-suppressed osteogenic differentiation through targeting CASP3 and activating PI3K/Akt signaling pathway. Int. J. Clin. Exp. Pathol. 2014, 7, 7249–7261. [Google Scholar] [PubMed]
- Hamada, Y.; Fujii, H.; Fukagawa, M. Role of oxidative stress in diabetic bone disorder. Bone 2009, 45 (Suppl. 1), S35–S38. [Google Scholar] [CrossRef] [PubMed]
- Alikhani, M.; Alikhani, Z.; Boyd, C.; MacLellan, C.M.; Raptis, M.; Liu, R.; Pischon, N.; Trackman, P.C.; Gerstenfeld, L.; Graves, D.T. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone 2007, 40, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, S.; Kato, S.; Yamagishi, S.; Inagaki, Y.; Ueda, S.; Arima, N.; Okawa, T.; Kojiro, M.; Nagata, K. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone. J. Bone Miner. Res. 2005, 20, 1647–1658. [Google Scholar] [CrossRef]
- Mercer, N.; Ahmed, H.; Etcheverry, S.B.; Vasta, G.R.; Cortizo, A.M. Regulation of advanced glycation end product (AGE) receptors and apoptosis by AGEs in osteoblast-like cells. Mol. Cell Biochem. 2007, 306, 87–94. [Google Scholar] [CrossRef]
- Katayama, Y.; Akatsu, T.; Yamamoto, M.; Kugai, N.; Nagata, N. Role of nonenzymatic glycosylation of type I collagen in diabetic osteopenia. J. Bone Miner. Res. 1996, 11, 931–937. [Google Scholar] [CrossRef]
- Sanguineti, R.; Storace, D.; Monacelli, F.; Federici, A.; Odetti, P. Pentosidine effects on human osteoblasts in vitro. Ann. N. Y. Acad. Sci. 2008, 1126, 166–172. [Google Scholar] [CrossRef]
- Okazaki, K.; Yamaguchi, T.; Tanaka, K.; Notsu, M.; Ogawa, N.; Yano, S.; Sugimoto, T. Advanced glycation end products (AGEs), but not high glucose, inhibit the osteoblastic differentiation of mouse stromal ST2 cells through the suppression of osterix expression, and inhibit cell growth and increasing cell apoptosis. Calcif. Tissue Int. 2012, 91, 286–296. [Google Scholar] [CrossRef]
- Santana, R.B.; Xu, L.; Chase, H.B.; Amar, S.; Graves, D.T.; Trackman, P.C. A role for advanced glycation end products in diminished bone healing in type 1 diabetes. Diabetes 2003, 52, 1502–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Mao, J.; Jiang, Y.; Xia, L.; Mao, L.; Wu, Y.; Ma, P.; Fang, B. AGEs Induce Apoptosis in Rat Osteoblast Cells by Activating the Caspase-3 Signaling Pathway Under a High-Glucose Environment In Vitro. Appl. Biochem. Biotechnol. 2016, 178, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Stolzing, A.; Sellers, D.; Llewelyn, O.; Scutt, A. Diabetes induced changes in rat mesenchymal stem cells. Cells Tissues Organs 2010, 191, 453–465. [Google Scholar] [CrossRef]
- Franke, S.; Rüster, C.; Pester, J.; Hofmann, G.; Oelzner, P.; Wolf, G. Advanced glycation end products affect growth and function of osteoblasts. Clin. Exp. Rheumatol. 2011, 29, 650–660. [Google Scholar]
- Hamada, Y.; Kitazawa, S.; Kitazawa, R.; Fujii, H.; Kasuga, M.; Fukagawa, M. Histomorphometric analysis of diabetic osteopenia in streptozotocin-induced diabetic mice: A possible role of oxidative stress. Bone 2007, 40, 1408–1414. [Google Scholar] [CrossRef]
