
Links between Immune Cells from the Periphery and the Brain in the Pathogenesis of Epilepsy: A Narrative Review

 , , and
, , and
Abstract
:1. Introduction
2. Review
2.1. Innate Immunity
Microglia and Monocytes
2.2. Differentiation between Microglia and Monocytes
2.2.1. Markers of Monocytes
2.2.2. Genetic Modulation Methods
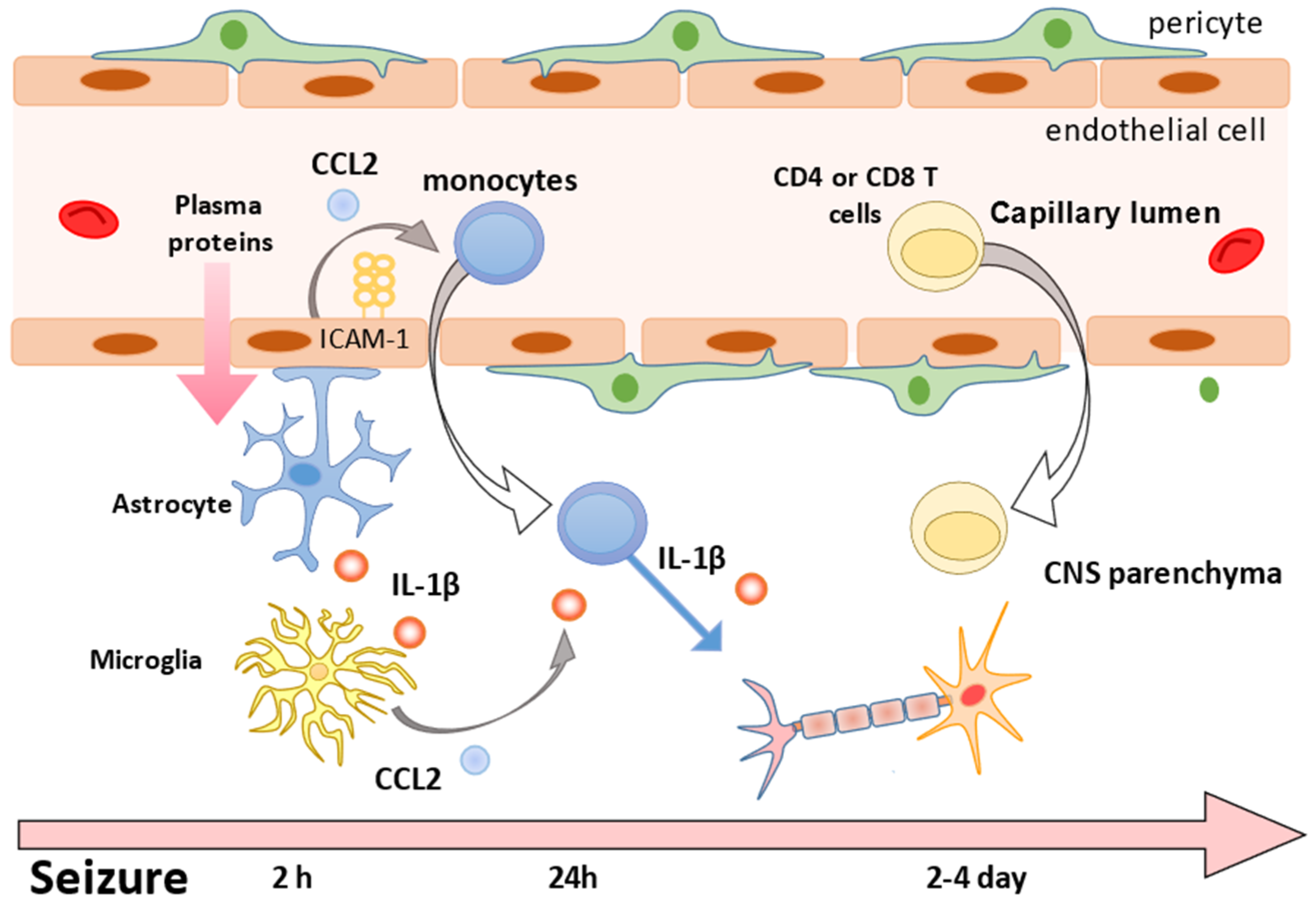
2.3. How Do Peripheral Monocytes Penetrate the Brain in the Pathogenesis of Epilepsy?
2.4. Disruptive and Non-Disruptive Changes in the BBB
2.4.1. Disruptive Changes in the BBB
2.4.2. Non-Disruptive Changes
2.5. Identification of Peripheral Monocytes in the Brain
2.6. Potential for Therapy by Controlling Monocytes
2.7. Association between the Clinical Picture and the Monocytes or Microglia
2.8. Adaptive Immunity
2.8.1. Experimental Evidence of the Invasion of Peripherally Adapted Immune Cells into the Brain
2.8.2. Association between the Clinical Picture and Cells of the T Lineage
2.9. Role of Pericytes in the Link between Peripheral Immune Cells and the Brain
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890. [Google Scholar] [CrossRef]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Hauser, W.A.; Mathern, G.; Moshé, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef]
- Ryvlin, P.; Nashef, L.; Tomson, T. Prevention of sudden unexpected death in epilepsy: A realistic goal? Epilepsia 2013, 54 (Suppl. 2), 23–28. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Hesdorffer, D.C.; Thurman, D.J.; Lhatoo, S.; Richerson, G. Sudden unexpected death in epilepsy: Epidemiology, mechanisms, and prevention. Lancet Neurol. 2016, 15, 1075–1088. [Google Scholar] [CrossRef]
- Vezzani, A.; Lang, B.; Aronica, E. Immunity and Inflammation in Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 6, a022699. [Google Scholar] [CrossRef] [Green Version]
- Granata, T.; Cross, H.; Theodore, W.; Avanzini, G. Immune-mediated epilepsies. Epilepsia 2011, 52 (Suppl. 3), 5–11. [Google Scholar] [CrossRef]
- Zattoni, M.; Mura, M.L.; Deprez, F.; Schwendener, R.A.; Engelhardt, B.; Frei, K.; Fritschy, J.M. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J. Neurosci. 2011, 31, 4037–4050. [Google Scholar] [CrossRef] [Green Version]
- Silverberg, J.; Ginsburg, D.; Orman, R.; Amassian, V.; Durkin, H.G.; Stewart, M. Lymphocyte infiltration of neocortex and hippocampus after a single brief seizure in mice. Brain. Behav. Immun. 2010, 24, 263–272. [Google Scholar] [CrossRef]
- Djukic, M.; Mildner, A.; Schmidt, H.; Czesnik, D.; Brück, W.; Priller, J.; Nau, R.; Prinz, M. Circulating monocytes engraft in the brain, differentiate into microglia and contribute to the pathology following meningitis in mice. Brain 2006, 129, 2394–2403. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. USA 2016, 113, E5665–E5674. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.S.; Peng, J.; Murugan, M.; Feng, L.J.; Liu, J.L.; Eyo, U.B.; Zhou, L.J.; Mogilevsky, R.; Wang, W.; Wu, L.J. Chemokine CCL2-CCR2 Signaling Induces Neuronal Cell Death via STAT3 Activation and IL-1β Production after Status Epilepticus. J. Neurosci. 2017, 37, 7878–7892. [Google Scholar] [CrossRef] [Green Version]
- Käufer, C.; Chhatbar, C.; Bröer, S.; Waltl, I.; Ghita, L.; Gerhauser, I.; Kalinke, U.; Löscher, W. Chemokine receptors CCR2 and CX3CR1 regulate viral encephalitis-induced hippocampal damage but not seizures. Proc. Natl. Acad. Sci. USA 2018, 115, E8929–E8938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Murugan, M.; Bosco, D.B.; Liu, Y.; Peng, J.; Worrell, G.A.; Wang, H.L.; Ta, L.E.; Richardson, J.R.; Shen, Y.; et al. Microglial proliferation and monocyte infiltration contribute to microgliosis following status epilepticus. Glia 2019, 67, 1434–1448. [Google Scholar] [CrossRef]
- Lu, J.Q.; Steve, T.A.; Wheatley, M.; Gross, D.W. Immune Cell Infiltrates in Hippocampal Sclerosis: Correlation with Neuronal Loss. J. Neuropathol. Exp. Neurol. 2017, 76, 206–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Robinson, A.P.; Ishii, T.; Duncan, D.S.; Alden, T.D.; Goings, G.E.; Ifergan, I.; Podojil, J.R.; Penaloza-MacMaster, P.; Kearney, J.A.; et al. Peripherally derived T regulatory and gammadelta T cells have opposing roles in the pathogenesis of intractable pediatric epilepsy. J. Exp. Med. 2018, 215, 1169–1186. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.C.; Garcia, A.J.; Mochizuki, A.Y.; Chang, J.W.; Reyes, S.D.; Salamon, N.; Prins, R.M.; Mathern, G.W.; Fallah, A. Evidence for Innate and Adaptive Immune Responses in a Cohort of Intractable Pediatric Epilepsy Surgery Patients. Front. Immunol. 2019, 10, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Shih, D.C.W.; Lim, A.; Paleja, B.; Ling, S.; Li Yun, L.; Li Poh, S.; Ngoh, A.; Arkachaisri, T.; Yeo, J.G.; et al. Pro-inflammatory, IL-17 pathways dominate the architecture of the immunome in pediatric refractory epilepsy. JCI Insight 2019, 5, e126337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekaart, D.W.M.; Anink, J.J.; Baayen, J.C.; Idema, S.; de Vries, H.E.; Aronica, E.; Gorter, J.A.; van Vliet, E.A. Activation of the innate immune system is evident throughout epileptogenesis and is associated with blood-brain barrier dysfunction and seizure progression. Epilepsia 2018, 59, 1931–1944. [Google Scholar] [CrossRef] [Green Version]
- Bosco, D.B.; Tian, D.S.; Wu, L.J. Neuroimmune interaction in seizures and epilepsy: Focusing on monocyte infiltration. FEBS J. 2020, 287, 4822–4837. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Marchi, N.; Banjara, M.; Janigro, D. Blood-brain barrier, bulk flow, and interstitial clearance in epilepsy. J. Neurosci. Methods 2016, 260, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Milesi, S.; Boussadia, B.; Plaud, C.; Catteau, M.; Rousset, M.C.; De Bock, F.; Schaeffer, M.; Lerner-Natoli, M.; Rigau, V.; Marchi, N. Redistribution of PDGFRβ cells and NG2DsRed pericytes at the cerebrovasculature after status epilepticus. Neurobiol. Dis. 2014, 71, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Oby, E.; Janigro, D. The blood-brain barrier and epilepsy. Epilepsia 2006, 47, 1761–1774. [Google Scholar] [CrossRef]
- Floris, S.; Blezer, E.L.; Schreibelt, G.; Döpp, E.; van der Pol, S.M.; Schadee-Eestermans, I.L.; Nicolay, K.; Dijkstra, C.D.; de Vries, H.E. Blood-brain barrier permeability and monocyte infiltration in experimental allergic encephalomyelitis: A quantitative MRI study. Brain 2004, 127, 616–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, H.D.; Hicks, R.R.; Smith, D.; McIntosh, T.K. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J. Neurosci. 1995, 15, 8223–8233. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- de Vries, E.E.; van den Munckhof, B.; Braun, K.P.; van Royen-Kerkhof, A.; de Jager, W.; Jansen, F.E. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2016, 63, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Merad, M. Regulation of microglia development and homeostasis. Glia 2013, 61, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Lund, H.; Pieber, M.; Parsa, R.; Han, J.; Grommisch, D.; Ewing, E.; Kular, L.; Needhamsen, M.; Espinosa, A.; Nilsson, E.; et al. Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat. Commun. 2018, 9, 4845. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, H.W. Definition of human blood monocytes. J. Leukoc. Biol. 2000, 67, 603–606. [Google Scholar] [CrossRef]
- Ong, S.M.; Teng, K.; Newell, E.; Chen, H.; Chen, J.; Loy, T.; Yeo, T.W.; Fink, K.; Wong, S.C. A Novel, Five-Marker Alternative to CD16-CD14 Gating to Identify the Three Human Monocyte Subsets. Front. Immunol. 2019, 10, 1761. [Google Scholar] [CrossRef] [Green Version]
- Ritzel, R.M.; Patel, A.R.; Grenier, J.M.; Crapser, J.; Verma, R.; Jellison, E.R.; McCullough, L.D. Functional differences between microglia and monocytes after ischemic stroke. J. Neuroinflamm. 2015, 12, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Lan, X.; Han, X.; Wang, J. Expression of Tmem119/Sall1 and Ccr2/CD69 in FACS-Sorted Microglia- and Monocyte/Macrophage-Enriched Cell Populations After Intracerebral Hemorrhage. Front. Cell. Neurosci. 2018, 12, 520. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.K.; Ji, K.; Min, K.; Joe, E.H. Brain inflammation and microglia: Facts and misconceptions. Exp. Neurobiol. 2013, 22, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, D.G.; Lue, L.F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimers Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Greter, M.; Lelios, I.; Croxford, A.L. Microglia Versus Myeloid Cell Nomenclature during Brain Inflammation. Front. Immunol. 2015, 6, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkstra, C.D.; Döpp, E.A.; Joling, P.; Kraal, G. The heterogeneity of mononuclear phagocytes in lymphoid organs: Distinct macrophage subpopulations in the rat recognized by monoclonal antibodies ED1, ED2 and ED3. Immunology 1985, 54, 589–599. [Google Scholar] [PubMed]
- Pey, P.; Pearce, R.K.; Kalaitzakis, M.E.; Griffin, W.S.; Gentleman, S.M. Phenotypic profile of alternative activation marker CD163 is different in Alzheimer’s and Parkinson’s disease. Acta Neuropathol. Commun. 2014, 2, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Plüddemann, A.; Martinez Estrada, F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, N.; Peng, J.; Murugan, M.; Wang, X.; Eyo, U.B.; Sun, D.; Ren, Y.; DiCicco-Bloom, E.; Young, W.; Dong, H.; et al. Spinal Microgliosis Due to Resident Microglial Proliferation Is Required for Pain Hypersensitivity after Peripheral Nerve Injury. Cell Rep. 2016, 16, 605–614. [Google Scholar] [CrossRef] [Green Version]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Banisadr, G.; Gosselin, R.D.; Mechighel, P.; Rostène, W.; Kitabgi, P.; Mélik Parsadaniantz, S. Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: Functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. J. Comp. Neurol. 2005, 492, 178–192. [Google Scholar] [CrossRef]
- Mizutani, M.; Pino, P.A.; Saederup, N.; Charo, I.F.; Ransohoff, R.M.; Cardona, A.E. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J. Immunol. 2012, 188, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Saederup, N.; Cardona, A.E.; Croft, K.; Mizutani, M.; Cotleur, A.C.; Tsou, C.L.; Ransohoff, R.M.; Charo, I.F. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE 2010, 5, e13693. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorter, J.A.; van Vliet, E.A.; Aronica, E. Status epilepticus, blood-brain barrier disruption, inflammation, and epileptogenesis. Epilepsy Behav. 2015, 49, 13–16. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; Otte, W.M.; Gorter, J.A.; Dijkhuizen, R.M.; Wadman, W.J. Longitudinal assessment of blood-brain barrier leakage during epileptogenesis in rats. A quantitative MRI study. Neurobiol. Dis. 2014, 63, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Mendes, N.F.; Pansani, A.P.; Carmanhães, E.R.F.; Tange, P.; Meireles, J.V.; Ochikubo, M.; Chagas, J.R.; da Silva, A.V.; Monteiro de Castro, G.; Le Sueur-Maluf, L. The Blood-Brain Barrier Breakdown During Acute Phase of the Pilocarpine Model of Epilepsy Is Dynamic and Time-Dependent. Front. Neurol. 2019, 10, 382. [Google Scholar] [CrossRef] [PubMed]
- Gorter, J.A.; Aronica, E.; van Vliet, E.A. The Roof is Leaking and a Storm is Raging: Repairing the Blood-Brain Barrier in the Fight Against Epilepsy. Epilepsy Curr. 2019, 19, 177–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-Mediated Blood-Brain Barrier Opening: Implications for Neuroprotection and Drug Delivery. J. Neurosci. 2016, 36, 7727–7739. [Google Scholar] [CrossRef] [PubMed]
- Librizzi, L.; Noè, F.; Vezzani, A.; de Curtis, M.; Ravizza, T. Seizure-induced brain-borne inflammation sustains seizure recurrence and blood-brain barrier damage. Ann. Neurol. 2012, 72, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, C.C.; Depino, A.M.; Prada, F.; Muraro, N.; Campbell, S.; Podhajcer, O.; Perry, V.H.; Anthony, D.C.; Pitossi, F.J. Reversible demyelination, blood-brain barrier breakdown, and pronounced neutrophil recruitment induced by chronic IL-1 expression in the brain. Am. J. Pathol. 2004, 165, 1827–1837. [Google Scholar] [CrossRef] [Green Version]
- Fabene, P.F.; Navarro Mora, G.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef]
- Vezzani, A.; Conti, M.; De Luigi, A.; Ravizza, T.; Moneta, D.; Marchesi, F.; De Simoni, M.G. Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: Functional evidence for enhancement of electrographic seizures. J. Neurosci. 1999, 19, 5054–5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arisi, G.M.; Foresti, M.L.; Katki, K.; Shapiro, L.A. Increased CCL2, CCL3, CCL5, and IL-1beta cytokine concentration in piriform cortex, hippocampus, and neocortex after pilocarpine-induced seizures. J. Neuroinflamm. 2015, 12, 129. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Moneta, D.; Richichi, C.; Aliprandi, M.; Burrows, S.J.; Ravizza, T.; Perego, C.; De Simoni, M.G. Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia 2002, 43 (Suppl. 5), 30–35. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.P.; Brennan, G.P.; Curran, M.; Kinney-Lang, E.; Dubé, C.; Rashid, F.; Ly, C.; Obenaus, A.; Baram, T.Z. Rapid, Coordinate Inflammatory Responses after Experimental Febrile Status Epilepticus: Implications for Epileptogenesis. Eneuro 2015, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.C.; Wallis, G.; Dahle, E.J.; McElroy, P.B.; Thomson, K.E.; Tesi, R.J.; Szymkowski, D.E.; West, P.J.; Smeal, R.M.; Patel, M.; et al. Hippocampal TNFα Signaling Contributes to Seizure Generation in an Infection-Induced Mouse Model of Limbic Epilepsy. Eneuro 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, C.H.; Eom, Y.W.; Yoo, M.H.; You, H.J.; Han, H.J.; Song, W.K.; Yoo, Y.J.; Chun, J.S.; Kim, J.H. Tumor necrosis factor-alpha generates reactive oxygen species via a cytosolic phospholipase A2-linked cascade. J. Biol. Chem. 2000, 275, 32357–32362. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.S.; Hu, S.; Ni, H.T.; Rowen, T.N.; Lokensgard, J.R.; Peterson, P.K. TNF-alpha-induced chemokine production and apoptosis in human neural precursor cells. J. Leukoc. Biol. 2005, 78, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Gibson, R.M.; Rothwell, N.J.; Le Feuvre, R.A. CNS injury: The role of the cytokine IL-1. Vet. J. 2004, 168, 230–237. [Google Scholar] [CrossRef]
- Osborn, L.; Hession, C.; Tizard, R.; Vassallo, C.; Luhowskyj, S.; Chi-Rosso, G.; Lobb, R. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell 1989, 59, 1203–1211. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzi, Y.; Caleo, M. Epilepsy, Seizures, and Inflammation: Role of the C-C Motif Ligand 2 Chemokine. DNA Cell Biol. 2016, 35, 257–260. [Google Scholar] [CrossRef]
- Izikson, L.; Klein, R.S.; Luster, A.D.; Weiner, H.L. Targeting monocyte recruitment in CNS autoimmune disease. Clin. Immunol. 2002, 103, 125–131. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Lu, J.; Ling, E.A.; Kaur, C. Monocyte chemoattractant protein-1 (MCP-1) produced via NF-kappaB signaling pathway mediates migration of amoeboid microglia in the periventricular white matter in hypoxic neonatal rats. Glia 2009, 57, 604–621. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.J.; Zhou, C.; Gravanis, I.; Rogove, A.D.; Wu, Y.P.; Bogenhagen, D.F.; Tsirka, S.E. Proteolytic activation of monocyte chemoattractant protein-1 by plasmin underlies excitotoxic neurodegeneration in mice. J. Neurosci. 2007, 27, 1738–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manley, N.C.; Bertrand, A.A.; Kinney, K.S.; Hing, T.C.; Sapolsky, R.M. Characterization of monocyte chemoattractant protein-1 expression following a kainate model of status epilepticus. Brain Res. 2007, 1182, 138–143. [Google Scholar] [CrossRef]
- Foresti, M.L.; Arisi, G.M.; Katki, K.; Montañez, A.; Sanchez, R.M.; Shapiro, L.A. Chemokine CCL2 and its receptor CCR2 are increased in the hippocampus following pilocarpine-induced status epilepticus. J. Neuroinflamm. 2009, 6, 40. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wang, X.; Mo, X.; Xi, Z.; Xiao, F.; Li, J.; Zhu, X.; Luan, G.; Wang, Y.; Li, Y.; et al. Expression of monocyte chemoattractant protein-1 in brain tissue of patients with intractable epilepsy. Clin. Neuropathol. 2008, 27, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Nordli, D.R., Jr.; Alden, T.D.; DiPatri, A., Jr.; Laux, L.; Kelley, K.; Rosenow, J.; Schuele, S.U.; Rajaram, V.; Koh, S. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J. Neuroinflamm. 2009, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronica, E.; Bauer, S.; Bozzi, Y.; Caleo, M.; Dingledine, R.; Gorter, J.A.; Henshall, D.C.; Kaufer, D.; Koh, S.; Loscher, W.; et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 2017, 58 (Suppl. 3), 27–38. [Google Scholar] [CrossRef] [PubMed]
- Vinet, J.; Vainchtein, I.D.; Spano, C.; Giordano, C.; Bordini, D.; Curia, G.; Dominici, M.; Boddeke, H.W.; Eggen, B.J.; Biagini, G. Microglia are less pro-inflammatory than myeloid infiltrates in the hippocampus of mice exposed to status epilepticus. Glia 2016, 64, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Akin, D.; Ravizza, T.; Maroso, M.; Carcak, N.; Eryigit, T.; Vanzulli, I.; Aker, R.G.; Vezzani, A.; Onat, F.Y. IL-1beta is induced in reactive astrocytes in the somatosensory cortex of rats with genetic absence epilepsy at the onset of spike-and-wave discharges, and contributes to their occurrence. Neurobiol. Dis. 2011, 44, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Dohgu, S.; Takata, F.; Machida, T.; Bölükbaşi Hatip, F.F.; Hatip-Al-Khatib, I.; Yamauchi, A.; Kataoka, Y. TNF-α-sensitive brain pericytes activate microglia by releasing IL-6 through cooperation between IκB-NFκB and JAK-STAT3 pathways. Brain Res. 2018, 1692, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Takata, F.; Dohgu, S.; Sakaguchi, S.; Sakai, K.; Yamanaka, G.; Iwao, T.; Matsumoto, J.; Kimura, I.; Sezaki, Y.; Tanaka, Y.; et al. Oncostatin-M-Reactive Pericytes Aggravate Blood-Brain Barrier Dysfunction by Activating JAK/STAT3 Signaling In Vitro. Neuroscience 2019, 422, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Jansson, D.; Smyth, L.C.; Dragunow, M. Brain Pericytes As Mediators of Neuroinflammation. Trends Pharmacol. Sci. 2017, 38, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, G.; Takamatsu, T.; Morichi, S.; Yamazaki, T.; Mizoguchi, I.; Ohno, K.; Watanabe, Y.; Ishida, Y.; Oana, S.; Suzuki, S.; et al. Interleukin-1β in peripheral monocytes is associated with seizure frequency in pediatric drug-resistant epilepsy. J. Neuroimmunol. 2021, 352, 577475. [Google Scholar] [CrossRef] [PubMed]
- Avignone, E.; Ulmann, L.; Levavasseur, F.; Rassendren, F.; Audinat, E. Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J. Neurosci. 2008, 28, 9133–9144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Liao, Y.; Morgan, S.; Mathur, R.; Feustel, P.; Mazurkiewicz, J.; Qian, J.; Chang, J.; Mathern, G.W.; Adamo, M.A.; et al. Noninflammatory Changes of Microglia Are Sufficient to Cause Epilepsy. Cell Rep. 2018, 22, 2080–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, J.G.; Boop, F.A.; Mrak, R.E.; Griffin, W.S. Increased neuronal beta-amyloid precursor protein expression in human temporal lobe epilepsy: Association with interleukin-1 alpha immunoreactivity. J. Neurochem. 1994, 63, 1872–1879. [Google Scholar] [CrossRef]
- Ravizza, T.; Gagliardi, B.; Noe, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Teng, Q.; Ghosh, C.; Fan, Q.; Nguyen, M.T.; Desai, N.K.; Bawa, H.; Rasmussen, P.; Masaryk, T.K.; Janigro, D. Blood-brain barrier damage, but not parenchymal white blood cells, is a hallmark of seizure activity. Brain Res. 2010, 1353, 176–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galea, I.; Bernardes-Silva, M.; