
Cyclin-Dependent Kinase Inhibitor BMI-1026 Induces Apoptosis by Downregulating Mcl-1 (L) and c-FLIP (L) and Inactivating p-Akt in Human Renal Carcinoma Cells

 ,
,
Abstract
:1. Introduction
2. Results
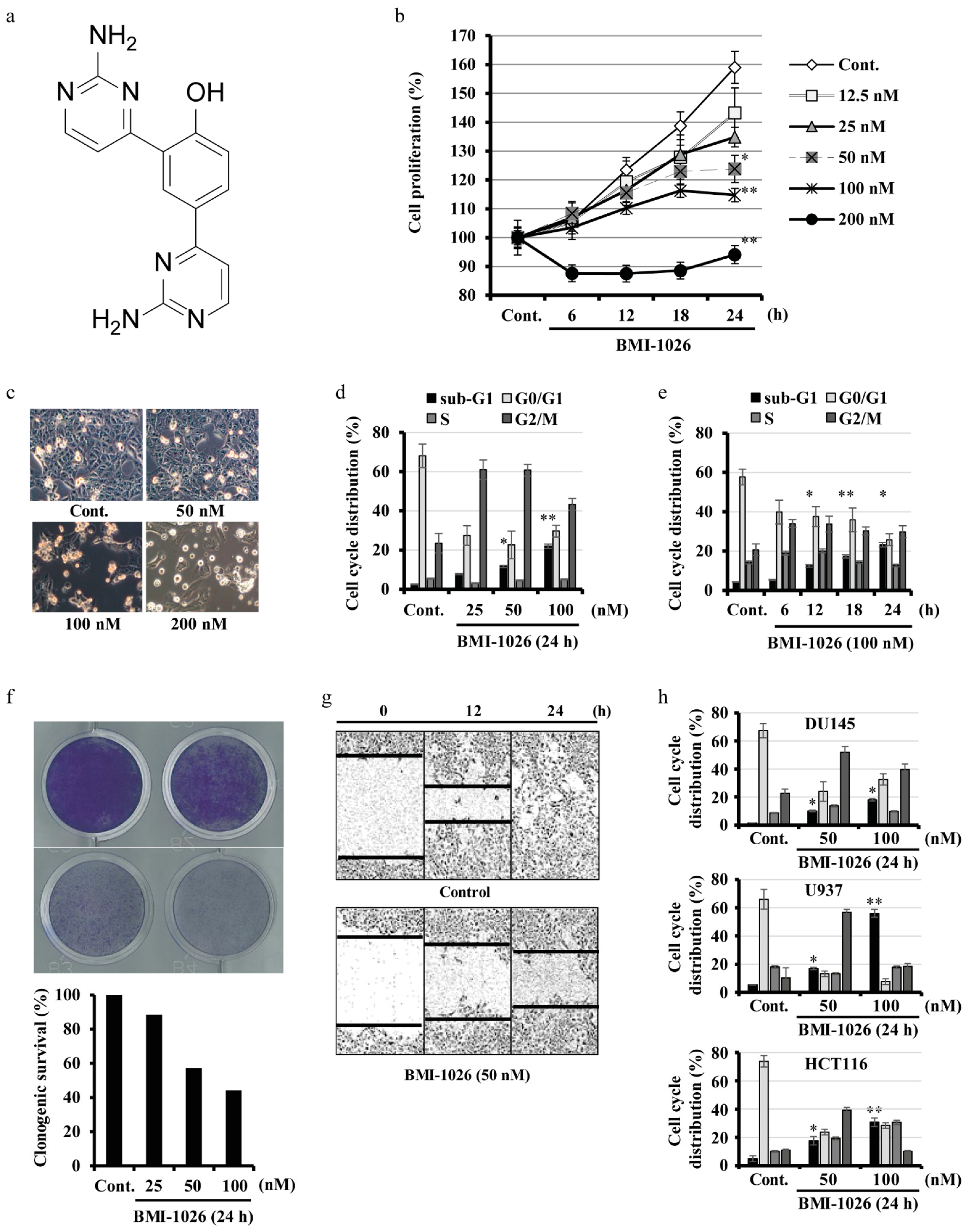
2.1. BMI-1026 Has Antiproliferative Effects and Induces Apoptosis in Various Human Cancer Cells
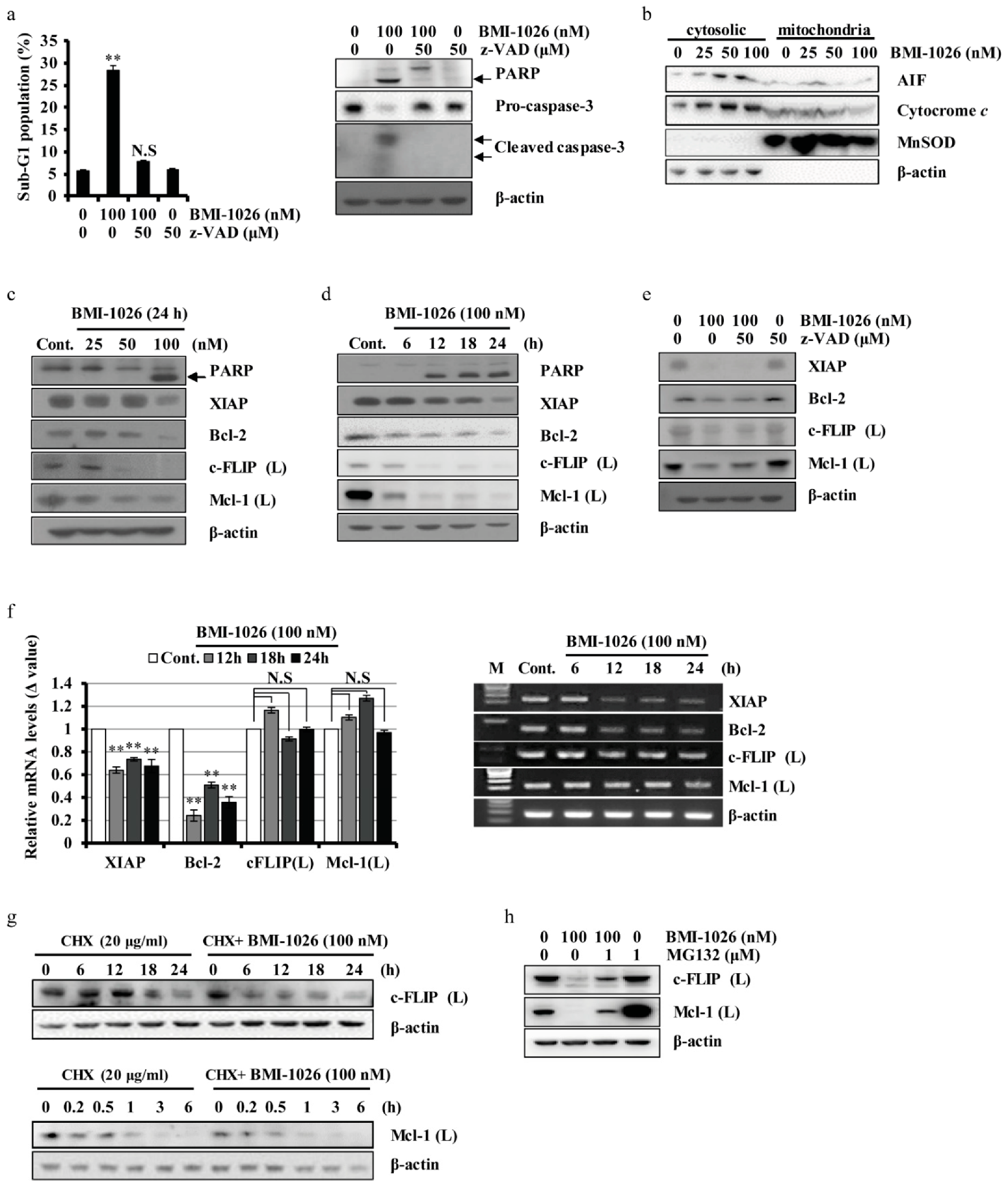
2.2. BMI-1026 Regulates Apoptosis-Related Proteins in Caki Cells
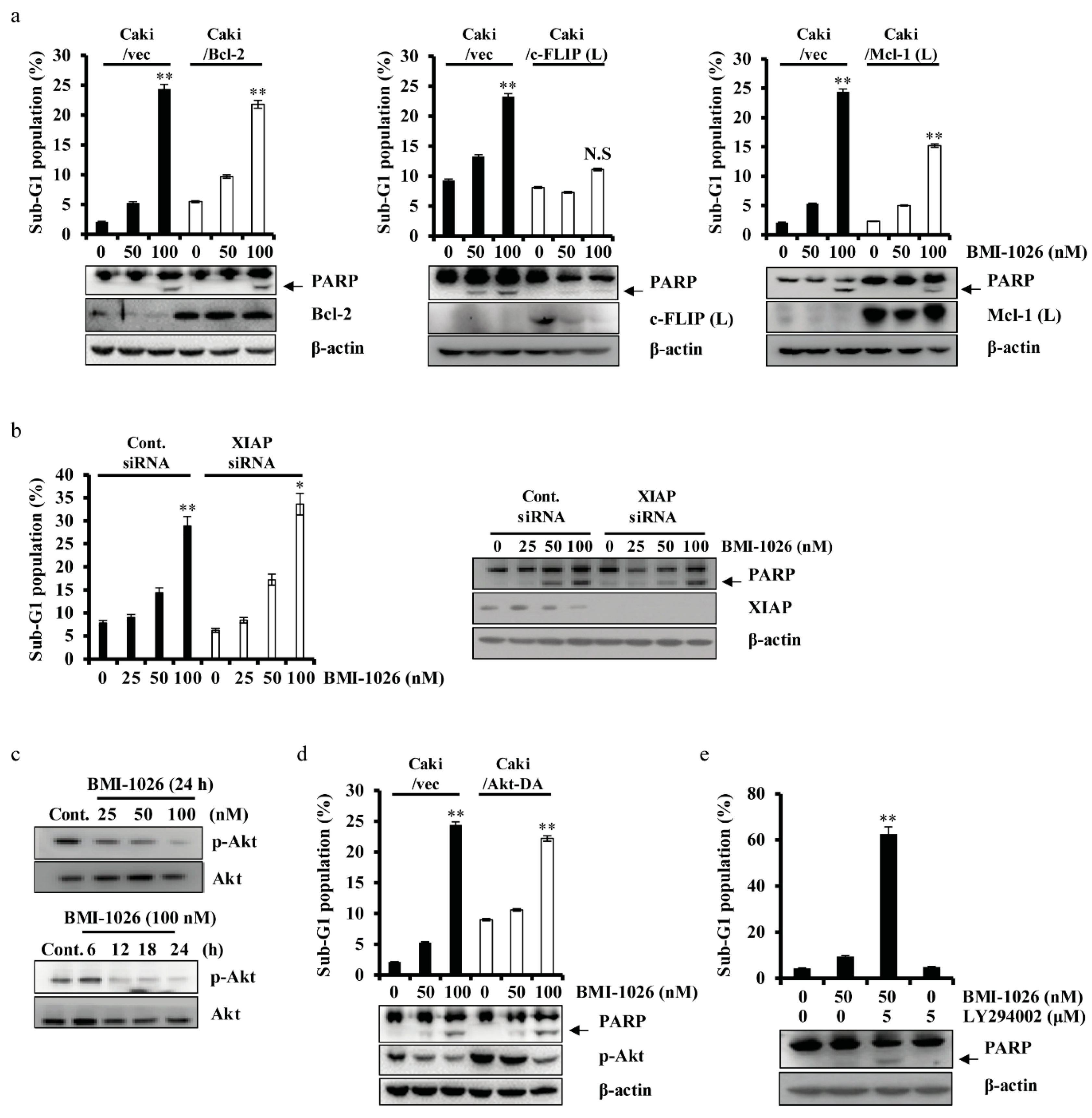
2.3. BMI-1026-Induced Apoptosis Is Associated with Various Apoptosis-Related Proteins in Caki Cells
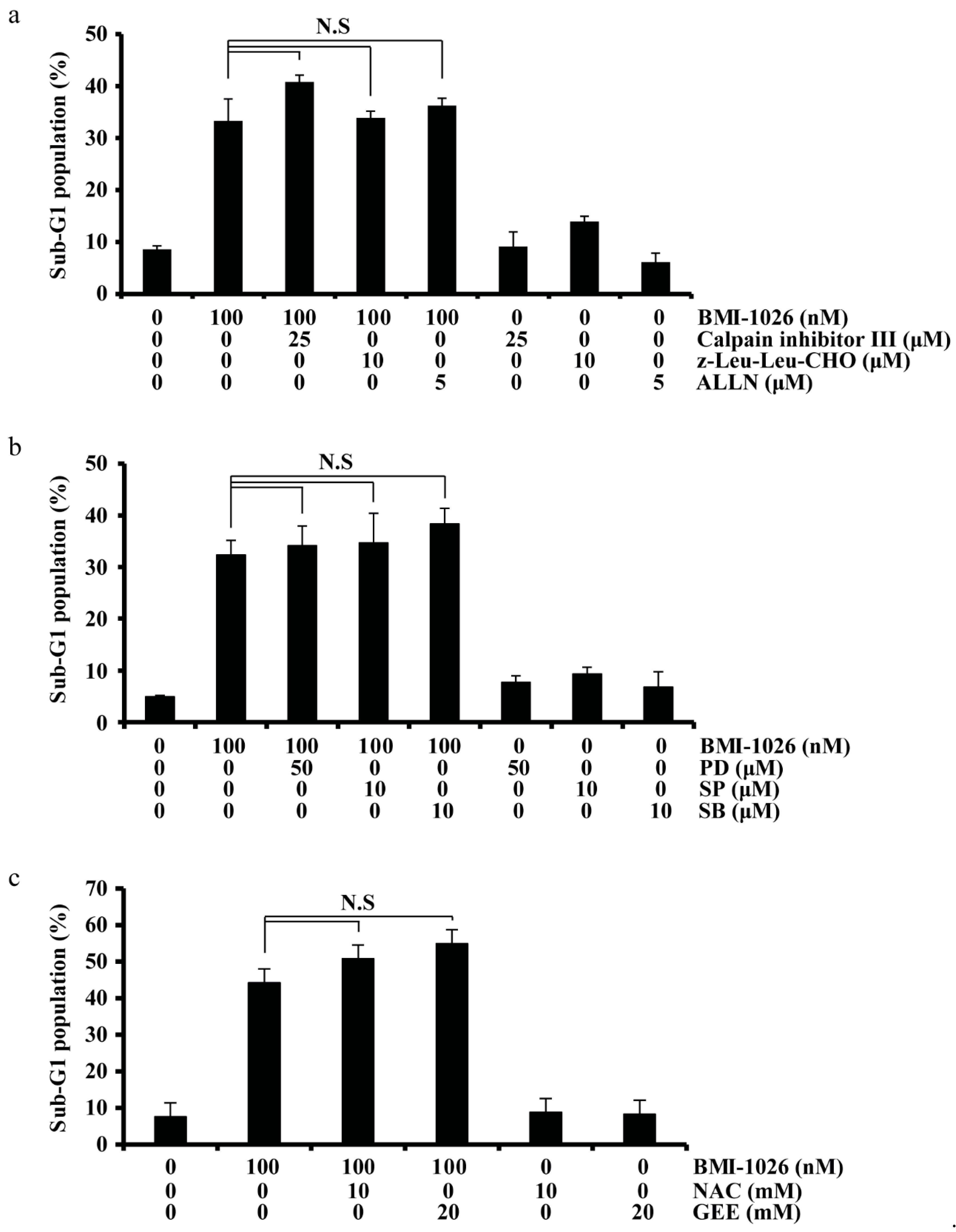
2.4. BMI-1026-Induced Apoptosis Is Independent of Calpain, MAPK, and ROS Signaling Pathways
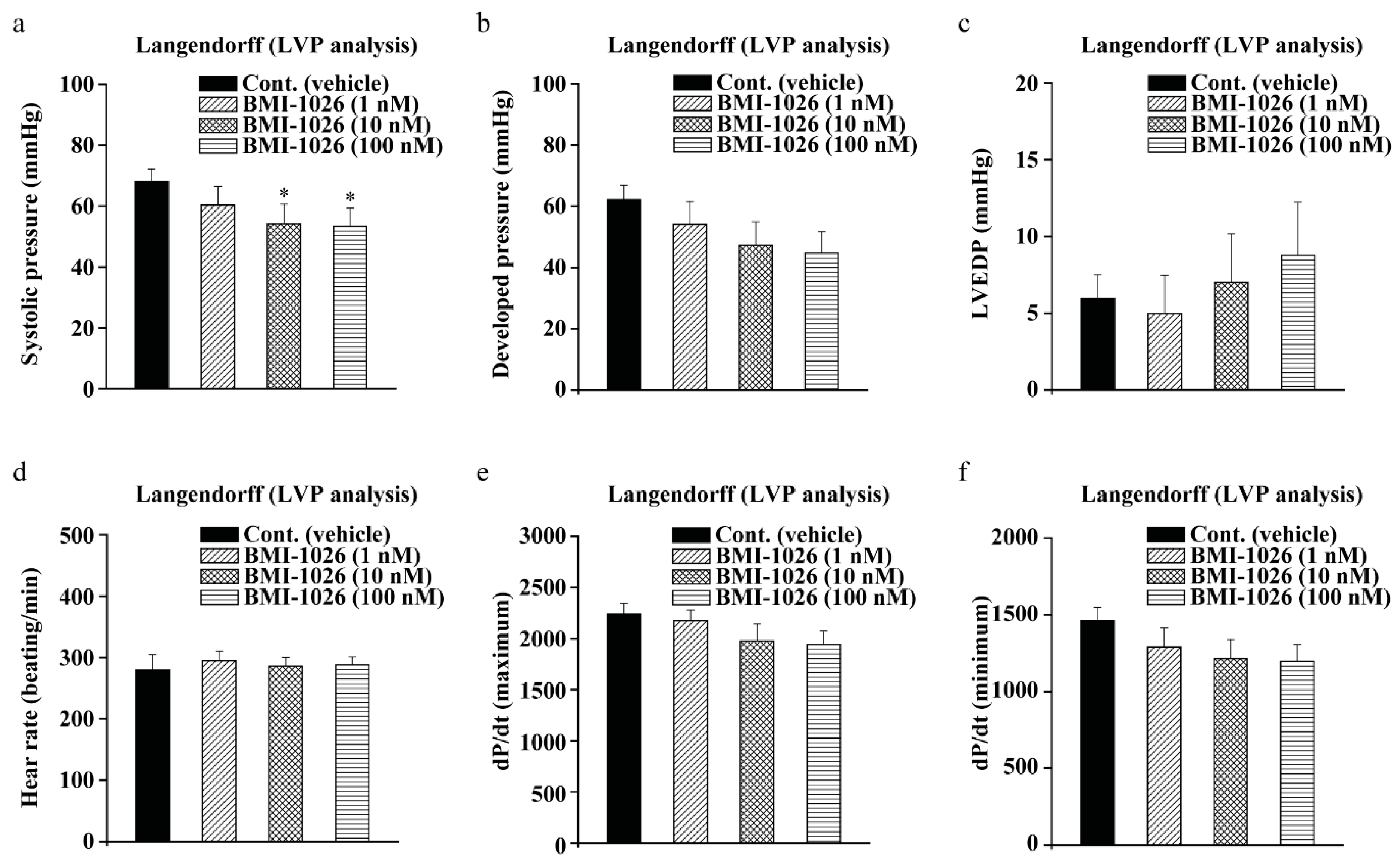
2.5. BMI-1026 Has Electrophysiological Safety
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Drugs and Materials
4.3. Western Blotting Analysis
4.4. Flow Cytometric Analysis
4.5. Analysis of Mitochondrial AIF and Cytochrome c Release
4.6. Reverse Transcription PCR
4.7. RNA Isolation and Reverse Transcriptase-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.8. Establishment of Stable Cell Lines Overexpressing Bcl-2, Mcl-1 (L), c-FLIP (L), and Constitutively Active Akt Constructs