- Li, J.; He, W.; Liao, B.; Yang, J. FFA-ROS-P53-mediated mitochondrial apoptosis contributes to reduction of osteoblastogenesis and bone mass in type 2 diabetes mellitus. Sci. Rep. 2015, 5, 12724. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, J.H. Activation of the PI3K/Akt pathway by oxidative stress mediates high glucose-induced increase of adipogenic differentiation in primary rat osteoblasts. J. Cell Biochem. 2013, 114, 2595–2602. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, C.; Bailey, C.J. Reconsideration of inhibitory effect of metformin on intestinal glucose absorption. J. Pharm. Pharmacol. 1991, 43, 120–121. [Google Scholar] [CrossRef]
- Wu, T.; Xie, C.; Wu, H.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Metformin reduces the rate of small intestinal glucose absorption in type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 290–293. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurman, L.; McCarthy, A.D.; Sedlinsky, C.; Gangoiti, M.V.; Arnol, V.; Bruzzone, L.; Cortizo, A.M. Metformin reverts deleterious effects of advanced glycation end-products (AGEs) on osteoblastic cells. Exp. Clin. Endocrinol. Diabetes 2008, 116, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Zhen, D.; Chen, Y.; Tang, X. Metformin reverses the deleterious effects of high glucose on osteoblast function. J. Diabetes Complicat. 2010, 24, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, I.; Yamamoto, M.; Yamaguchi, T.; Sugimoto, T. Effects of metformin and pioglitazone on serum pentosidine levels in type 2 diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 2011, 119, 362–365. [Google Scholar] [CrossRef]
- Mirmiranpour, H.; Mousavizadeh, M.; Noshad, S.; Ghavami, M.; Ebadi, M.; Ghasemiesfe, M.; Nakhjavani, M.; Esteghamati, A. Comparative effects of pioglitazone and metformin on oxidative stress markers in newly diagnosed type 2 diabetes patients: A randomized clinical trial. J. Diabetes Complicat. 2013, 27, 501–507. [Google Scholar] [CrossRef]
- Gao, Y.; Li, Y.; Xue, J.; Jia, Y.; Hu, J. Effect of the anti-diabetic drug metformin on bone mass in ovariectomized rats. Eur. J. Pharmacol. 2010, 635, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xu, X.; Yang, Z.; Liu, Y.; Wu, X.; Huang, Z.; Liu, J.; Huang, Z.; Kong, G.; Ding, J.; et al. Metformin Alleviates the Bone Loss Induced by Ketogenic Diet: An In Vivo Study in Mice. Calcif. Tissue Int. 2019, 104, 59–69. [Google Scholar] [CrossRef]
- Tseng, C.H. Metformin use is associated with a lower risk of osteoporosis/vertebral fracture in Taiwanese patients with type 2 diabetes mellitus. Eur. J. Endocrinol. 2020, 184, 299–310. [Google Scholar] [CrossRef]
- Zhu, X.Z.; Zheng, X.Y. Metformin Prevents Nonunion after Three-Cannulated-Screw Fixation in Displaced Femoral Neck Fractures: A Retrospective Study. Biomed. Res. Int. 2016, 2016, 5682541. [Google Scholar] [CrossRef]
- Bahrambeigi, S.; Yousefi, B.; Rahimi, M.; Shafiei-Irannejad, V. Metformin; an old antidiabetic drug with new potentials in bone disorders. Biomed. Pharmacother. 2019, 109, 1593–1601. [Google Scholar] [CrossRef]
- Meng, J.; Ma, X.; Wang, N.; Jia, M.; Bi, L.; Wang, Y.; Li, M.; Zhang, H.; Xue, X.; Hou, Z.; et al. Activation of GLP-1 Receptor Promotes Bone Marrow Stromal Cell Osteogenic Differentiation through β-Catenin. Stem Cell Rep. 2016, 6, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Li, S.; Wang, N.; Xue, P.; Li, Y. Liraglutide, a glucagon-like peptide-1 receptor agonist, suppresses osteoclastogenesis through the inhibition of NF-κB and MAPK pathways via GLP-1R. Biomed. Pharmacother. 2020, 130, 110523. [Google Scholar] [CrossRef] [PubMed]
