
Role of Adaptor Protein Myeloid Differentiation 88 (MyD88) in Post-Subarachnoid Hemorrhage Inflammation: A Systematic Review


 ,
,  ,
, 
Abstract
:1. Introduction
2. Myeloid Differentiation Primary-Response Gene 88 (MyD88)
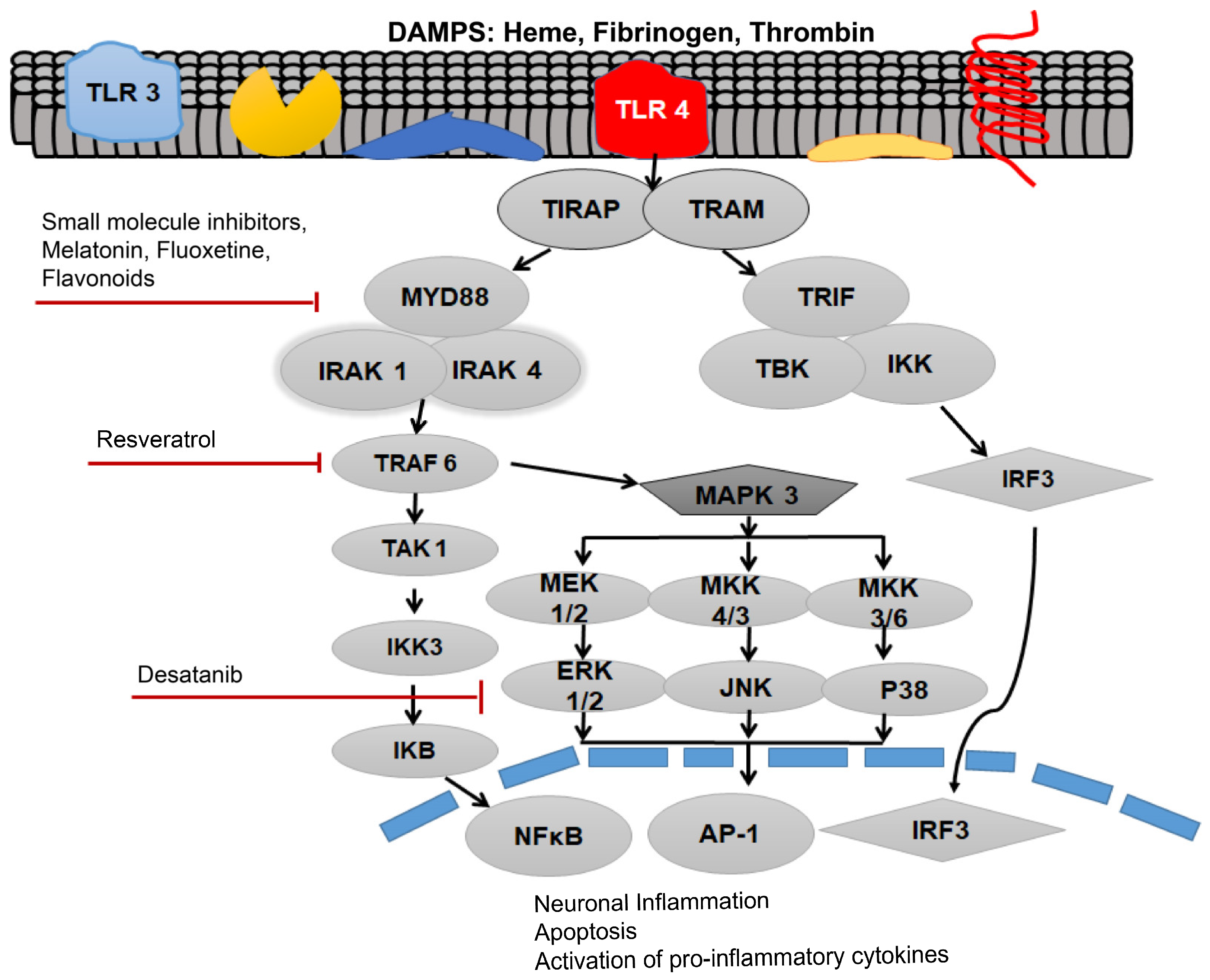
3. Role of TLR4-MyD88 in Post-SAH Inflammation
4. MyD88 as a Therapeutic Target of Post-SAH Inflammation
4.1. Progesterone
4.2. Small Molecule Inhibitors
4.3. Aptamers
4.4. Polyphenols
4.5. Melatonin
4.6. Monoclonal Antibodies
4.7. Pentoxifylline
4.8. Astaxanthin
4.9. Fluoxetine
5. Discussion
6. Conclusions
7. Methodology
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2014, 10, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Alafaci, C.; Macdonald, R.L. Management of aneurysmal subarachnoid hemorrhage: State of the art and future perspectives. Surg. Neurol. Int. 2017, 8, 11. [Google Scholar] [CrossRef]
- Korja, M.; Kaprio, J. Controversies in epidemiology of intracranial aneurysms and SAH. Nat. Rev. Neurol. 2015, 12, 50–55. [Google Scholar] [CrossRef]
- Gągało, I.; Rusiecka, I.; Kocic, I. Tyrosine Kinase Inhibitor as a new Therapy for Ischemic Stroke and other Neurologic Diseases: Is there any Hope for a Better Outcome? Curr. Neuropharmacol. 2015, 13, 836–844. [Google Scholar] [CrossRef]
- Etminan, N.; Rinkel, G.J. Unruptured intracranial aneurysms: Development, rupture and preventive management. Nat. Rev. Neurol. 2016, 12, 699–713. [Google Scholar]
- Macdonald, R.L.; Diringer, M.N.; Citerio, G. Understanding the disease: Aneurysmal subarachnoid hemorrhage. Intensiv. Care Med. 2014, 40, 1940–1943. [Google Scholar] [CrossRef]
- Chaudhry, S.R.; Frede, S.; Seifert, G.; Kinfe, T.M.; Niemelä, M.; Lamprecht, A.; Muhammad, S. Temporal profile of serum mitochondrial DNA (mtDNA) in patients with aneurysmal subarachnoid hemorrhage (aSAH). Mitochondrion 2019, 47, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, S.R.; Güresir, A.; Stoffel-Wagner, B.; Fimmers, R.; Kinfe, T.M.; Dietrich, D.; Lamprecht, A.; Vatter, H.; Güresir, E.; Muhammad, S. Systemic High-Mobility Group Box-1: A Novel Predictive Biomarker for Cerebral Vasospasm in Aneurysmal Subarachnoid Hemorrhage*. Crit. Care Med. 2018, 46, e1023–e1028. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.R.; Hafez, A.; Jahromi, B.R.; Kinfe, T.M.; Lamprecht, A.; Niemelä, M.; Muhammad, S. Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2018, 19, 2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, S.; Lehecka, M.; Niemelä, M.; Muhammad, S. Sterile Inflammation, Potential Target in Aneurysmal Subarachnoid Hemorrhage. World Neurosurg. 2019, 123, 159–160. [Google Scholar] [CrossRef]
- Muhammad, S.; Chaudhry, S.R.; Kahlert, U.D.; Lehecka, M.; Korja, M.; Niemelä, M.; Hänggi, D. Targeting High Mobility Group Box 1 in Subarachnoid Hemorrhage: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 2709. [Google Scholar] [CrossRef] [Green Version]
- Cahill, J.; Calvert, J.W.; Zhang, J.H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2006, 26, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Cahill, J.; Zhang, J.H. Subarachnoid Hemorrhage: Is It Time for a New Direction? Stroke 2008, 40 (Suppl. 1), S86–S87. [Google Scholar] [CrossRef] [Green Version]