4.9. siRNA
4.10. Recording of LVP in Rats
4.11. Clonogenic Assay
4.12. Cell Imaging System
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Cdk1 | cyclin-dependent kinase 1 |
| AIF | apoptosis-inducing factor |
| XIAP | X-linked inhibitor of apoptosis protein |
| c-FLIP | cellular FADD-like IL-1β-converting enzyme inhibitory protein |
| SD | Standard deviation |
| TdP | torsade de pointes |
| hERG | human ether-à-go-go-related gene |
| LVP | left ventricular pressure |
| z-VAD-fmk | benzyloxy carbony-Val-Ala-Asp-fluoromethyl ketone |
| PARP | poly(ADP-ribose) polymerase |
| PCR | polymerase chain reaction |
| siRNA | small interfering RNA |
| PUMA | p53 upregulated modulator of apoptosis |
| ALLN | Ac-Leu-Leu-Nle-CHO |
| MAPK | mitogen-activated protein kinase |
| p-Akt | phospho-Akt |
| ATCC | the American Type Culture Collection |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | fetal bovine serum |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| PBS | phosphate-buffered saline |
| RT-qPCR | RNA isolation and reverse transcriptase-quantitative polymerase chain reaction |
| ANOVA | analysis of variance |
References
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharm. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [Green Version]