- Mansur, S.A.; Mieczkowska, A.; Flatt, P.R.; Chappard, D.; Irwin, N.; Mabilleau, G. The GLP-1 Receptor Agonist Exenatide Ameliorates Bone Composition and Tissue Material Properties in High Fat Fed Diabetic Mice. Front. Endocrinol. Lausanne 2019, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Sun, H.; Yu, J.; Wang, X.; Liu, D.; Zhao, L.; Sun, L.; Zhao, H.; Tao, B.; Liu, J. Glucagon-like peptide-1 receptor agonist Liraglutide has anabolic bone effects in ovariectomized rats without diabetes. PLoS ONE 2015, 10, e0132744. [Google Scholar] [CrossRef]
- Ma, X.; Meng, J.; Jia, M.; Bi, L.; Zhou, Y.; Wang, Y.; Hu, J.; He, G.; Luo, X. Exendin-4, a glucagon-like peptide-1 receptor agonist, prevents osteopenia by promoting bone formation and suppressing bone resorption in aged ovariectomized rats. J. Bone Miner. Res. 2013, 28, 1641–1652. [Google Scholar] [CrossRef] [Green Version]
- Driessen, J.H.; Henry, R.M.; van Onzenoort, H.A.; Lalmohamed, A.; Burden, A.M.; Prieto-Alhambra, D.; Neef, C.; Leufkens, H.G.; de Vries, F. Bone fracture risk is not associated with the use of glucagon-like peptide-1 receptor agonists: A population-based cohort analysis. Calcif. Tissue Int. 2015, 97, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Mulvihill, E.E.; Varin, E.M.; Gladanac, B.; Campbell, J.E.; Ussher, J.R.; Baggio, L.L.; Yusta, B.; Ayala, J.; Burmeister, M.A.; Matthews, D.; et al. Cellular Sites and Mechanisms Linking Reduction of Dipeptidyl Peptidase-4 Activity to Control of Incretin Hormone Action and Glucose Homeostasis. Cell Metab. 2017, 25, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Yang, H.; Wang, Y.; Yan, X.; Li, D.; Cao, Z.; Ning, Y.; Zhang, C. Anagliptin stimulates osteoblastic cell differentiation and mineralization. Biomed. Pharmacother. 2020, 129, 109796. [Google Scholar] [CrossRef]
- Charoenphandhu, N.; Suntornsaratoon, P.; Sa-Nguanmoo, P.; Tanajak, P.; Teerapornpuntakit, J.; Aeimlapa, R.; Chattipakorn, N.; Chattipakorn, S. Dipeptidyl Peptidase-4 Inhibitor, Vildagliptin, Improves Trabecular Bone Mineral Density and Microstructure in Obese, Insulin-Resistant, Pre-diabetic Rats. Can. J. Diabetes 2018, 42, 545–552. [Google Scholar] [CrossRef]
- Guerra-Menéndez, L.; Sádaba, M.C.; Puche, J.E.; Lavandera, J.L.; de Castro, L.F.; de Gortázar, A.R.; Castilla-Cortázar, I. IGF-I increases markers of osteoblastic activity and reduces bone resorption via osteoprotegerin and RANK-ligand. J. Transl. Med. 2013, 11, 271. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Sheng, Z.Y.; Tang, C.L.; Chen, L.; Pan, L.; Chen, J.Y. High cholesterol diet increases osteoporosis risk via inhibiting bone formation in rats. Acta Pharmacol. Sin. 2011, 32, 1498–1504. [Google Scholar] [CrossRef] [Green Version]
- Monami, M.; Dicembrini, I.; Antenore, A.; Mannucci, E. Dipeptidyl peptidase-4 inhibitors and bone fractures: A meta-analysis of randomized clinical trials. Diabetes Care 2011, 34, 2474–2476. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; An, J.H.; Park, H.; Yang, J.Y.; Choi, H.J.; Kim, S.W.; Park, Y.J.; Kim, S.Y.; Yim, M.; Baek, W.Y.; et al. Positive regulation of osteogenesis by bile acid through FXR. J. Bone Miner. Res. 2013, 28, 2109–2121. [Google Scholar] [CrossRef]