- Etminan, N.; Macdonald, R.L. Medical Complications After Aneurysmal Subarachnoid Hemorrhage: An Emerging Contributor to Poor Outcome. World Neurosurg. 2015, 83, 303–304. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef] [PubMed]
- Rahimifard, M.; Maqbool, F.; Moeini-Nodeh, S.; Niaz, K.; Abdollahi, M.; Braidy, N.; Nabavi, S.M. Targeting the TLR4 signaling pathway by polyphenols: A novel therapeutic strategy for neuroinflammation. Ageing Res. Rev. 2017, 36, 11–19. [Google Scholar] [CrossRef]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O.J.N. Hesperetin, a citrus flavonoid, attenuates LPS-induced neuroinflammation, apoptosis and memory impairments by modulating TLR4/NF-κB signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [Green Version]
- Janssens, S.; Beyaert, R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem. Sci. 2002, 27, 474–482. [Google Scholar] [CrossRef]
- Gewies, A.; Ruland, J.; Kotlyarov, A.; Gaestel, M.; Procaccia, S.; Seger, R.; Yasuda, S.; Sugiura, H.; Yamagata, K.; Le, N.-T.; et al. MyD88, Myeloid Differentiation Primary Response Gene 88. In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer: New York, NY, USA, 2012; pp. 1149–1159. [Google Scholar]
- Cheng, X.; Yang, Y.-L.; Yang, H.; Wang, Y.-H.; Du, G.-H. Kaempferol alleviates LPS-induced neuroinflammation and BBB dysfunction in mice via inhibiting HMGB1 release and down-regulating TLR4/MyD88 pathway. Int. Immunopharmacol. 2018, 56, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10, 1–83. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Luo, Q.; Zhao, Y.-H.; Chen, X. Toll-like receptor-4 pathway as a possible molecular mechanism for brain injuries after subarachnoid hemorrhage. Int. J. Neurosci. 2020, 130, 953–964. [Google Scholar] [CrossRef]
- Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin Is an Endogenous Toll-Like Receptor 4 Ligand—Relevance to Subarachnoid Hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028–5046. [Google Scholar] [CrossRef] [Green Version]
- Karimy, J.K.; Reeves, B.C.; Kahle, K.T. Targeting TLR4-dependent inflammation in post-hemorrhagic brain injury. Expert Opin. Ther. Targets 2020, 24, 525–533. [Google Scholar] [CrossRef]
- Sokół, B.; Wąsik, N.; Jankowski, R.; Hołysz, M.; Więckowska, B.; Jagodziński, P. Soluble Toll-Like Receptors 2 and 4 in Cerebrospinal Fluid of Patients with Acute Hydrocephalus following Aneurysmal Subarachnoid Haemorrhage. PLoS ONE 2016, 11, e0156171. [Google Scholar]
- Karimy, J.K.; Zhang, J.; Kurland, D.B.; Theriault, B.C.; Duran, D.; Stokum, J.A.; Furey, C.G.; Zhou, X.; Mansuri, M.S.; Montejo, J.; et al. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat. Med. 2017, 23, 997–1003. [Google Scholar] [CrossRef]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef]
- Fang, H.; Wang, P.-F.; Zhou, Y.; Wang, Y.-C.; Yang, Q.-W. Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J. Neuroinflamm. 2013, 10, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddahi, A.; Ansar, S.; Chen, Q.; Edvinsson, L. Metabolism, Blockade of the MEK/ERK Pathway with a Raf Inhibitor Prevents Activation of Pro-Inflammatory Mediators in Cerebral Arteries and Reduction in Cerebral Blood Flow after Subarachnoid Hemorrhage in a Rat Model. Br. J. Pharmacol. 2010, 31, 144–154. [Google Scholar]
- Suzuki, H.; Hasegawa, Y.; Chen, W.; Kanamaru, K.; Zhang, J.H. Recombinant osteopontin in cerebral vasospasm after subarachnoid hemorrhage. Ann. Neurol. 2010, 68, 650–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wu, M.; Wu, C.-Y.; Xia, G.-Q. Role of progesterone in TLR4-MyD88-dependent signaling pathway in pre-eclampsia. Acta Acad. Med. Wuhan 2013, 33, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zuo, G.; Shi, X.-Y.; Zhang, J.; Fang, Q.; Chen, G. Progesterone Administration Modulates Cortical TLR4/NF-κB Signaling Pathway after Subarachnoid Hemorrhage in Male Rats. Mediat. Inflamm. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Zhang, D.; Wei, Y.; Ni, H.; Liang, W.; Zhang, H.; Hao, S.; Jin, W.; Li, K.; Hang, C.-H. Inhibition of myeloid differentiation primary response protein 88 provides neuroprotection in early brain injury following experimental subarachnoid hemorrhage. Sci. Rep. 2017, 7, 15797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, G.; Moraga, A.; Cuartero, M.I.; García-Culebras, A.; Peña-Martínez, C.; Pradillo, J.M.; Hernández-Jiménez, M.; Sacristán, S.; Ayuso, M.I.; Gonzalo-Gobernado, R.J.M.T. TLR4-binding DNA aptamers show a protective effect against acute stroke in animal models. Mol. Ther. 2018, 26, 2047–2059. [Google Scholar] [CrossRef] [Green Version]
- Jakus, P.B.; Kalman, N.; Antus, C.; Radnai, B.; Tucsek, Z.; Gallyas, F., Jr.; Sumegi, B.; Veres, B.J. TRAF6 is functional in inhibition of TLR4-mediated NF-κB activation by resveratrol. J. Nutr. Chem. 2013, 24, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhuang, Z.; Lu, Y.; Tao, T.; Zhou, Y.; Liu, G.; Wang, H.; Zhang, D.; Wu, L.; Dai, H.; et al. Curcumin Mitigates Neuro-Inflammation by Modulating Microglia Polarization Through Inhibiting TLR4 Axis Signaling Pathway Following Experimental Subarachnoid Hemorrhage. Front. Neurosci. 2019, 13, 1223. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.Y.; Ye, Z.N.; Zhuang, Z.; Gao, Y.; Tang, C.; Zhou, C.H.; Wang, C.X.; Zhang, X.S.; Xie, G.B.; Liu, J.P.; et al. Biochanin A Reduces Inflammatory Injury and Neuronal Apoptosis following Subarachnoid Hemorrhage via Suppression of the TLRs/TIRAP/MyD88/NF-kappaB Pathway. Behav. Neurol. 2018, 2018, 1960106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-S.; Li, W.; Wu, Q.; Wu, L.-Y.; Ye, Z.-N.; Liu, J.-P.; Zhuang, Z.; Zhou, M.-L.; Zhang, X.; Hang, C.-H. Resveratrol Attenuates Acute Inflammatory Injury in Experimental Subarachnoid Hemorrhage in Rats via Inhibition of TLR4 Pathway. Int. J. Mol. Sci. 2016, 17, 1331. [Google Scholar] [CrossRef] [Green Version]
- Shao, A.W.; Wu, H.J.; Chen, S.; Ammar, A.B.; Zhang, J.M.; Hong, Y. Resveratrol attenuates early brain injury after subarachnoid hemorrhage through inhibition of NF-κB-dependent inflammatory/MMP-9 pathway. CNS Neurosci. Ther. 2014, 20, 182–185. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and inflammation-Story of a double-edged blade. J. Pineal Res. 2018, 65, e12525. [Google Scholar] [CrossRef] [Green Version]
- Tarocco, A.; Caroccia, N.; Morciano, G.; Wieckowski, M.R.; Ancora, G.; Garani, G.; Pinton, P. Melatonin as a master regulator of cell death and inflammation: Molecular mechanisms and clinical implications for newborn care. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Posa, L.; de Gregorio, D.; Gobbi, G.; Comai, S. Targeting Melatonin MT2 Receptors: A Novel Pharmacological Avenue for Inflammatory and Neuropathic Pain. Curr. Med. Chem. 2018, 25, 3866–3882. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, L.; You, W.; Ji, C.; Chen, G. Melatonin alleviates secondary brain damage and neurobehavioral dysfunction after experimental subarachnoid hemorrhage: Possible involvement of TLR4-mediated inflammatory pathway. J. Pineal Res. 2013, 55, 399–408. [Google Scholar] [CrossRef]
- Tweedie, D.; Sambamurti, K.; Greig, N.H. TNF-α Inhibition as a Treatment Strategy for Neurodegenerative Disorders: New Drug Candidates and Targets. Curr. Alzheimer Res. 2007, 4, 378–385. [Google Scholar] [CrossRef]
- Xia, D.Y.; Zhang, H.S.; Wu, L.Y.; Zhang, X.S.; Zhou, M.L.; Hang, C.H. Pentoxifylline Alleviates Early Brain Injury After Experimental Subarachnoid Hemorrhage in Rats: Possibly via Inhibiting TLR 4/NF-κB Signaling Pathway. Neurochem. Res. 2017, 42, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, Y.; Wu, Q.; Dai, H.; Li, W.; Lv, S.; Zhou, X.; Zhang, X.; Hang, C.; Wang, J. Astaxanthin mitigates subarachnoid hemorrhage injury primarily by increasing sirtuin 1 and inhibiting the Toll-like receptor 4 signaling pathway. FASEB J. 2018, 33, 722–737. [Google Scholar] [CrossRef]
- Liu, F.Y.; Cai, J.; Wang, C.; Ruan, W.; Guan, G.P.; Pan, H.Z.; Li, J.R.; Qian, C.; Chen, J.S.; Wang, L.; et al. Fluoxetine attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage: A possible role for the regulation of TLR4/MyD88/NF-kappaB signaling pathway. J. Neuroinflamm. 2018, 15, 347. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, X.-S.; Zhang, Z.-H.; Zhou, X.-M.; Gao, Y.-Y.; Liu, G.-J.; Wang, H.; Wu, L.-Y.; Li, W.; Hang, C.-H. Peroxiredoxin 2 activates microglia by interacting with Toll-like receptor 4 after subarachnoid hemorrhage. J. Neuroinflamm. 2018, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Okada, T. Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural Regen. Res. 2017, 12, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Rivest, S. MyD88 signaling in brain endothelial cells is essential for the neuronal activity and glucocorticoid release during systemic inflammation. Mol. Psychiatry 2008, 13, 480–497. [Google Scholar] [CrossRef]
- Ryu, K.-Y.; Lee, H.-j.; Woo, H.; Kang, R.-J.; Han, K.-M.; Park, H.; Lee, S.M.; Lee, J.-Y.; Jeong, Y.J.; Nam, H.-W.; et al. Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling. J. Neuroinflamm. 2019, 16, 1–36. [Google Scholar] [CrossRef]
- Olson, M.A.; Lee, M.S.; Kissner, T.L.; Alam, S.; Waugh, D.S.; Saikh, K.U. Discovery of small molecule inhibitors of MyD88-dependent signaling pathways using a computational screen. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Pérez, S.; Hernández-Palma, L.A.; Oregon-Romero, E.; Anaya-Macías, B.U.; García-Arellano, S.; González-Estevez, G.; Muñoz-Valle, J.F. Downregulation of Inflammatory Cytokine Release from IL-1β and LPS-Stimulated PBMC Orchestrated by ST2825, a MyD88 Dimerisation Inhibitor. Molecules 2020, 25, 4322. [Google Scholar] [CrossRef]
- Pierce, J.W.; Schoenleber, R.; Jesmok, G.; Best, J.; Moore, S.A.; Collins, T.; Gerritsen, M.E. Novel Inhibitors of Cytokine-induced IκBα Phosphorylation and Endothelial Cell Adhesion Molecule Expression Show Anti-inflammatory Effects in Vivo. J. Biol. Chem. 1997, 272, 21096–21103. [Google Scholar] [CrossRef] [Green Version]
- Kawakita, F.; Fujimoto, M.; Liu, L.; Nakano, F.; Nakatsuka, Y.; Suzuki, H. Effects of Toll-Like Receptor 4 Antagonists Against Cerebral Vasospasm After Experimental Subarachnoid Hemorrhage in Mice. Mol. Neurobiol. 2016, 54, 6624–6633. [Google Scholar] [CrossRef]
- Zusso, M.; Lunardi, V.; Franceschini, D.; Pagetta, A.; Lo, R.; Stifani, S.; Frigo, A.C.; Giusti, P.; Moro, S. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J. Neuroinflamm. 2019, 16, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pea, F.; Pavan, F.; Nascimben, E.; Benetton, C.; Scotton, P.G.; Vaglia, A.; Furlanut, M. Levofloxacin Disposition in Cerebrospinal Fluid in Patients with External Ventriculostomy. Antimicrob. Agents Chemother. 2003, 47, 3104–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khattar, N.K.; James, R.F. Heparin: The Silver Bullet of Aneurysmal Subarachnoid Hemorrhage? Front. Neurol. 2018, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Hayman, E.G.; Patel, A.P.; James, R.F.; Simard, J.M. Heparin and Heparin-Derivatives in Post-Subarachnoid Hemorrhage Brain Injury: A Multimodal Therapy for a Multimodal Disease. Molecules 2017, 22, 724. [Google Scholar] [CrossRef] [Green Version]
- Yi, N.Y.; Newman, D.R.; Zhang, H.; Morales Johansson, H.M.; Sannes, P.L. Heparin and LPS-induced COX-2 expression in airway cells: A link between its anti-inflammatory effects and GAG sulfation. Exp. Lung Res. 2015, 41, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Dai, Y.; Zhang, X.; Hu, Y.-C.; Zhang, D.; Li, W.; Zhang, X.-S.; Zhu, J.-H.; Zhou, M.-L.; Hang, C.-H. Expression and cell distribution of myeloid differentiation primary response protein 88 in the cerebral cortex following experimental subarachnoid hemorrhage in rats: A pilot study. Brain Res. 2013, 1520, 134–144. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, J.H. Recent Advances in Stem Cell Research in Subarachnoid Hemorrhage. Stem Cells Dev. 2020, 29, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Rivera, P.D.; Hanamsagar, R.; Kan, M.J.; Tran, P.K.; Stewart, D.; Jo, Y.C.; Gunn, M.; Bilbo, S.D. Removal of microglial-specific MyD88 signaling alters dentate gyrus doublecortin and enhances opioid addiction-like behaviors. Brain Behav. Immun. 2019, 76, 104–115. [Google Scholar] [CrossRef]


{kind=link}
