
Estrogen Receptor Functions and Pathways at the Vascular Immune Interface


{kind=link}
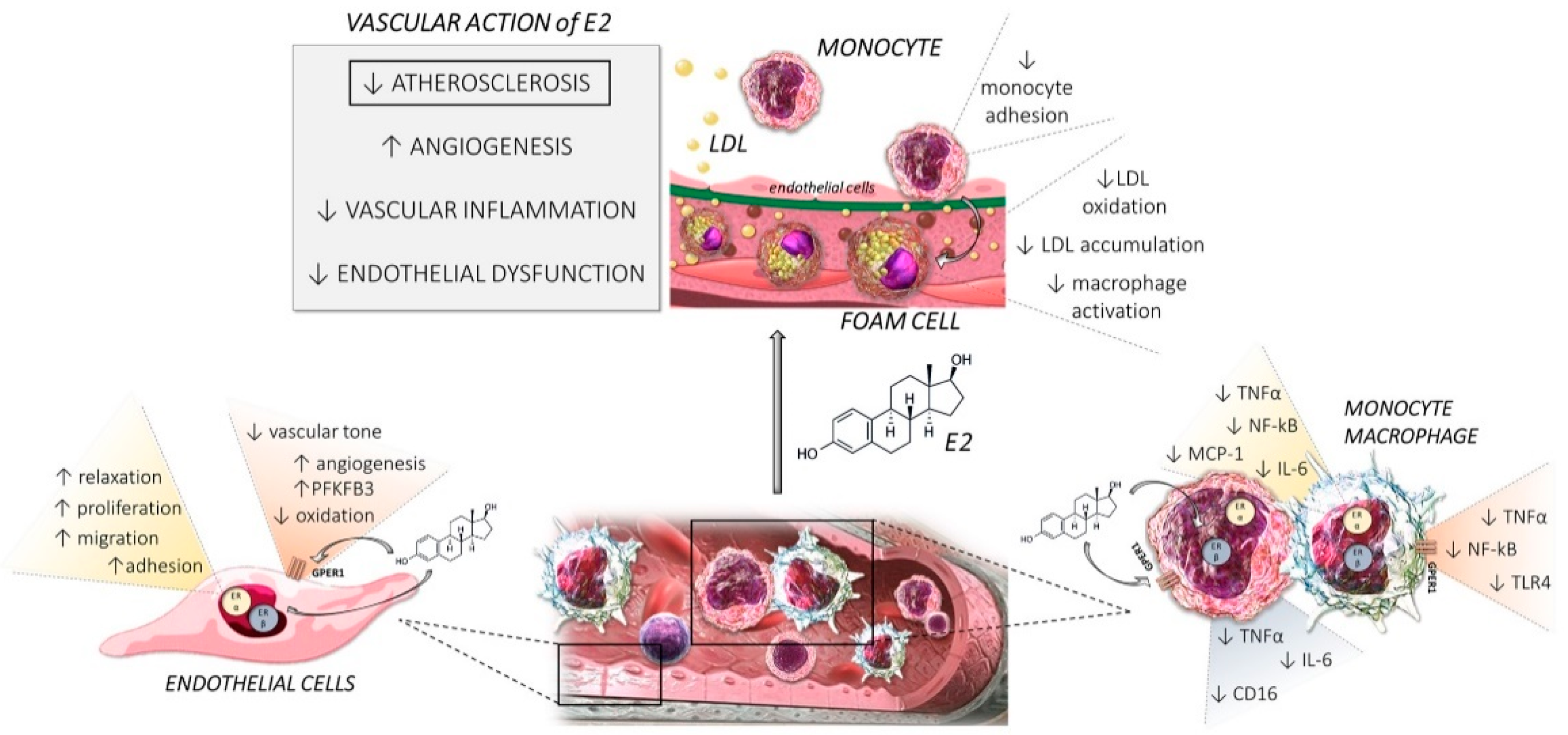
Abstract
:1. The Vascular Immune Interface
2. Cardiovascular Risk in Women
3. Estrogen Receptors in the Cardiovascular System
4. Sex Differences and Estrogenic Pathways Regulate Endothelial Angiogenesis
5. Estrogenic Pathways in the Monocyte-Macrophage System
5.1. Patterns of Estrogen Receptor Expression in Monocytes and Macrophages
5.2. Estrogenic Pathways at the Vascular Immune Interface
5.3. Estrogens: Regulators of the Immune Function of the Monocyte-Macrophage System
5.4. Estrogens: Regulators of Macrophage Immunophenotypes
6. A Possible Role for Estrogens in Constraining Myelopoiesis and Cardiovascular Risk
7. Intricacies of ER Pharmacological Modulation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar]
- Schnitzler, J.G.; Dallinga-Thie, G.M.; Kroon, J. The role of (modified) lipoproteins in vascular function: A duet between monocytes and the endothelium. Curr. Med. Chem. 2019, 26, 1594–1609. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Taleb, S.; Mallat, Z.; Tedgui, A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 969–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.L.; Tai, J.J.; Wong, W.C.; Han, H.; Sem, X.; Yeap, W.H.; Kourilsky, P.; Wong, S.C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef] [Green Version]
- Cignarella, A.; Tedesco, S.; Cappellari, R.; Fadini, G.P. The continuum of monocyte phenotypes: Experimental evidence and prognostic utility in assessing cardiovascular risk. J. Leukoc. Biol. 2018, 103, 1021–1028. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Bonacina, F.; Guinamard, R.R.; Norata, G.D. Immunometabolic function of cholesterol in cardiovascular disease and beyond. Cardiovasc. Res. 2019, 115, 1393–1407. [Google Scholar] [CrossRef]
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70. [Google Scholar]
- Xing, D.; Nozell, S.; Chen, Y.F.; Hage, F.; Oparil, S. Estrogen and mechanisms of vascular protection. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 289–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarkson, T.B. Estrogen effects on arteries vary with stage of reproductive life and extent of subclinical atherosclerosis progression. Menopause 2018, 25, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, A.; Michos, E.D.; Samad, Z.; Ballantyne, C.M.; Virani, S.S. The use of sex-specific factors in the assessment of women’s cardiovascular risk. Circulation 2020, 141, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.H.E.M.; Rosano, G.; Cifkova, R.; Chieffo, A.; van Dijken, D.; Hamoda, H.; Kunadian, V.; Laan, E.; Lambrinoudaki, I.; Maclaran, K.; et al. Cardiovascular health after menopause transition, pregnancy disorders, and other gynaecologic conditions: A consensus document from European cardiologists, gynaecologists, and endocrinologists. Eur. Heart J. 2021, 42, 967–984. [Google Scholar] [CrossRef]
- Honold, L.; Nahrendorf, M. Resident and monocyte-derived macrophages in cardiovascular disease. Circ. Res. 2018, 122, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Toniolo, A.; Fadini, G.P.; Tedesco, S.; Cappellari, R.; Vegeto, E.; Maggi, A.; Avogaro, A.; Bolego, C.; Cignarella, A. Alternative activation of human macrophages is rescued by estrogen treatment in vitro and impaired by menopausal status. J. Clin. Endocrinol. Metab. 2015, 100, E50–E58. [Google Scholar] [CrossRef] [Green Version]
- Menazza, S.; Murphy, E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ. Res. 2016, 118, 994–1007. [Google Scholar] [CrossRef]
- Murphy, A.J.; Guyre, P.M.; Wira, C.R.; Pioli, P.A. Estradiol regulates expression of estrogen receptor ERα46 in human macrophages. PLoS ONE 2009, 4, e5539. [Google Scholar] [CrossRef] [Green Version]
- Pelekanou, V.; Kampa, M.; Kiagiadaki, F.; Deli, A.; Theodoropoulos, P.; Agrogiannis, G.; Patsouris, E.; Tsapis, A.; Castanas, E.; Notas, G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J. Leukoc. Biol. 2016, 99, 333–347. [Google Scholar] [CrossRef]
- Stygar, D.; Westlund, P.; Eriksson, H.; Sahlin, L. Identification of wild type and variants of oestrogen receptors in polymorphonuclear and mononuclear leucocytes. Clin. Endocrinol. 2006, 64, 74–81. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prossnitz, E.R.; Arterburn, J.B. International Union of Basic and Clinical Pharmacology. XCVII. G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey, S.H.; Cohen, J.A.; Brosnihan, K.B.; Gallagher, P.E.; Chappell, M.C. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology 2009, 150, 3753–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, Q.K. Reciprocality between estrogen biology and calcium signaling in the cardiovascular system. Front. Endocrinol. 2020, 11, 568203. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; Nguyen, M.T.; Henstridge, D.C.; Nguyen, A.K.; Beaven, S.W.; Watt, M.J.; Hevener, A.L. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERα-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E304–E319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Bian, Z.; Lu, P.; Karas, R.H.; Bao, L.; Cox, D.; Hodgin, J.; Shaul, P.W.; Thoren, P.; Smithies, O.; et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science 2002, 295, 505–508. [Google Scholar] [CrossRef]
- Meyer, M.R.; Amann, K.; Field, A.S.; Hu, C.; Hathaway, H.J.; Kanagy, N.L.; Walker, M.K.; Barton, M.; Prossnitz, E.R. Deletion of G protein-coupled estrogen receptor increases endothelial vasoconstriction. Hypertension 2012, 59, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Bolego, C.; Cignarella, A.; Sanvito, P.; Pelosi, V.; Pellegatta, F.; Puglisi, L.; Pinna, C. The acute estrogenic dilation of rat aorta is mediated solely by selective estrogen receptor-α agonists and is abolished by estrogen deprivation. J. Pharmacol. Exp. Ther. 2005, 313, 1203–1208. [Google Scholar] [CrossRef]
- Meyer, M.R.; Fredette, N.C.; Barton, M.; Prossnitz, E.R. Endothelin-1 but not angiotensin II contributes to functional aging in murine carotid arteries. Life Sci. 2014, 118, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Tran, Q.K.; Firkins, R.; Giles, J.; Francis, S.; Matnishian, V.; Tran, P.; VerMeer, M.; Jasurda, J.; Burgard, M.A.; Gebert-Oberle, B. Estrogen enhances linkage in the vascular endothelial calmodulin network via a feedforward mechanism at the G protein-coupled estrogen receptor 1. J. Biol. Chem. 2016, 291, 10805–10823. [Google Scholar] [CrossRef] [Green Version]
- Boscaro, C.; Trenti, A.; Baggio, C.; Scapin, C.; Trevisi, L.; Cignarella, A.; Bolego, C. Sex differences in the pro-angiogenic response of human endothelial cells: Focus on PFKFB3 and FAK activation. Front. Pharmacol. 2020, 11, 587221. [Google Scholar] [CrossRef]
- Mudrovcic, N.; Arefin, S.; Van Craenenbroeck, A.H.; Kublickiene, K. Endothelial maintenance in health and disease: Importance of sex differences. Pharmacol. Res. 2017, 119, 48–60. [Google Scholar] [CrossRef]
- Morales, D.E.; McGowan, K.A.; Grant, D.S.; Maheshwari, S.; Bhartiya, D.; Cid, M.C.; Kleinman, H.K.; Schnaper, H.W. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation 1995, 91, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Krasinski, K.; Spyridopoulos, I.; Asahara, T.; van der Zee, R.; Isner, J.M.; Losordo, D.W. Estradiol accelerates functional endothelial recovery after arterial injury. Circulation 1997, 95, 1768–1772. [Google Scholar] [CrossRef]