- Dancey, J.; Sausville, E.A. Issues and progress with protein kinase inhibitors for cancer treatment. Nat. Rev. Drug Discov. 2003, 2, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Seong, Y.S.; Min, C.; Li, L.; Yang, J.Y.; Kim, S.Y.; Cao, X.; Kim, K.; Yuspa, S.H.; Chung, H.H.; Lee, K.S. Characterization of a novel cyclin-dependent kinase 1 inhibitor, BMI-1026. Cancer Res. 2003, 63, 7384–7391. [Google Scholar]
- Seo, H.J.; Park, H.J.; Choi, H.S.; Hwang, S.Y.; Park, J.S.; Seong, Y.S. BMI-1026 treatment can induce SAHF formation by activation of Erk1/2. BMB Rep. 2008, 41, 523–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niiya, F.; Tatsumoto, T.; Lee, K.S.; Miki, T. Phosphorylation of the cytokinesis regulator ECT2 at G2/M phase stimulates association of the mitotic kinase Plk1 and accumulation of GTP-bound RhoA. Oncogene 2006, 25, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Choi, T.-S. Effects of BMI-1026, a potent CDK inhibitor, on murine oocyte maturation and metaphase II arrest. Reprod. Dev. Biol. 2007, 31, 71–76. [Google Scholar]
- Niiya, F.; Xie, X.; Lee, K.S.; Inoue, H.; Miki, T. Inhibition of cyclin-dependent kinase 1 induces cytokinesis without chromosome segregation in an ECT2 and MgcRacGAP-dependent manner. J. Biol. Chem. 2005, 280, 36502–36509. [Google Scholar] [CrossRef] [Green Version]
- Sarazan, R.D.; Mittelstadt, S.; Guth, B.; Koerner, J.; Zhang, J.; Pettit, S. Cardiovascular function in nonclinical drug safety assessment: Current issues and opportunities. Int. J. Toxicol. 2011, 30, 272–286. [Google Scholar] [CrossRef]
- Redfern, W.S.; Carlsson, L.; Davis, A.S.; Lynch, W.G.; MacKenzie, I.; Palethorpe, S.; Siegl, P.K.; Strang, I.; Sullivan, A.T.; Wallis, R.; et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. [Google Scholar] [CrossRef]
- Norton, K.; Iacono, G.; Vezina, M. Assessment of the pharmacological effects of inotropic drugs on left ventricular pressure and contractility: An evaluation of the QA interval as an indirect indicator of cardiac inotropism. J. Pharm. Toxicol. Methods 2009, 60, 193–197. [Google Scholar] [CrossRef]
- Ben-David, J.; Zipes, D.P. Torsades de pointes and proarrhythmia. Lancet 1993, 341, 1578–1582. [Google Scholar] [CrossRef]
- Keating, M.T.; Sanguinetti, M.C. Molecular genetic insights into cardiovascular disease. Sci. N. Y. 1996, 272, 681–685. [Google Scholar] [CrossRef]
- Le Bras, M.; Rouy, I.; Brenner, C. The modulation of inter-organelle cross-talk to control apoptosis. Med. Chem. 2006, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [Green Version]
- Hikisz, P.; Kilianska, Z.M. PUMA, a critical mediator of cell death--one decade on from its discovery. Cell Mol. Biol. Lett. 2012, 17, 646–669. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Shin, K.J.; Bae, S.S.; Hwang, Y.A.; Seo, J.K.; Ryu, S.H.; Suh, P.G. 2,2′,4,6,6′-pentachlorobiphenyl induces apoptosis in human monocytic cells. Toxicol. Appl. Pharmacol. 2000, 169, 1–7. [Google Scholar] [CrossRef]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar]
- Reed, J.C. Bcl-2 and the regulation of programmed cell death. J. Cell Biol. 1994, 124, 1–6. [Google Scholar] [CrossRef]
- Bae, J.; Donigian, J.R.; Hsueh, A.J. Tankyrase 1 interacts with Mcl-1 proteins and inhibits their regulation of apoptosis. J. Biol. Chem. 2003, 278, 5195–5204. [Google Scholar] [CrossRef] [Green Version]
- Safa, A.R.; Day, T.W.; Wu, C.H. Cellular FLICE-like inhibitory protein (C-FLIP): A novel target for cancer therapy. Curr. Cancer Drug Targets 2008, 8, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Schimmer, A.D.; Dalili, S.; Batey, R.A.; Riedl, S.J. Targeting XIAP for the treatment of malignancy. Cell Death Differ. 2006, 13, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N.; et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003, 4, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Sci. N. Y. 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Rahmani, M.; Dai, Y.; Conrad, D.; Krystal, G.; Dent, P.; Grant, S. The lethal effects of pharmacological cyclin-dependent kinase inhibitors in human leukemia cells proceed through a phosphatidylinositol 3-kinase/Akt-dependent process. Cancer Res. 2003, 63, 1822–1833. [Google Scholar]
- Dutta, S.; Chang, K.C.; Beattie, K.A.; Sheng, J.; Tran, P.N.; Wu, W.W.; Wu, M.; Strauss, D.G.; Colatsky, T.; Li, Z. Optimization of an In silico Cardiac Cell Model for Proarrhythmia Risk Assessment. Front. Physiol. 2017, 8, 616. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequences * |
|---|---|
| XIAP sense | 5′-GCTTGCAAGAGCTGGATTTT-3′ |
| XIAP antisense | 5′-TGGCTTCCAATCCGTGAG-3′ |
| Bcl-2 sense | 5′-CAGGTATGCACCCAGAGTGA-3′ |
| Bcl-2 antisense | 5′-GTCTCT GAAGACGCTGCTCA-3′ |
| c-FLIP (L) sense | 5′-TGCTGAAGTCATCCATCAGG-3′ |
| c-FLIP (L) antisense | 5′-ATTCCTAGGGGC TTGCTCT-3′ |
| Mcl-1 (L) sense | 5′-GCGACTGGCAAAGCTTGGCCTCAA-3′ |
| Mcl-1 (L) antisense | 5′-CAACTCTAGAAACTGGTTTTGGTG-3′ |
| β-actin sense | 5′-AATCTGGCACCACACCTTCTA-3′ |
| β-actin antisense | 5′-ATAGCACAGCCTGGATAGCAA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.E.; Lee, J.; Park, J.W.; Kang, H.; Nam, Y.R.; Kwon, T.K.; Kim, K.-S.; Kim, S. Cyclin-Dependent Kinase Inhibitor BMI-1026 Induces Apoptosis by Downregulating Mcl-1 (L) and c-FLIP (L) and Inactivating p-Akt in Human Renal Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 4268. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084268
Kim DE, Lee J, Park JW, Kang H, Nam YR, Kwon TK, Kim K-S, Kim S. Cyclin-Dependent Kinase Inhibitor BMI-1026 Induces Apoptosis by Downregulating Mcl-1 (L) and c-FLIP (L) and Inactivating p-Akt in Human Renal Carcinoma Cells. International Journal of Molecular Sciences. 2021; 22(8):4268. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084268
Chicago/Turabian StyleKim, Dong Eun, Jinho Lee, Jong Wook Park, Hyunsu Kang, Yu Ri Nam, Taeg Kyu Kwon, Ki-Suk Kim, and Shin Kim. 2021. "Cyclin-Dependent Kinase Inhibitor BMI-1026 Induces Apoptosis by Downregulating Mcl-1 (L) and c-FLIP (L) and Inactivating p-Akt in Human Renal Carcinoma Cells" International Journal of Molecular Sciences 22, no. 8: 4268. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084268