| Pharmacological Agent | Drug Target | Dose | Animal Model | Mechanism | Reference |
|---|---|---|---|---|---|
| ApTLR#1R | TLR4 | 0.1 and 1 nmol i.p. 6 h,12 h and 24 h | CCA and MCA ligation of Mice | Bind to TLR4, avoiding the ligand Binding | [42] |
| ApTLR#4F | TLR4 | 0.1 and 1 nmol i.p. 6 h,12 h and 24 h | CCA and MCA ligation of Mice | Bind to TLR4, avoiding the ligand Binding | [42] |
| Astaxanthin | MyD88, NFKB | 0.1 and 0.2 mmol i.p. 12 h and 24 h | Prechiasmatic cistern SAH | Inhibit the NFKB activity | [46] |
| Ciprofloxacin & Levofloxacin | MD-2 | 100, 200, 500 mg/kg i.v. 24 h | Prechiasmatic cistern SAH | Blocks the dimerization of TLR4 by interacting with hydrophobic region of MD-2, inhibiting the MD-2 TLR4 interaction | [53,65] |
| Curcumin | MyD88, TLR4 | 200 mg/kg i.p. 15 min | Prechiasmatic cistern SAH | Targets TLR4, preventing TLR4 dimerization upon ligand binding | [44] |
| Dasatinib | TLR4, MyD 88, ERK-AKT | 20 mg/kg i.p for 4 days 20 mg/kg orally for 2 weeks | LPS induced neuro-inflammation | Mechanism of action is unknown but suggested to bind and inhibit TLR4, pERK, and pAKT | [59] |
| Fluoxetine | TLR4 | 10 mg/kg i.v 6 h and 12 h | CCA ligation of mice | Unknown mechanism | [55] |
| Melatonin | TLR4/Myd88 | 0.5 or 1 mg/kg i.p. | LPS induced neuro-inflammation | Inhibit the TLR 4 and MyD88 cascade | [51] |
| Pentoxyfyline | TLR4 | 60 mg/kg i.p. 6 h and 24 h | Prechiasmatic cistern SAH | Inhibits the TLR4 signaling | [53] |
| Progesterone | MyD88 | 16 mg/kg 1,6 and 24 h | Prechiasmatic cistern SAH | Inhibit the MyD88 | [40] |
| Resveratrol | TLR4/TRAF6 | 60 mg/kg i.p. 6 h and 24 h | Prechiasmatic cistern SAH | Interferes with TLR4 dimerization and mitigates TRAF6 ubiquitination and activation of downstream mediators | [43] |
| ST2825 | MYD88 | 10, 30, 50 mmol i.p. | - | Mimics and directly binds to the TIR domain on MyD88, preventing MyD88 homodimerization and further signaling | [62] |
| T6167923 | MyD88 | 10, 30, 50 mmol i.p. | - | Mimics and directly binds to the TIR domain on MyD88, preventing MyD88 | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, H.; Khan, M.A.; Kahlert, U.D.; Niemelä, M.; Hänggi, D.; Chaudhry, S.R.; Muhammad, S. Role of Adaptor Protein Myeloid Differentiation 88 (MyD88) in Post-Subarachnoid Hemorrhage Inflammation: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 4185. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084185
Ahmed H, Khan MA, Kahlert UD, Niemelä M, Hänggi D, Chaudhry SR, Muhammad S. Role of Adaptor Protein Myeloid Differentiation 88 (MyD88) in Post-Subarachnoid Hemorrhage Inflammation: A Systematic Review. International Journal of Molecular Sciences. 2021; 22(8):4185. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084185
Chicago/Turabian StyleAhmed, Hammad, Mahtab Ahmad Khan, Ulf Dietrich Kahlert, Mika Niemelä, Daniel Hänggi, Shafqat Rasul Chaudhry, and Sajjad Muhammad. 2021. "Role of Adaptor Protein Myeloid Differentiation 88 (MyD88) in Post-Subarachnoid Hemorrhage Inflammation: A Systematic Review" International Journal of Molecular Sciences 22, no. 8: 4185. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084185