- Donnan, J.R.; Grandy, C.A.; Chibrikov, E.; Marra, C.A.; Aubrey-Bassler, K.; Johnston, K.; Swab, M.; Hache, J.; Curnew, D.; Nguyen, H.; et al. Comparative safety of the sodium glucose co-transporter 2 (SGLT2) inhibitors: A systematic review and meta-analysis. BMJ Open 2019, 9, e022577. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Park, C.; Lee, Y.K.; Ha, Y.C.; Jang, S.; Shin, C.S. Risk of fractures and diabetes medications: A nationwide cohort study. Osteoporos. Int. 2016, 27, 2709–2715. [Google Scholar] [CrossRef]
- Zou, W.; Rohatgi, N.; Chen, T.H.; Schilling, J.; Abu-Amer, Y.; Teitelbaum, S.L. PPAR-γ regulates pharmacological but not physiological or pathological osteoclast formation. Nat. Med. 2016, 22, 1203–1205. [Google Scholar] [CrossRef]
- Wan, Y.; Chong, L.W.; Evans, R.M. PPAR-gamma regulates osteoclastogenesis in mice. Nat. Med. 2007, 13, 1496–1503. [Google Scholar] [CrossRef]
- Di Somma, C.; Colao, A.; Di Sarno, A.; Klain, M.; Landi, M.L.; Facciolli, G.; Pivonello, R.; Panza, N.; Salvatore, M.; Lombardi, G. Bone marker and bone density responses to dopamine agonist therapy in hyperprolactinemic males. J. Clin. Endocrinol. Metab. 1998, 83, 807–813. [Google Scholar] [CrossRef]
- Hanami, K.; Nakano, K.; Saito, K.; Okada, Y.; Yamaoka, K.; Kubo, S.; Kondo, M.; Tanaka, Y. Dopamine D2-like receptor signaling suppresses human osteoclastogenesis. Bone 2013, 56, 1–8. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Class | Effect on Bone | Mechanism of Action | Model/Study Type | References |
|---|---|---|---|---|
| Biguanides (e.g., metformin) |
|
| in vitro | [130,131] reviewed by [139] |
| - | clinical | [132,133] | |
| - | rat/mouse | [134,135] | |
| - | clinical | [136,137] | |
| GLP-1 receptor agonists |
|
| in vitro | [140,141] |
| - | rat/mouse | [142,143,144] | |
| - | clinical | [145] | |
| DPP4 inhibitors |
|
| in vitro | [147] |
|
| rat | [148] | |
| - | meta-analysis | [151] | |
| SGLT2 Inhibitors |
| - | clinical | [153] |
| - | clinical | [154,155] | |
| - | mouse | [156] | |
| Thiazolidinediones |
| - | clinical | [157] |
|
| in vitro | [158,159] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donat, A.; Knapstein, P.-R.; Jiang, S.; Baranowsky, A.; Ballhause, T.-M.; Frosch, K.-H.; Keller, J. Glucose Metabolism in Osteoblasts in Healthy and Pathophysiological Conditions. Int. J. Mol. Sci. 2021, 22, 4120. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084120
Donat A, Knapstein P-R, Jiang S, Baranowsky A, Ballhause T-M, Frosch K-H, Keller J. Glucose Metabolism in Osteoblasts in Healthy and Pathophysiological Conditions. International Journal of Molecular Sciences. 2021; 22(8):4120. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084120
Chicago/Turabian StyleDonat, Antonia, Paul-Richard Knapstein, Shan Jiang, Anke Baranowsky, Tobias-Malte Ballhause, Karl-Heinz Frosch, and Johannes Keller. 2021. "Glucose Metabolism in Osteoblasts in Healthy and Pathophysiological Conditions" International Journal of Molecular Sciences 22, no. 8: 4120. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084120