
Celecoxib as a Valuable Adjuvant in Cutaneous Melanoma Treated with Trametinib

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Melanoma Co-Culture Bioassays
Cell Co-Culture and Reagents
2.2. Cytotoxicity Assay
2.3. Experimental Design
3. Results
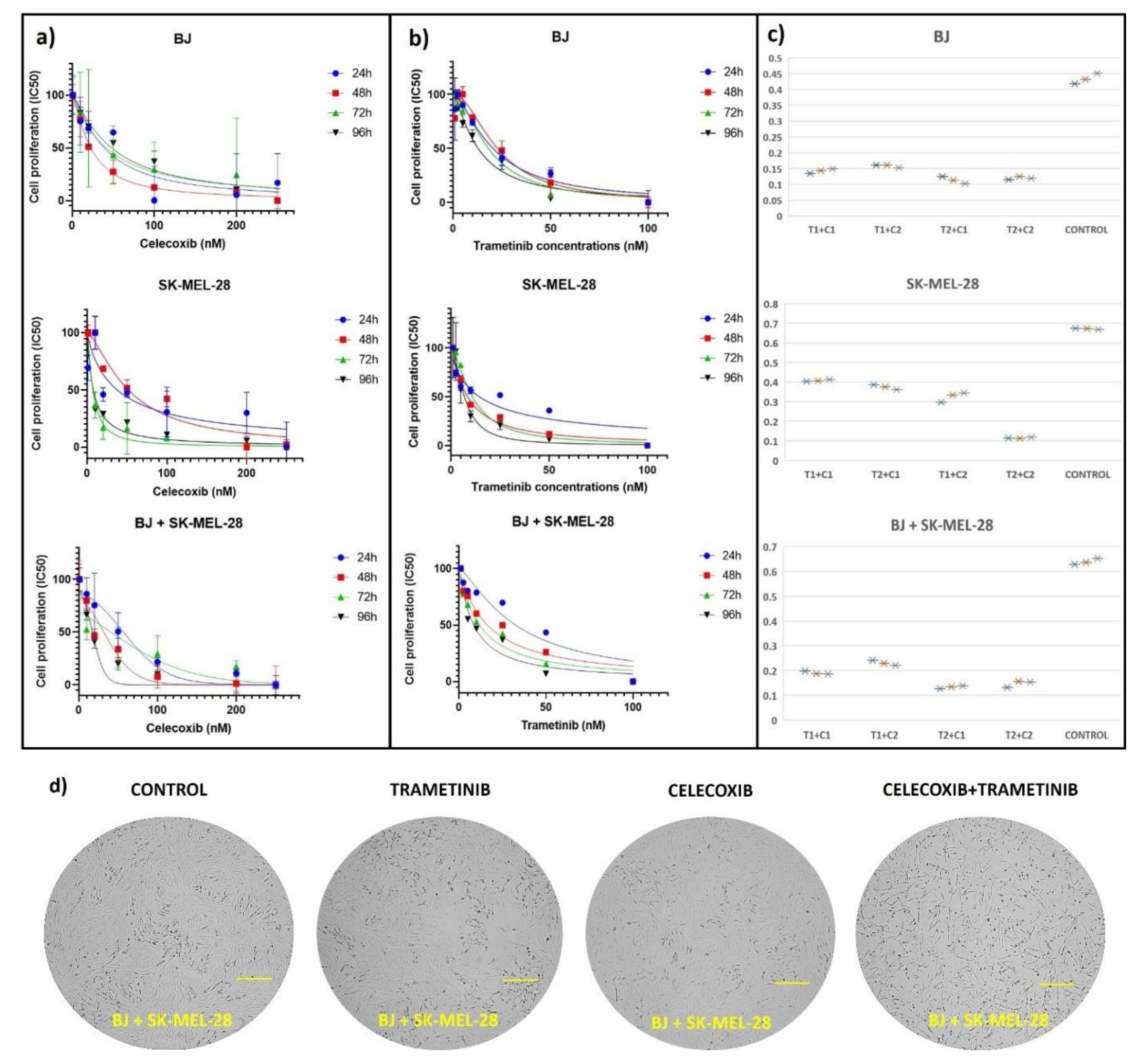
3.1. Cell Viability Assay
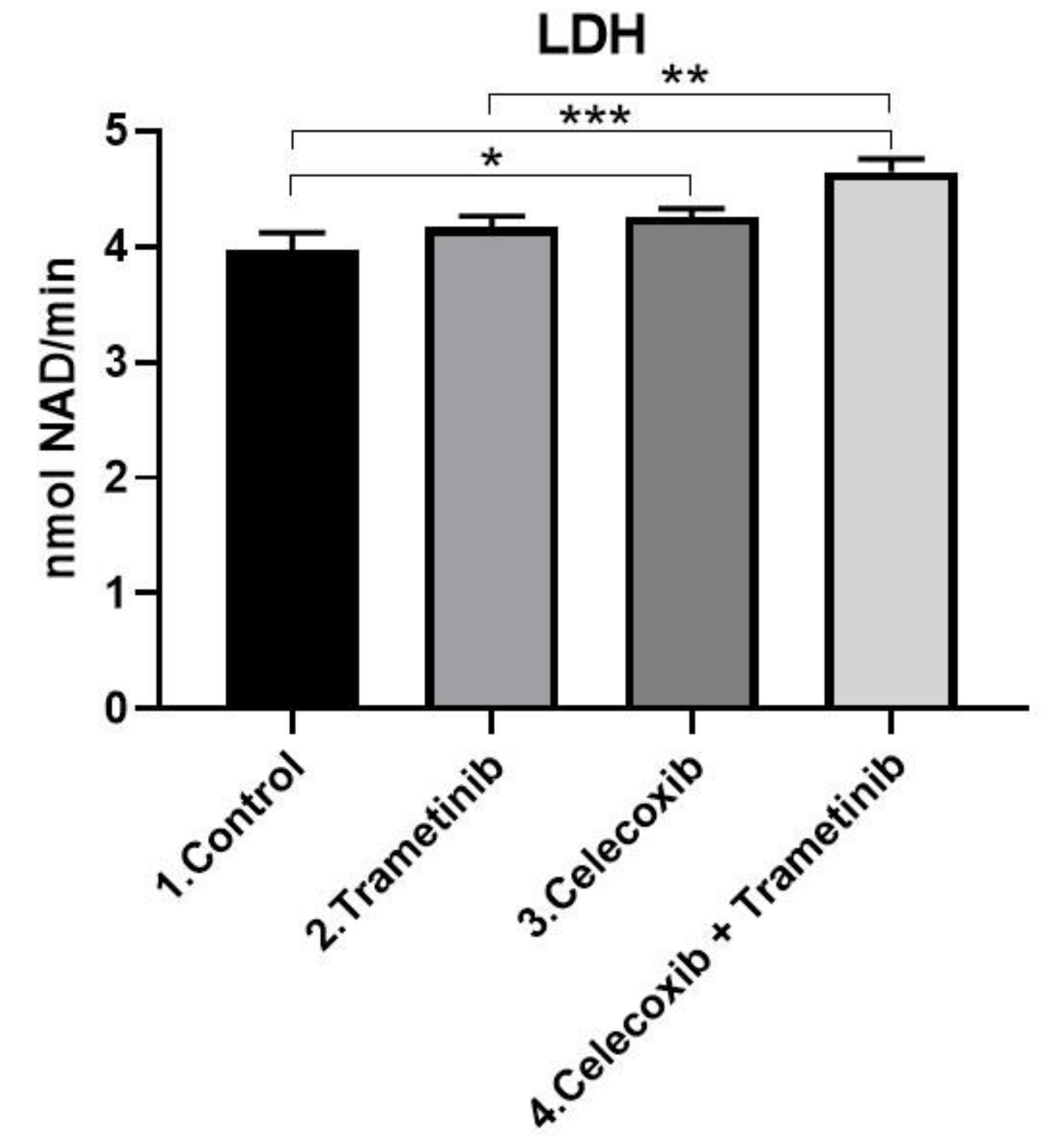
3.2. Cell Membrane Integrity Assay
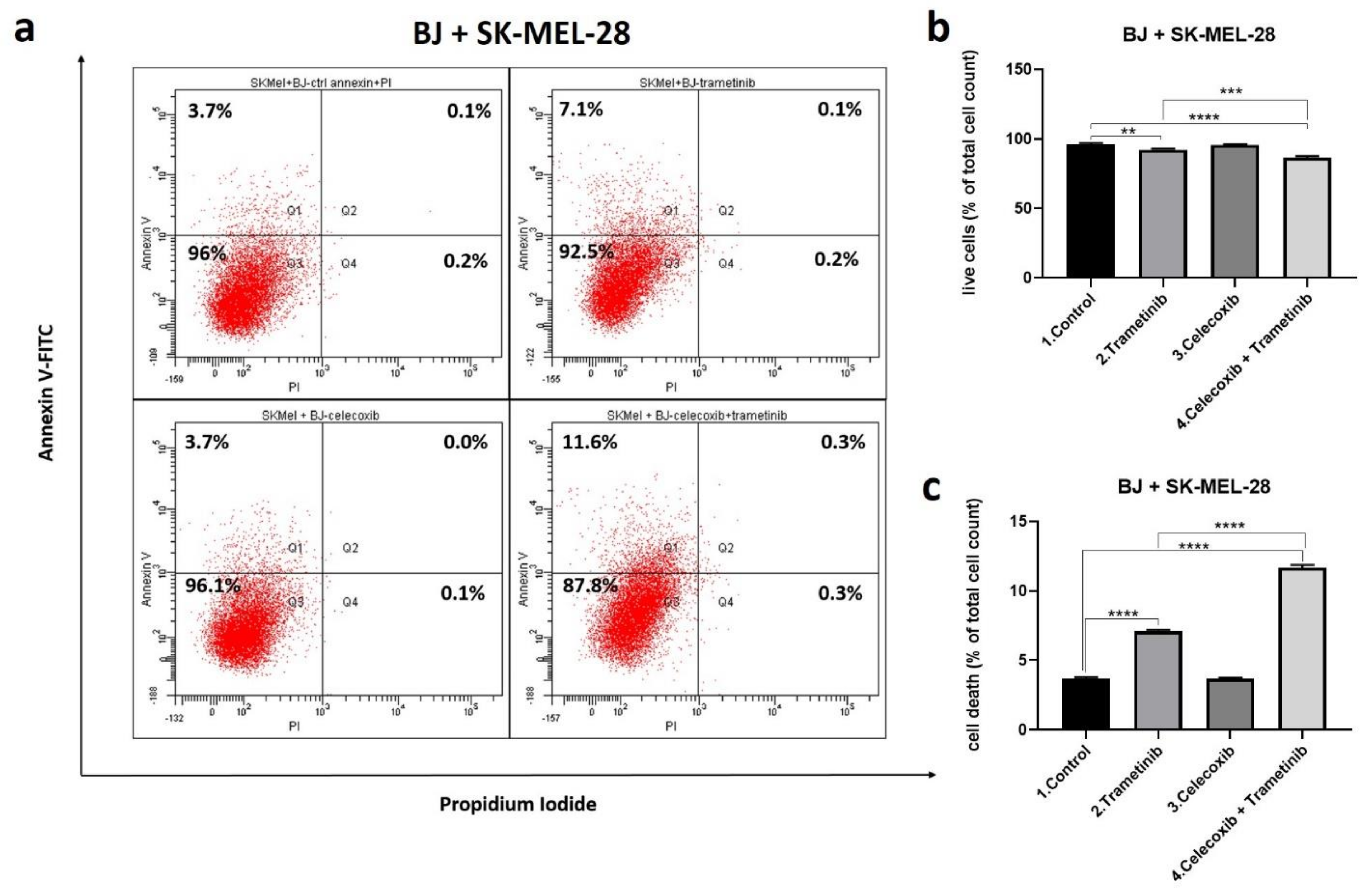
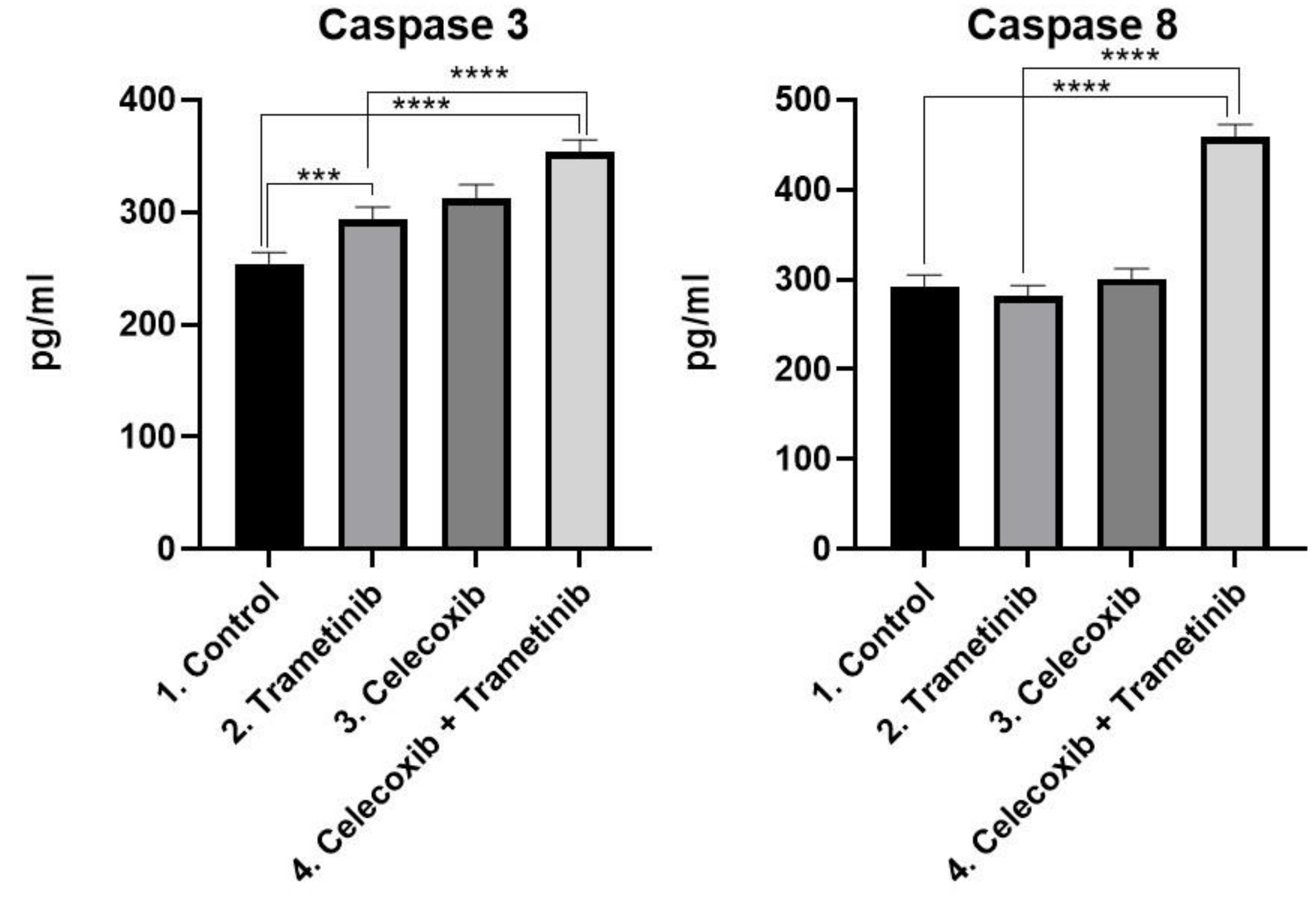
3.3. Cell Death Mechanism
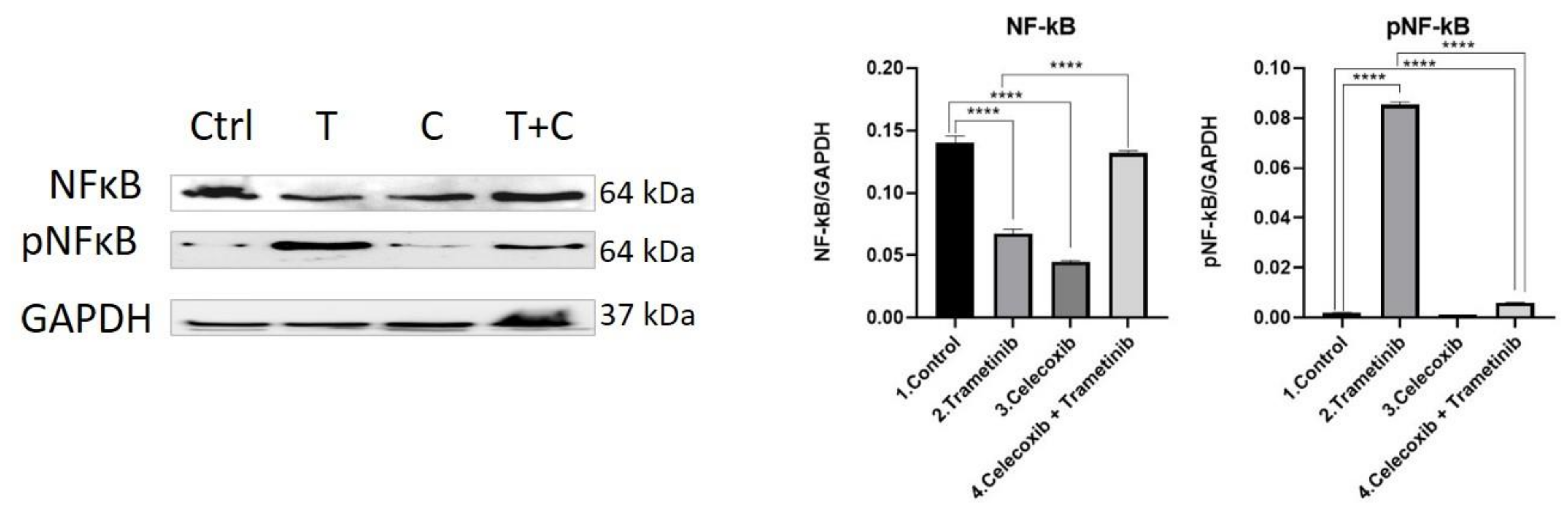
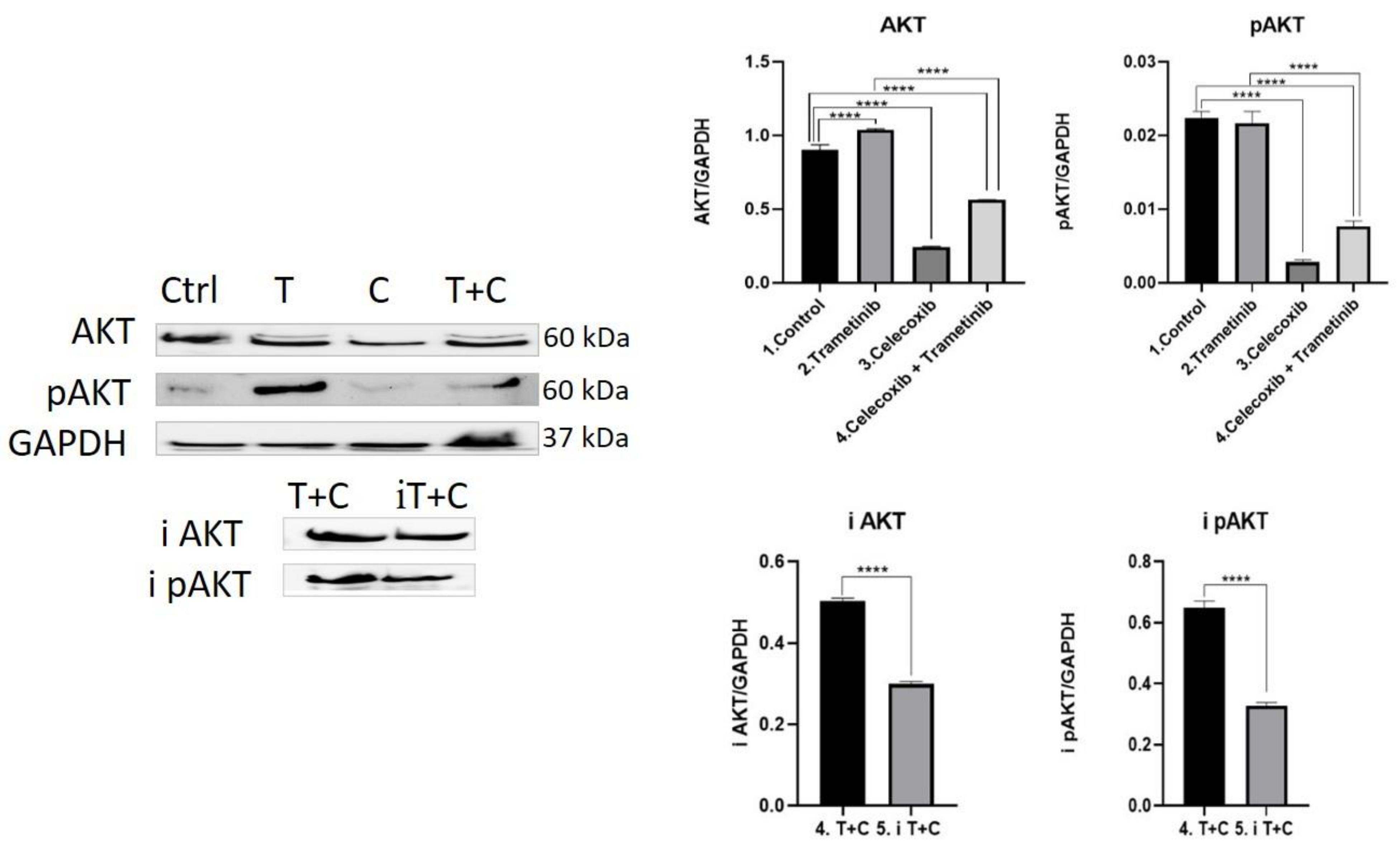
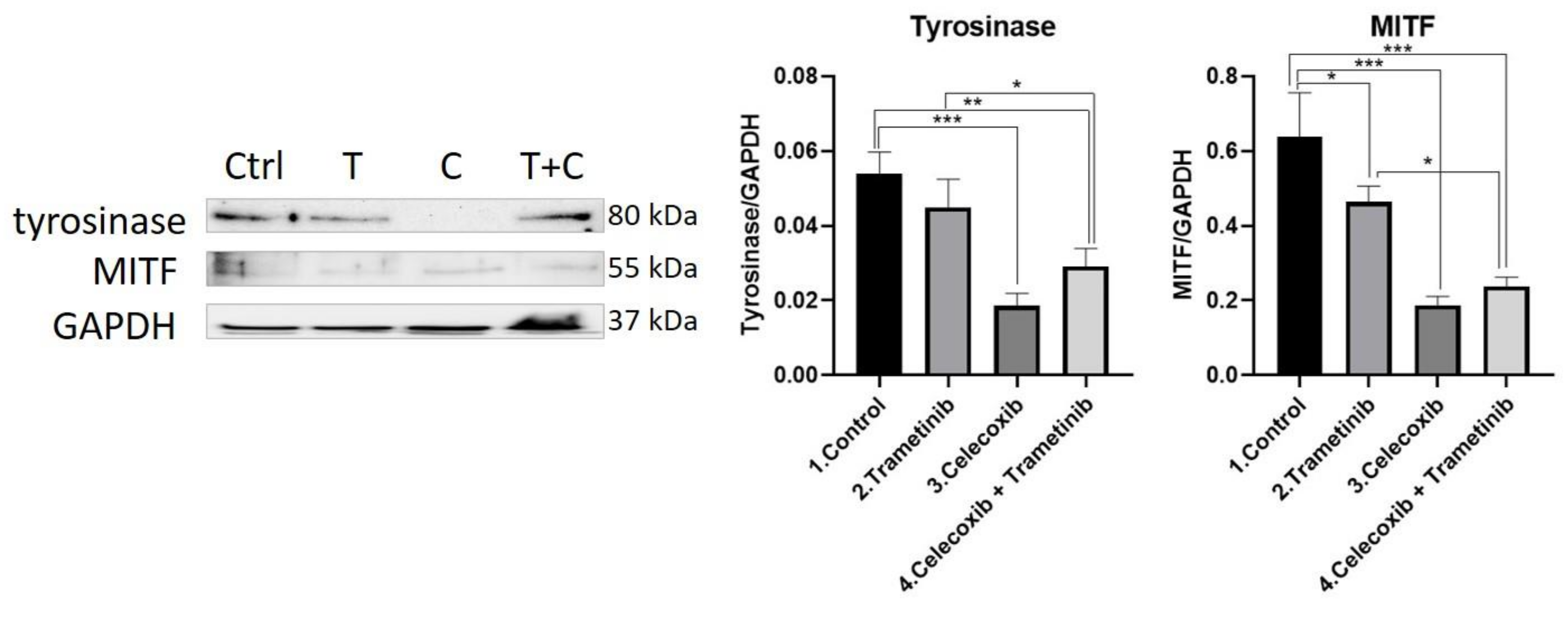
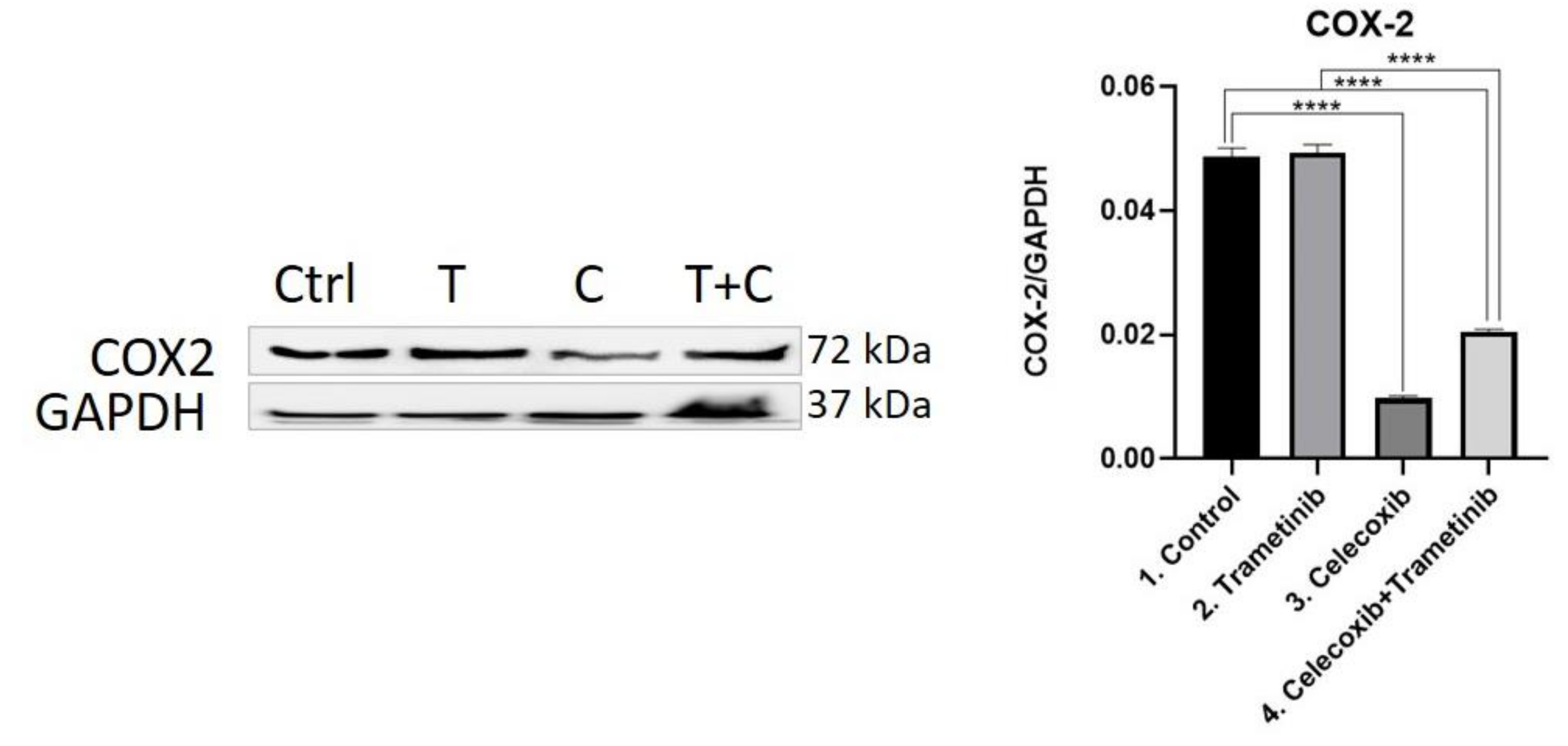
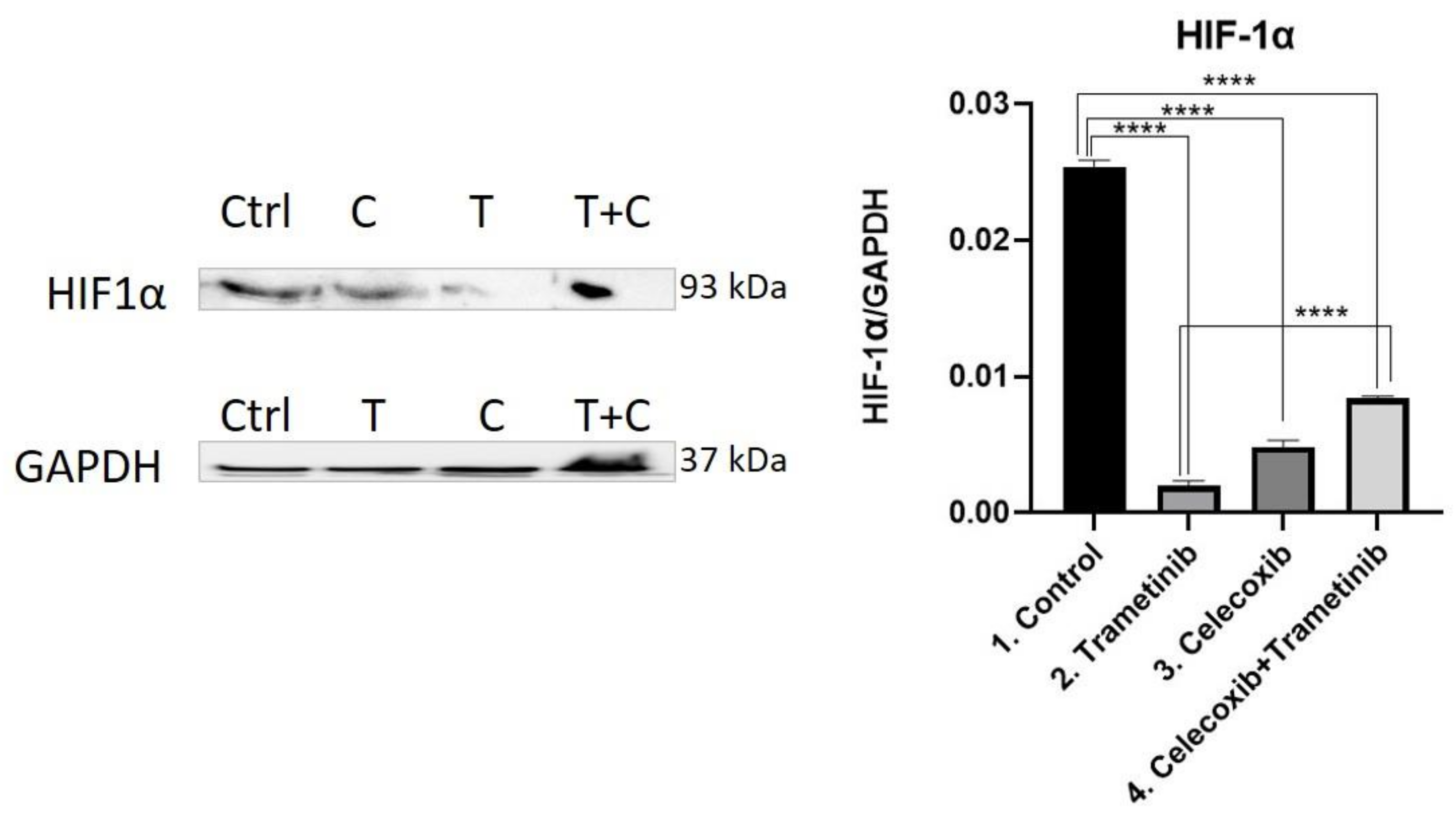
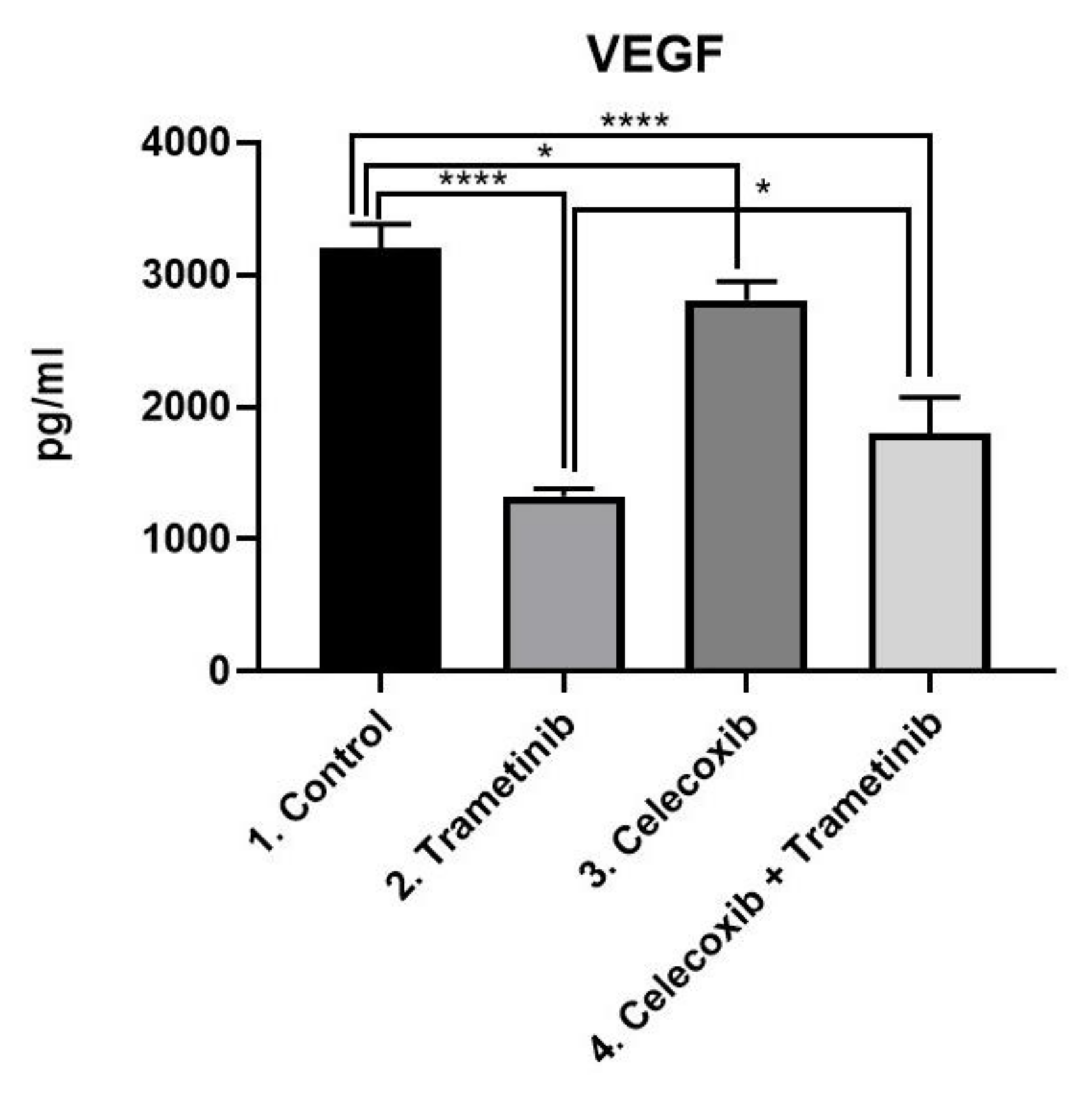
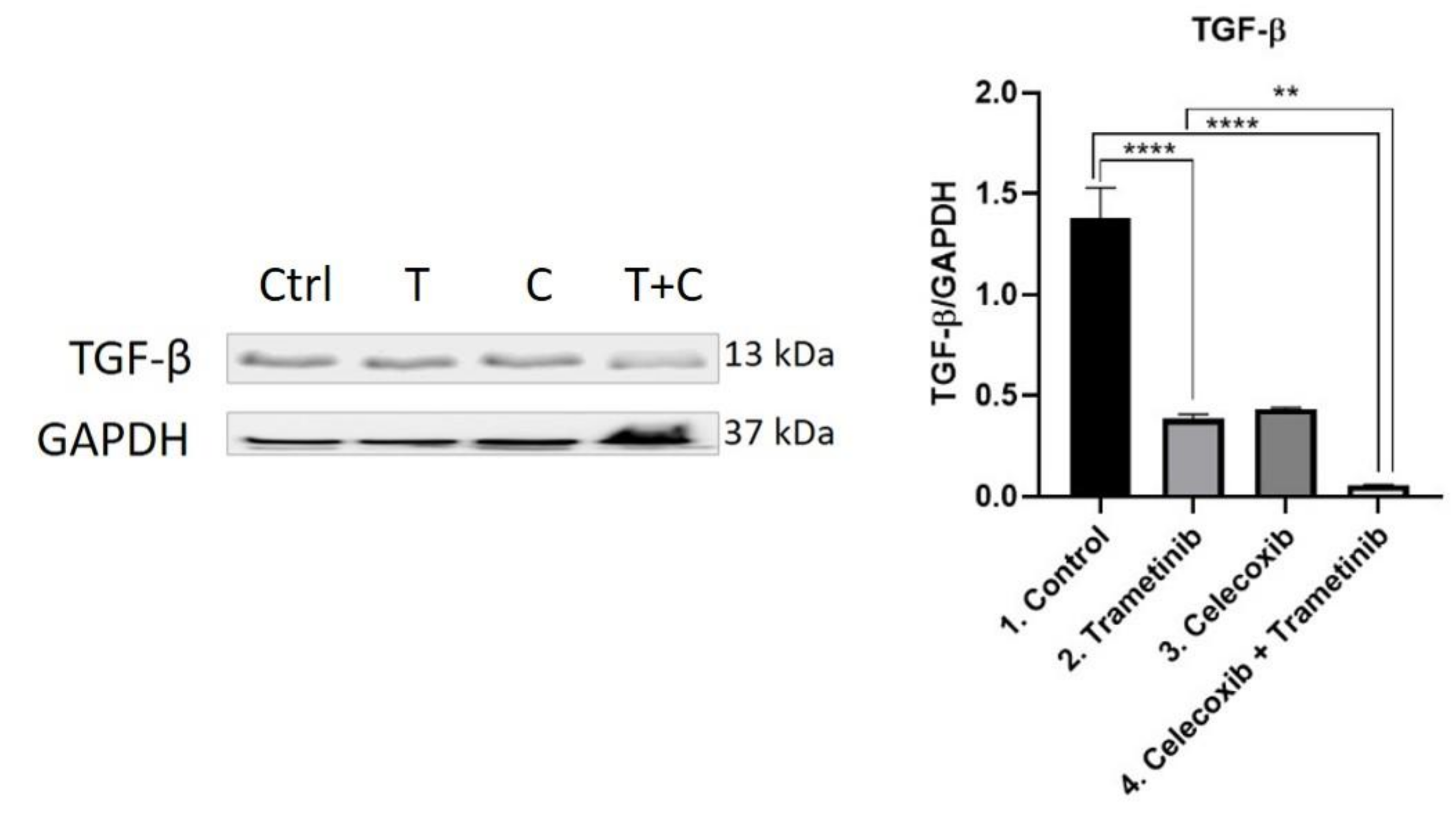
3.4. Inflammation, Melanogenesis, Angiogenesis and Signaling Molecules Assessment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| C | celecoxib |
| T | trametinib |
| T + C | celecoxib + trametinib |
| NF-κB | nuclear factor-kappa B |
| p NF-κB | phosphorylated nuclear factor-kappa B |
| PI3K | phosphatidylinositol 3-kinase |
| mTOR | mechanistic target of rapamycin |
| panAKT | protein kinase B |
| pan pAKT | phosphorylated protein kinase B |
| IKK | IkB kinase |
| MAPK | mitogen activated protein kinase |
| STAT3 | signal transducer and activator of transcription 3 |
| PTEN | phosphatase and tensin homolog |
| MITF | microphthalmia transcription factor |
| COX-2 | cyclooxygenase-2 |
| PGE2 | prostaglandin E2 |
| VEGF | vascular endothelial growth factor |
| HIF-1α | hypoxia inducible factor–1 alpha |
| TGF- β | transforming growth factor beta |
| LDH | lactate dehydrogenase |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| PD-1/PD-L1 | programmed ccell death–1/programmed cell death-ligand 1 |
| TAM | TAM receptor protein tyrosine kinases (TYRO3, AXL, MER) |
| α-MSH | alpha-melanocyte stimulating hormone |
| CAFs | cancer-associated fibroblasts |
| FSP-1 | fibroblast specific protein 1 |
| FAP-α | fibroblast activation protein-alpha |
| α-SMA | α-smooth muscle actin |
| PDGFR | platelet-derived growth factor receptor |
| YAP/PAX 3 | yes-associated protein 65/paired box gene 3 |
| TEAD/SMAD | transcriptional enhancer factor TEF-1 domain family member 1 |
| SIRT1 | family of proteins that are the main signal transducers for the receptors of the TGF-β |
References
- Roesch, A.; Melanoma, B.C. Braun-Falco’s Dermatology; Springer: Berlin/Heidelberg, Germany, 2020; pp. 1–7. [Google Scholar]
- Whiteman, D.C.; Green, A.C.; Olsen, C.M. The growing burden of invasive melanoma: Projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J. Investig. Dermatol. 2016, 136, 1161–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy. Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, S.; Xu, T.; Xiong, M.; Zhou, X.; Wang, Y.; Haydu, L.E.; Ross, M.I.; Gershenwald, J.E.; Prieto, V.G.; Cormier, J.N.; et al. Role of immune response, inflammation and tumor immune response–related cytokines/chemokines in melanoma progression. J. Invest. Dermatol. 2019, 139, 2352–2358. [Google Scholar] [CrossRef]
- Schadendorf, D.; van Akkooi, A.C.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Al Emran, A.; Tseng, H.Y.; Coleman, M.C.; Tiffen, J.; Cook, S.; McGuire, H.; Gallagher, S.; Feng, C.; Hersey, P. Do innate killing mechanisms activated by in-flammasomes have a role in treating melanoma? Pigm. Cell Melanoma Res. 2020, 33, 660–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, N.; Della Corte, M.; Pelaia, C.; Piazzetta, G.; Lobello, N.; Del Duca, E.; Bennardo, L.; Nisticò, S.P. Primary Mucosal Melanoma Presenting with a Unilateral Nasal Obstruction of the Left Inferior Turbinate. Medicina 2021, 57, 359. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef]
- Hayes, T.K.; Luo, F.; Cohen, O.; Goodale, A.B.; Lee, Y.; Pantel, S.; Bagul, M.; Piccioni, F.; Root, D.E.; Garraway, L.A.; et al. A Functional Landscape of Resistance to MEK1/2 and CDK4/6 Inhibition in NRAS-Mutant Melanoma. Cancer Res. 2019, 79, 2352–2366. [Google Scholar] [CrossRef] [Green Version]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varedi, A.; Rahman, H.; Kumar, D.; Catrow, J.L.; Cox, J.E.; Liu, T.; Florell, S.R.; Boucher, K.M.; Okwundu, N.; Burnett, W.J.; et al. ASA Suppresses PGE2 in Plasma and Melanocytic Nevi of Human Subjects at Increased Risk for Melanoma. Pharmaceut 2020, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Minisini, A.M.; Pascoletti, G.; Intersimone, D.; Poletto, E.; Driol, P.; Spizzo, R.; Scott, C.A.; Puglisi, F.; Fasola, G.; Di Loreto, C. Expression of thymidine phosphorylase and cyclooxygenase-2 in melanoma. Melanoma Res. 2013, 23, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.R.; Siegelin, M.D.; Rompel, R.; Enk, A.H.; Gaiser, T. COX-2 expression in malignant melanoma: A novel prognostic marker? Melanoma Res. 2009, 19, 8–16. [Google Scholar] [CrossRef]