- Concina, P.; Sordello, S.; Barbacanne, M.A.; Elhage, R.; Pieraggi, M.T.; Fournial, G.; Plouet, J.; Bayard, F.; Arnal, J.F. The mitogenic effect of 17β-estradiol on in vitro endothelial cell proliferation and on in vivo reendothelialization are both dependent on vascular endothelial growth factor. J. Vasc. Res. 2000, 37, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.J.; Gips, S.J.; Moldovan, N.; Wilhide, C.C.; Milliken, E.E.; Hoang, A.T.; Hruban, R.H.; Silverman, H.S.; Dang, C.V.; Goldschmidt-Clermont, P.J. 17β-estradiol inhibits apoptosis of endothelial cells. Biochem. Biophys. Res. Commun. 1997, 237, 372–381. [Google Scholar] [CrossRef]
- Spyridopoulos, I.; Sullivan, A.B.; Kearney, M.; Isner, J.M.; Losordo, D.W. Estrogen-receptor-mediated inhibition of human endothelial cell apoptosis—Estradiol as a survival factor. Circulation 1997, 95, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Scorticati, C.; Mannella, P.; Fadiel, A.; Giretti, M.S.; Fu, X.D.; Baldacci, C.; Garibaldi, S.; Caruso, A.; Fornari, L.; et al. Estrogen receptor α interacts with Gα13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway. Mol. Endocrinol. 2006, 20, 1756–1771. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, A.M.; Flamini, M.I.; Zullino, S.; Gopal, S.; Genazzani, A.R.; Simoncini, T. Estrogen receptor-α promotes endothelial cell motility through focal adhesion kinase. Mol. Hum. Reprod. 2011, 17, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Johns, A.; Freay, A.D.; Fraser, W.; Korach, K.S.; Rubanyi, G.M. Disruption of estrogen receptor gene prevents 17β estradiol-induced angiogenesis in transgenic mice. Endocrinology 1996, 137, 4511–4513. [Google Scholar] [CrossRef] [Green Version]
- Losordo, D.W.; Isner, J.M. Estrogen and angiogenesis: A review. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Missiaen, R.; Morales-Rodriguez, F.; Eelen, G.; Carmeliet, P. Targeting endothelial metabolism for anti-angiogenesis therapy: A pharmacological perspective. Vascul. Pharmacol. 2017, 90, 8–18. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [Green Version]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Ferri, N.; Cignarella, A.; Trevisi, L.; Bolego, C. The glycolytic enzyme PFKFB3 is involved in estrogen-mediated angiogenesis via GPER1. J. Pharmacol. Exp. Ther. 2017, 361, 398–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boscaro, C.; Carotti, M.; Albiero, M.; Trenti, A.; Fadini, G.P.; Trevisi, L.; Sandonà, D.; Cignarella, A.; Bolego, C. Non-genomic mechanisms in the estrogen regulation of glycolytic protein levels in endothelial cells. FASEB J. 2020, 34, 12768–12784. [Google Scholar] [CrossRef] [PubMed]
- Neves, K.B.; Montezano, A.C.; Lang, N.N.; Touyz, R.M. Vascular toxicity associated with anti-angiogenic drugs. Clin. Sci. 2020, 134, 2503–2520. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.R.; Wray, S. 17-β-Estradiol regulates expression of genes that function in macrophage activation and cholesterol homeostasis. J. Steroid Biochem. Mol. Biol. 2002, 81, 203–216. [Google Scholar] [CrossRef]
- Bolego, C.; Cignarella, A.; Staels, B.; Chinetti-Gbaguidi, G. Macrophage function and polarization in cardiovascular disease: A role of estrogen signaling? Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Härkönen, P.L.; Väänänen, H.K. Monocyte-macrophage system as a target for estrogen and selective estrogen receptor modulators. Annu. N. Y. Acad. Sci. 2006, 1089, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Campesi, I.; Marino, M.; Montella, A.; Pais, S.; Franconi, F. Sex differences in estrogen receptor α and β levels and activation status in LPS-stimulated human macrophages. J. Cell Physiol. 2017, 232, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Frazier-Jessen, M.R.; Kovacs, E.J. Estrogen modulation of JE/monocyte chemoattractant protein-1 mRNA expression in murine macrophages. J. Immunol. 1995, 154, 1838–1845. [Google Scholar]
- Friedrich, E.B.; Clever, Y.P.; Wassmann, S.; Hess, C.; Nickenig, G. 17β-estradiol inhibits monocyte adhesion via down-regulation of Rac1 GTPase. J. Mol. Cell Cardiol. 2006, 40, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Suzuki, A.; Mizuno, K.; Asada, Y.; Ino, Y.; Kuwayama, T.; Tamakoshi, K.; Mizutani, S.; Tomoda, Y. Effects of 17β-estradiol and progesterone on migration of human monocytic THP-1 cells stimulated by minimally oxidized low-density lipoprotein in vitro. Cardiovasc. Res. 1997, 34, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Huber, L.A.; Scheffler, E.; Poll, T.; Ziegler, R.; Dresel, H.A. 17 beta-estradiol inhibits LDL oxidation and cholesteryl ester formation in cultured macrophages. Free Radic. Res. Commun. 1990, 8, 167–173. [Google Scholar] [CrossRef]
- Ribas, V.; Drew, B.G.; Le, J.A.; Soleymani, T.; Daraei, P.; Sitz, D.; Mohammad, L.; Henstridge, D.C.; Febbraio, M.A.; Hewitt, S.C.; et al. Myeloid-specific estrogen receptor α deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc. Natl. Acad. Sci. USA 2011, 108, 16457–16462. [Google Scholar] [CrossRef] [Green Version]
- Hodgin, J.B.; Krege, J.H.; Reddick, R.L.; Korach, K.S.; Smithies, O.; Maeda, N. Estrogen receptor α is a major mediator of 17β-estradiol’s atheroprotective effects on lesion size in Apoe−/− mice. J. Clin. Investig. 2001, 107, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Cignarella, A.; Engel, T.; von Eckardstein, A.; Kratz, M.; Lorkowski, S.; Lueken, A.; Assmann, G.; Cullen, P. Pharmacological regulation of cholesterol efflux in human monocyte-derived macrophages in the absence of exogenous cholesterol acceptors. Atherosclerosis 2005, 179, 229–236. [Google Scholar] [CrossRef]
- Shibata, N.; Glass, C.K. Regulation of macrophage function in inflammation and atherosclerosis. J. Lipid Res. 2009, 50, S277–S281. [Google Scholar] [CrossRef] [Green Version]
- Shahoei, S.H.; Nelson, E.R. Nuclear receptors, cholesterol homeostasis and the immune system. J. Steroid Biochem. Mol. Biol. 2019, 191, 105364. [Google Scholar] [CrossRef]
- Unanue, E.R. Cooperation between mononuclear phagocytes and lymphocytes in immunity. N. Engl. J. Med. 1980, 303, 977–985. [Google Scholar] [CrossRef]
- Burger, D.; Dayer, J.M. Cytokines, acute-phase proteins, and hormones—IL-1 and TNF-α production in contact-mediated activation of monocytes by T lymphocytes. Annu. N. Y. Acad. Sci. 2002, 966, 464–473. [Google Scholar] [CrossRef]
- Mohammad, I.; Starskaia, I.; Nagy, T.; Guo, J.; Yatkin, E.; Väänänen, K.; Watford, W.T.; Chen, Z. Estrogen receptor α contributes to T cell-mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci. Signal. 2018, 11, eaap9415. [Google Scholar] [CrossRef] [Green Version]
- Benedek, G.; Zhang, J.; Nguyen, H.; Kent, G.; Seifert, H.; Vandenbark, A.A.; Offner, H. Novel feedback loop between M2 macrophages/microglia and regulatory B cells in estrogen-protected EAE mice. J. Neuroimmunol. 2017, 305, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Bongen, E.; Lucian, H.; Khatri, A.; Fragiadakis, G.K.; Bjornson, Z.B.; Nolan, G.P.; Utz, P.J.; Khatri, P. Sex differences in the blood transcriptome identify robust changes in immune cell proportions with aging and influenza infection. Cell Rep. 2019, 29, 1961–1973.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, R.; Cheung, A.S.; Pang, K.; Saffery, R.; Novakovic, B. Sexual dimorphism in innate immunity: The role of sex hormones and epigenetics. Front. Immunol. 2021, 11, 604000. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, C.M.; Toriola, A.T.; Koepl, L.M.; Sandifer, T.; Poole, E.M.; Duggan, C.; McTiernan, A.; Issa, J.P. Metabolic, hormonal and immunological associations with global DNA methylation among postmenopausal women. Epigenetics 2012, 7, 1020–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Prossnitz, E.R. Targeting the G protein-coupled estrogen receptor (GPER) in obesity and diabetes. Endocr. Metab. Sci. 2021, 2, 100080. [Google Scholar] [CrossRef]
- Calippe, B.; Douin-Echinard, V.; Delpy, L.; Laffargue, M.; Lélu, K.; Krust, A.; Pipy, B.; Bayard, F.; Arnal, J.F.; Guéry, J.C.; et al. 17β-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor α signaling in macrophages in vivo. J. Immunol. 2010, 185, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
- Campbell, L.; Emmerson, E.; Williams, H.; Saville, C.R.; Krust, A.; Chambon, P.; Mace, K.A.; Hardman, M.J. Estrogen receptor-alpha promotes alternative macrophage activation during cutaneous repair. J. Investig. Dermatol. 2014, 134, 2447–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Trevisi, L.; Bolego, C.; Cignarella, A. Estrogen, angiogenesis, immunity and cell metabolism: Solving the puzzle. Int. J. Mol. Sci. 2018, 19, 859. [Google Scholar] [CrossRef] [Green Version]
- Straub, R.H. The complex role of estrogens in inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.P.; Feng, W.; Xing, D.; Weathington, N.M.; Blalock, J.E.; Chen, Y.F.; Oparil, S. Estrogen modulates inflammatory mediator expression and neutrophil chemotaxis in injured arteries. Circulation 2004, 110, 1664–1669. [Google Scholar] [CrossRef]
- Murphy, A.J.; Guyre, P.M.; Pioli, P.A. Estradiol suppresses NF-κB activation through coordinated regulation of let-7a and miR-125b in primary human macrophages. J. Immunol. 2010, 184, 5029–5037. [Google Scholar] [CrossRef] [PubMed]
- Soucy, G.; Boivin, G.; Labrie, F.; Rivest, S. Estradiol is required for a proper immune response to bacterial and viral pathogens in the female brain. J. Immunol. 2005, 174, 6391–6398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghisletti, S.; Meda, C.; Maggi, A.; Vegeto, E. 17β-estradiol inhibits inflammatory gene expression by controlling NF-κB intracellular localization. Mol. Cell Biol. 2005, 25, 2957–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, K.C.; Curran, E.M.; Judy, B.M.; Lubahn, D.B.; Estes, D.M. Estrogen receptor-α deficiency promotes increased TNF-α secretion and bacterial killing by murine macrophages in response to microbial stimuli in vitro. J. Leukoc. Biol. 2004, 75, 1166–1172. [Google Scholar] [CrossRef] [Green Version]
- Pioli, P.A.; Jensen, A.L.; Weaver, L.K.; Amiel, E.; Shen, Z.; Shen, L.; Wira, C.R.; Guyre, P.M. Estradiol attenuates lipopolysaccharide-induced CXC chemokine ligand 8 production by human peripheral blood monocytes. J. Immunol. 2007, 179, 6284–6290. [Google Scholar] [CrossRef] [Green Version]
- Kramer, P.R.; Kramer, S.F.; Guan, G. 17β-Estradiol regulates cytokine release through modulation of CD16 expression in monocytes and monocyte-derived macrophages. Arthritis Rheum. 2004, 50, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.R.; Winger, V.; Kramer, S.F. 17β-Estradiol utilizes the estrogen receptor to regulate CD16 expression in monocytes. Mol. Cell Endocrinol. 2007, 279, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Shimizu, T.; Yu, H.P.; Hsieh, Y.C.; Choudhry, M.A.; Bland, K.I.; Chaudry, I.H. Estrogen receptor-α predominantly mediates the salutary effects of 17β-estradiol on splenic macrophages following trauma-hemorrhage. Am. J. Physiol. Cell Physiol. 2007, 293, C978–C984. [Google Scholar] [CrossRef]
- Xing, D.; Oparil, S.; Yu, H.; Gong, K.; Feng, W.; Black, J.; Chen, Y.F.; Nozell, S. Estrogen modulates NFκB signaling by enhancing IκBα levels and blocking p65 binding at the promoters of inflammatory genes via estrogen receptor-β. PLoS ONE 2012, 7, e36890. [Google Scholar] [CrossRef]
- Liu, Y.; Mei, C.; Liu, H.; Wang, H.; Zeng, G.; Lin, J.; Xu, M. Modulation of cytokine expression in human macrophages by endocrine-disrupting chemical bisphenol-A. Biochem. Biophys. Res. Commun. 2014, 451, 592–598. [Google Scholar] [CrossRef]
- Pepe, G.; Braga, D.; Renzi, T.A.; Villa, A.; Bolego, C.; D’Avila, F.; Barlassina, C.; Maggi, A.; Locati, M.; Vegeto, E. Self-renewal and phenotypic conversion are the main physiological responses of macrophages to the endogenous estrogen surge. Sci. Rep. 2017, 7, 44270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J. Macrophage polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Bowling, M.R.; Xing, D.; Kapadia, A.; Chen, Y.F.; Szalai, A.J.; Oparil, S.; Hage, F.G. Estrogen effects on vascular inflammation are age dependent: Role of estrogen receptors. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1477–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopinska-Gluszak, U.; Waligora, J.; Grzela, T.; Gluszak, M.; Jozwiak, J.; Radomski, D.; Roszkowski, P.I.; Malejczyk, J. Effect of estrogen/progesterone hormone replacement therapy on natural killer cell cytotoxicity and immunoregulatory cytokine release by peripheral blood mononuclear cells of postmenopausal women. J. Reprod. Immunol. 2006, 69, 65–75. [Google Scholar] [CrossRef]
- Corcoran, M.P.; Meydani, M.; Lichtenstein, A.H.; Schaefer, E.J.; Dillard, A.; Lamon-Fava, S. Sex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal women. J. Endocrinol. 2010, 206, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoni, E.; Guignard, L.; Knaup Reymond, M.; Perreau, M.; Roth-Kleiner, M.; Calandra, T.; Roger, T. Estradiol and progesterone strongly inhibit the innate immune response of mononuclear cells in newborns. Infect. Immun. 2011, 79, 2690–2698. [Google Scholar] [CrossRef] [Green Version]
- Pechenino, A.S.; Lin, L.; Mbai, F.N.; Lee, A.R.; He, X.M.; Stallone, J.N.; Knowlton, A.A. Impact of aging vs. estrogen loss on cardiac gene expression: Estrogen replacement and inflammation. Physiol. Genom. 2011, 43, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Boardman, H.M.; Hartley, L.; Eisinga, A.; Main, C.; Roqué i Figuls, M.; Bonfill Cosp, X.; Gabriel Sanchez, R.; Knight, B. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst. Rev. 2015, 3, CD002229. [Google Scholar] [CrossRef] [PubMed]
- Stossi, F.; Madak-Erdoğan, Z.; Katzenellenbogen, B. Macrophage-elicited loss of estrogen receptor-α in breast cancer cells via involvement of MAPK and c-Jun at the ESR1 genomic locus. Oncogene 2012, 31, 1825–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Q.; Gao, R.; Peng, C.; Yi, J.; Liu, L.; Yang, S.; Li, D.; Hu, J.; Luo, T.; Mei, M.; et al. Bisphenol A promotes hepatic lipid deposition involving Kupffer cells M1 polarization in male mice. J. Endocrinol. 2017, 234, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, T.; Li, L. Stem cells and their niche: An inseparable relationship. Development 2007, 134, 2001–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Aguilera, A.; Arranz, L.; Martín-Pérez, D.; García-García, A.; Stavropoulou, V.; Kubovcakova, L.; Isern, J.; Martín-Salamanca, S.; Langa, X.; Skoda, R.C.; et al. Estrogen signaling selectively induces apoptosis of hematopoietic progenitors and myeloid neoplasms without harming steady-state hematopoiesis. Cell Stem Cell 2014, 15, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Nakada, D.; Oguro, H.; Levi, B.P.; Ryan, N.; Kitano, A.; Saitoh, Y.; Takeichi, M.; Wendt, G.R.; Morrison, S.J. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature 2014, 505, 555–558. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Albiero, M.; Ciciliot, S.; Tedesco, S.; Menegazzo, L.; D’Anna, M.; Scattolini, V.; Cappellari, R.; Zuccolotto, G.; Rosato, A.; Cignarella, A.; et al. Diabetes-associated myelopoiesis drives stem cell mobilopathy through an OSM-p66Shc signaling pathway. Diabetes 2019, 68, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Kraakman, M.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.S.; Abdel-Latif, A.; Smyth, S.S.; et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014, 19, 821–835. [Google Scholar] [CrossRef] [Green Version]
- Fest, J.; Ruiter, T.R.; Groot Koerkamp, B.; Rizopoulos, D.; Ikram, M.A.; van Eijck, C.H.J.; Stricker, B.H. The neutrophil-to-lymphocyte ratio is associated with mortality in the general population: The Rotterdam Study. Eur. J. Epidemiol. 2019, 34, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Gaunt, S.D.; Pierce, K.R. Myelopoiesis and marrow adherent cells in estradiol-treated mice. Vet. Pathol. 1985, 22, 403–408. [Google Scholar] [CrossRef]
- Gaunt, S.D.; Pierce, K.R. Effects of estradiol on hematopoietic and marrow adherent cells of dogs. Am. J. Vet. Res. 1986, 47, 906–909. [Google Scholar]
- Farris, G.M.; Benjamin, S.A. Inhibition of myelopoiesis by conditioned medium from cultured canine thymic cells exposed to estrogen. Am. J. Vet. Res. 1993, 54, 1366–1373. [Google Scholar] [PubMed]
- Thurmond, T.S.; Murante, F.G.; Staples, J.E.; Silverstone, A.E.; Korach, K.S.; Gasiewicz, T.A. Role of estrogen receptor α in hematopoietic stem cell development and B lymphocyte maturation in the male mouse. Endocrinology 2000, 141, 2309–2318. [Google Scholar] [CrossRef]
- Chapple, R.H.; Hu, T.; Tseng, Y.J.; Liu, L.; Kitano, A.; Luu, V.; Hoegenauer, K.A.; Iwawaki, T.; Li, Q.; Nakada, D. ERα promotes murine hematopoietic regeneration through the Ire1α-mediated unfolded protein response. eLife 2018, 7, e31159. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, F.F.; Zhang, X.; Coppin, E.; Vasamsetti, S.B.; Modugu, G.; Schloss, M.J.; Rohde, D.; McAlpine, C.S.; Iwamoto, Y.; Libby, P.; et al. Bone marrow endothelial cells regulate myelopoiesis in diabetes mellitus. Circulation 2020, 142, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.P. Regulation of signal transduction pathways by estrogen and progesterone. Annu. Rev. Physiol. 2005, 67, 335–376. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prossnitz, E.R.; Sklar, L.A.; Oprea, T.I.; Arterburn, J.B. GPR30: A novel therapeutic target in estrogen-related disease. Trends Pharmacol. Sci. 2008, 29, 116–123. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Zuckerman, S.H.; Bryan, N. Inhibition of LDL oxidation and myeloperoxidase dependent tyrosyl radical formation by the selective estrogen receptor modulator raloxifene (LY139481 HCL). Atherosclerosis 1996, 126, 65–75. [Google Scholar] [CrossRef]
- Yu, J.; Eto, M.; Akishita, M.; Okabe, T.; Ouchi, Y. A selective estrogen receptor modulator inhibits TNF-α-induced apoptosis by activating ERK1/2 signaling pathway in vascular endothelial cells. Vascul. Pharmacol. 2009, 51, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Lv, Y.; Tang, Z.; Nie, F.; Huang, P.; Pei, Q.; Guo, R. Bazedoxifene plays a protective role against inflammatory injury of endothelial cells by targeting CD40. Cardiovasc Ther. 2020, 2020, 1795853. [Google Scholar] [CrossRef] [PubMed]
- Bolego, C.; Vegeto, E.; Pinna, C.; Maggi, A.; Cignarella, A. Selective agonists of estrogen receptor isoforms: New perspectives for cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2192–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolego, C.; Rossoni, G.; Fadini, G.P.; Vegeto, E.; Pinna, C.; Albiero, M.; Boscaro, E.; Agostini, C.; Avogaro, A.; Gaion, R.M.; et al. Selective estrogen receptor-α agonist provides widespread heart and vascular protection with enhanced endothelial progenitor cell mobilization in the absence of uterotrophic action. FASEB J. 2010, 24, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Hu, C.; Staquicini, D.I.; Brigman, J.L.; Liu, M.; Mauvais-Jarvis, F.; Pasqualini, R.; Arap, W.; Arterburn, J.B.; Hathaway, H.J.; et al. Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Abot, A.; Fontaine, C.; Buscato, M.; Solinhac, R.; Flouriot, G.; Fabre, A.; Drougard, A.; Rajan, S.; Laine, M.; Milon, A.; et al. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor α modulation, uncoupling nuclear and membrane activation. EMBO Mol. Med. 2014, 6, 1328–1346. [Google Scholar] [CrossRef]
- Buscato, M.; Davezac, M.; Zahreddine, R.; Adlanmerini, M.; Métivier, R.; Fillet, M.; Cobraiville, G.; Moro, C.; Foidart, J.M.; Lenfant, F.; et al. Estetrol prevents Western diet-induced obesity and atheroma independently of hepatic estrogen receptor α. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E19–E29. [Google Scholar] [CrossRef]
- Abderrahman, B.; Maximov, P.Y.; Curpan, R.F.; Hanspal, J.S.; Fan, P.; Xiong, R.; Tonetti, D.A.; Thatcher, G.R.J.; Jordan, V.C. Pharmacology and molecular mechanisms of clinically relevant estrogen estetrol and estrogen mimic BMI-135 for the treatment of endocrine-resistant breast cancer. Mol. Pharmacol. 2020, 98, 364–381. [Google Scholar] [CrossRef]
- Coelingh Bennink, H.J.; Verhoeven, C.; Zimmerman, Y.; Visser, M.; Foidart, J.M.; Gemzell-Danielsson, K. Clinical effects of the fetal estrogen estetrol in a multiple-rising-dose study in postmenopausal women. Maturitas 2016, 91, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Wang, L.; Nelson, A.T.; Han, C.; He, S.; Henn, M.A.; Menon, K.; Chen, J.J.; Baek, A.E.; Vardanyan, A.; et al. 27-Hydroxycholesterol acts on myeloid immune cells to induce T cell dysfunction, promoting breast cancer progression. Cancer Lett. 2020, 493, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Houben, T.; Bitorina, A.V.; Oligschlaeger, Y.; Jeurissen, M.L.; Rensen, S.; Köhler, S.E.; Westerterp, M.; Lütjohann, D.; Theys, J.; Romano, A.; et al. Sex-opposed inflammatory effects of 27-hydroxycholesterol are mediated via differences in estrogen signaling. J. Pathol. 2020, 251, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Umetani, M.; Domoto, H.; Gormley, A.K.; Yuhanna, I.S.; Cummins, C.L.; Javitt, N.B.; Korach, K.S.; Shaul, P.W.; Mangelsdorf, D.J. 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat. Med. 2007, 13, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Kitazato, K.T.; Kinouchi, T.; Yagi, K.; Tada, Y.; Satomi, J.; Kageji, T.; Nagahiro, S. Activation of estrogen receptor-α and of angiotensin-converting enzyme 2 suppresses ischemic brain damage in oophorectomized rats. Hypertension 2011, 57, 1161–1166. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dama, A.; Baggio, C.; Boscaro, C.; Albiero, M.; Cignarella, A. Estrogen Receptor Functions and Pathways at the Vascular Immune Interface. Int. J. Mol. Sci. 2021, 22, 4254. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084254
Dama A, Baggio C, Boscaro C, Albiero M, Cignarella A. Estrogen Receptor Functions and Pathways at the Vascular Immune Interface. International Journal of Molecular Sciences. 2021; 22(8):4254. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084254
Chicago/Turabian StyleDama, Aida, Chiara Baggio, Carlotta Boscaro, Mattia Albiero, and Andrea Cignarella. 2021. "Estrogen Receptor Functions and Pathways at the Vascular Immune Interface" International Journal of Molecular Sciences 22, no. 8: 4254. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084254