- Ferreira, M.; Krykbaeva, I.; Damsky, W.; Kluger, H.M.; Bosenberg, M. Evaluating the role of the COX2/PGE2 pathway in anti-melanoma immunity. Int. J. Clin. Oncol. 2019, 37 (Suppl. 15), e14114. [Google Scholar] [CrossRef]
- Mao, Y.; Poschke, I.; Wennerberg, E.; De Coaña, Y.P.; Brage, S.E.; Schultz, I.; Hansson, J.; Masucci, G.; Lundqvist, A.; Kiessling, R. Melanoma-educated CD14+ cells acquire a mye-loid-derived suppressor cell phenotype through COX-2–dependent mechanisms. Cancer Res. 2013, 73, 3877–3887. [Google Scholar] [CrossRef] [Green Version]
- Goulet, A.C.; Einsphar, J.G.; Alberts, D.S.; Beas, A.; Burk, C.; Bhattacharyya, A.K.; Bangert, J.; Harmon, J.M.; Fujiwara, H.; Koki, A.; et al. Analysis of cyclooxygenase 2 (COX-2) ex-pression during malignant melanoma progression. Cancer Biol Ther. 2003, 2, 713–718. [Google Scholar] [CrossRef]
- Bhatt, R.S.; Merchan, J.; Parker, R.; Wu, H.K.; Zhang, L.; Seery, V.; Heymach, J.V.; Atkins, M.B.; McDermott, D.; Sukhatme, V.P. A phase 2 pilot trial of low-dose, continuous infusion, or “metronomic” paclitaxel and oral celecoxib in patients with metastatic melanoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2010, 116, 1751–1756. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, I.; Nakajima, K.; Shono, K.; Mizobuchi, Y.; Fujihara, T.; Shikata, E.; Yamaguchi, T.; Kitazato, K.; Sampetrean, O.; Saya, H.; et al. Downregulation of PD-L1 via FKBP5 by celecoxib augments antitumor effects of PD-1 blockade in a malignant glioma model. Neuro-Oncol. Adv. 2020, 2, vdz058. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Thorn, C.F.; Bertagnolli, M.M.; Grosser, T.; Altman, R.B.; Klein, T.E. Celecoxib pathways: Pharmacokinetics and pharma-codynamics. Pharm. Genom. 2012, 22, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, P.; Kuphal, S.; Spruss, T.; Hellerbrand, C.; Bosserhoff, A.K. Wild-type KRAS is a novel therapeutic target for melanoma contributing to primary and acquired resistance to BRAF inhibition. Oncogene 2018, 37, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.S.; Bonazzi, V.F.; Boyle, G.M.; Palmer, J.M.; Symmons, J.; Lanagan, C.M.; Schmidt, C.W.; Herington, A.C.; Ballotti, R.; Pollock, P.M.; et al. miR-514a regulates the tumour suppressor NF1 and modulates BRAFi sensitivity in melanoma. Oncotarget 2015, 6, 17753. [Google Scholar] [CrossRef] [Green Version]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2017, 8, 22262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozar, I.; Cesi, G.; Margue, C.; Philippidou, D.; Kreis, S. Impact of BRAF kinase inhibitors on the miRNomes and transcriptomes of melanoma cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2980–2992. [Google Scholar] [CrossRef]
- Liu, X.; Wu, J.; Qin, H.; Xu, J. The role of autophagy in the resistance to BRAF inhibition in BRAF-Mutated melanoma. Target. Oncol. 2018, 13, 437–446. [Google Scholar] [CrossRef]
- Zelenay, S.; Van Der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Saji, S.; Hirose, M.; Toi, M. Novel sensitizing agents: Potential contribution of COX-2 inhibitor for endocrine therapy of breast cancer. Breast Cancer 2004, 11, 129–133. [Google Scholar] [CrossRef]
- Harris, R.E. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology 2009, 17, 55–67. [Google Scholar] [CrossRef]
- Patil, V.M.; Noronha, V.; Joshi, A.; Abhyankar, A.; Menon, N.; Dhumal, S.; Prabhash, K. Beyond conventional chemotherapy, tar-geted therapy and immunotherapy in squamous cell cancer of the oral cavity. Oral. Oncol. 2020, 105, 104673. [Google Scholar] [CrossRef]
- Hedberg, M.L.; Peyser, N.D.; Bauman, J.E.; Gooding, W.E.; Li, H.; Bhola, N.E.; Zhu, T.R.; Zeng, Y.; Brand, T.M.; Kim, M.O.; et al. Use of nonsteroidal anti-inflammatory drugs predicts improved patient survival for PIK3CA-altered head and neck cancer. J. Exp. Med. 2019, 216, 419–427. [Google Scholar] [CrossRef]
- Tudor, D.V.; Bâldea, I.; Lupu, M.; Kacso, T.; Kutasi, E.; Hopârtean, A.; Stretea, R.; Filip, A.G. COX-2 as a potential biomarker and therapeutic target in melanoma. Cancer Biol. Med. 2020, 17, 20. [Google Scholar] [PubMed]
- Kim, N.; Kim, C.H.; Ahn, D.W.; Lee, K.S.; Cho, S.J.; Park, J.H.; Lee, M.K.; Kim, J.S.; Jung, H.C.; Song, I.S. Anti-gastric cancer effects of celecoxib, a selective COX-2 inhibitor, through inhibition of Akt signaling. J. Gastroenterol. Hepatol. 2009, 24, 480–487. [Google Scholar] [CrossRef]
- Rosas, C.; Sinning, M.; Ferreira, A.; Fuenzalida, M.; Lemus, D. Celecoxib decreases growth and angiogenesis and promotes apoptosis in a tumor cell line resistant to chemotherapy. Biol. Res. 2014, 47, 27. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.M.; Knopf, J.; Reiner, E.; Bossuyt, V.; Epstein, L.; DiGiovanna, M.; Chung, G.; Silber, A.; Sanft, T.; Hofstatter, E.; et al. Mutation based treatment recommendations from next generation sequencing data: A comparison of web tools. Oncotarget 2016, 7, 22064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, T.J.; Janssen, A.; Schmidt, R.; Geisslinger, G.; Grösch, S. Targeting the beta-catenin/APC pathway: A novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB J. 2005, 19, 1353–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setiawati, A.; Setiawati, A. Celecoxib, a COX-2 Selective Inhibitor, Induces Cell Cycle Arrest at the G2/M Phase in HeLa Cer-vical Cancer Cells. Asian Pac. J. Cancer Prev. 2016, 17, 1655–1660. [Google Scholar] [CrossRef]
- Liu, M.; Li, C.M.; Chen, Z.F.; Ji, R.; Guo, Q.H.; Li, Q.; Zhang, H.L.; Zhou, Y.N. Celecoxib regulates apoptosis and autophagy via the PI3K/Akt signaling pathway in SGC-7901 gastric cancer cells. Int. J. Molec. Med. 2014, 33, 1451–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [Green Version]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dréno, B.; Fargnoli, M.C.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics–Update 2019. Eur. J. Cancer. 2020, 126, 141–158. [Google Scholar] [CrossRef] [Green Version]
- Agarwala, S.S.; Keilholz, U.; Gilles, E.; Bedikian, A.Y.; Wu, J.; Kay, R.; Stein, C.A.; Itri, L.M.; Suciu, S.; Eggermont, A.M. LDH correlation with survival in advanced melanoma from two large, randomised trials (Oblimersen GM301 and EORTC 18951). Eur. J. Cancer. 2009, 45, 1807–1814. [Google Scholar] [CrossRef]
- Pritchard, R.; Rodríguez-Enríquez, S.; Pacheco-Velázquez, S.C.; Bortnik, V.; Moreno-Sánchez, R.; Ralph, S. Celecoxib inhibits mi-tochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apop-totic signalling pathway. Biochem. Pharmacol. 2018, 154, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Jendrossek, V.; Handrick, R.; Belka, C. Celecoxib activates a novel mitochondrial apoptosis signaling pathway. FASEB J. 2003, 17, 1547–1549. [Google Scholar] [CrossRef]
- Bundscherer, A.; Hafner, C.; Maisch, T.; Becker, B.; Landthaler, M.; Vogt, T. Antiproliferative and proapoptotic effects of rapamycin and celecoxib in malignant melanoma cell lines. Oncol. Rep. 2008, 19, 547–553. [Google Scholar] [CrossRef]
- Sadhu, S.S.; Wang, S.; Averineni, R.K.; Seefeldt, T.; Yang, Y.; Guan, X. In-vitro and in-vivo inhibition of melanoma growth and me-tastasis by the drug combination of celecoxib and dacarbazine. Melanoma Res. 2016, 26, 572–579. [Google Scholar] [CrossRef]
- Irvine, M.; Stewart, A.; Pedersen, B.; Boyd, S.; Kefford, R.; Rizos, H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 2018, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275–3289. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced proteolytic shedding of receptor tyrosine kinases is a post-translational mechanism of kinase inhibitor resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef] [Green Version]
- Botti, G.; Fratangelo, F.; Cerrone, M.; Liguori, G.; Cantile, M.; Anniciello, A.M.; Scala, S.; D’Alterio, C.; Trimarco, C.; Ianaro, A.; et al. COX-2 expression positively correlates with PD-L1 expression in human melanoma cells. J. Trans. Med. 2017, 15, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.; Chen, J.; Lu, J.; Yi, L.; Tong, X.; Kang, L.; Pei, S.; Ouyang, Y.; Jiang, L.; Ding, Y.; et al. Roles of inflammation factors in melanogenesis. Mol. Med. Rep. 2020, 21, 1421–1430. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Shin, J.Y.; Kim, M.R.; Hann, S.K.; Oh, S.H. siRNA-mediated knock-down of COX-2 in melanocytes suppresses melano-genesis. Exp Dermatol. 2012, 21, 420–425. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.P.; Rana, S.; Ferguson, J.; Rowling, E.J.; Flaherty, K.T.; Wargo, J.A.; Marais, R.; Wellbrock, C. A PAX3/BRN2 rheostat controls the dynamics of BRAF mediated MITF regulation in MITFhigh/AXLlow melanoma. Pigment Cell Melanoma Res. 2019, 32, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Fosslien, E. Molecular pathology of cyclooxygenase-2 in cancer-induced angiogenesis. Ann. Clin. Lab. Sci. 2001, 31, 325–348. [Google Scholar] [PubMed]
- Sui, W.; Zhang, Y.; Wang, Z.; Wang, Z.; Jia, Q.; Wu, L.; Zhang, W. Antitumor effect of a selective COX-2 inhibitor, celecoxib, may be attributed to angiogenesis inhibition through modulating the PTEN/PI3K/Akt/HIF-1 pathway in an H22 murine hepatocar-cinoma model. Oncol. Rep. 2014, 31, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482. [Google Scholar]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer pro-gression. Nat. Med. 2011, 17, 320. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Yang, K.; Andl, T.; Wickett, R.R.; Zhang, Y. Perspective of targeting cancer-associated fibroblasts in melanoma. J. Cancer. 2015, 6, 717. [Google Scholar] [CrossRef] [Green Version]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Räsänen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef]
- Miskolczi, Z.; Smith, M.P.; Rowling, E.J.; Ferguson, J.; Barriuso, J.; Wellbrock, C. Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene 2018, 37, 3166–3182. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, S.J.; Wang, R.K.; Duncan, D.D.; Kulesz-Martin, M.; Lee, K. Imaging the mechanical stiffness of skin lesions by in vivo acousto-optical elastography. Opt. Express 2006, 14, 9770–9779. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting drivers of non-mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellbrock, C.; Arozarena, I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. 2015, 28, 390–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Li, Y.; Nishimura, E.K.; Xin, H.; Zhou, A.; Guo, Y.; Dong, L.; Denning, M.F.; Nickoloff, B.J.; Cui, R. Inhibition of PAX3 by TGF-beta modulates melanocyte viability. Mol. Cell 2008, 32, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Cha, B.K.; Kim, Y.S.; Hwang, K.E.; Cho, K.H.; Oh, S.H.; Kim, B.R.; Jun, H.Y.; Yoon, K.H.; Jeong, E.T.; Kim, H.R. Celecoxib and sulindac inhibit TGF-β1-induced epitheli-al-mesenchymal transition and suppress lung cancer migration and invasion via downregulation of sirtuin 1. Oncotarget 2016, 7, 57213. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Jin, K.; Jiang, T.; Wang, L.; Shen, S.; Luo, Z.; Tuo, Y.; Liu, X.; Hu, Y.; Pang, Z. Celecoxib normalizes the tumor microenvironment and enhances small nanotherapeutics delivery to A549 tumors in nude mice. Sci. Rep. 2017, 7, 1–2. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Schrump (MD): National Institutes of Health Clinical Center (US). Identifier: NCT01341496, Epigenetically-Modified Autologous Tumor Cell Vaccs and ISCOMATRIX(TM) Adjuvant with Metronomic Oral Cyclophosphamide and Celecoxib in Pts Undergoing Resection of Sarcomas, Melanomas, Germ Cell Tumors, or Epithelial Malignancies Metastatic to Lungs, Pleura or Mediastinum. Available online: https://clinicaltrials.gov/ct2/show/NCT01341496?term=cox-2&cond=Melanoma&rank=6 (accessed on 25 April 2011).
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention–Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef] [PubMed]
- European Celecoxib Trial in Primary Breast Cancer—Clinical Trials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02429427?term=celecoxib&cond=cancer&draw=2&rank=1 (accessed on 29 April 2015).
- Celecoxib in Preventing Non-Small Cell Lung Cancer in Tobacco Smokers—Clinical Trials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00020878?term=celecoxib&cond=cancer&draw=2&rank=4 (accessed on 27 January 2003).
- Escuin-Ordinas, H.; Atefi, M.; Fu, Y.; Cass, A.; Ng, C.; Huang, R.R.; Yashar, S.; Comin-Anduix, B.; Avramis, E.; Cochran, A.J.; et al. COX-2 inhibition prevents the appearance of cutaneous squamous cell carcinomas accelerated by BRAF inhibitors. Mol. Oncol. 2014, 8, 250–260. [Google Scholar] [CrossRef]
- Paton, E.L.; Turner, J.A.; Schlaepfer, I.R. Overcoming Resistance to Therapies Targeting the MAPK Pathway in BRAF-Mutated Tumours. J Oncol. 2020, 2020, 1079827. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, J.; Huang, S.Q.; Su, H.H.; Zhou, S.F. Genetic polymorphism of the human cytochrome P450 2C9 gene and its clinical significance. Curr. Drug Metab. 2009, 10, 781–834. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, S.K.; Nguyen, D.; Bowen, C.; Richards-Peterson, L.; Skordos, K.W. The metabolic drug-drug interaction profile of Dabrafenib: In vitro investigations and quantitative extrapolation of the P450-mediated DDI risk. Drug Metab. Dispos. 2014, 42, 1180–1190. [Google Scholar] [CrossRef] [Green Version]
- Asgari, M.M.; Maruti, S.S.; White, E. A large cohort study of nonsteroidal anti-inflammatory drug use and melanoma incidence. JNCI J. Natl. Cancer Inst. 2008, 100, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Gamba, C.A.; Swetter, S.M.; Stefanick, M.L.; Kubo, J.; Desai, M.; Spaunhurst, K.M.; Sinha, A.A.; Asgari, M.M.; Sturgeon, S.; Tang, J.Y. Aspirin is associated with lower melanoma risk among postmenopausal Caucasian women: The Women’s Health Initiative. Cancer 2013, 119, 1562–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, J.R.; Grossman, D. Aspirin and other NSAIDs as chemoprevention agents in melanoma. Cancer Prev. Res. 2014, 7, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tudor, D.V.; Bâldea, I.; Olteanu, D.E.; Fischer-Fodor, E.; Piroska, V.; Lupu, M.; Călinici, T.; Decea, R.M.; Filip, G.A. Celecoxib as a Valuable Adjuvant in Cutaneous Melanoma Treated with Trametinib. Int. J. Mol. Sci. 2021, 22, 4387. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094387
Tudor DV, Bâldea I, Olteanu DE, Fischer-Fodor E, Piroska V, Lupu M, Călinici T, Decea RM, Filip GA. Celecoxib as a Valuable Adjuvant in Cutaneous Melanoma Treated with Trametinib. International Journal of Molecular Sciences. 2021; 22(9):4387. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094387
Chicago/Turabian StyleTudor, Diana Valentina, Ioana Bâldea, Diana Elena Olteanu, Eva Fischer-Fodor, Virag Piroska, Mihai Lupu, Tudor Călinici, Roxana Maria Decea, and Gabriela Adriana Filip. 2021. "Celecoxib as a Valuable Adjuvant in Cutaneous Melanoma Treated with Trametinib" International Journal of Molecular Sciences 22, no. 9: 4387. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094387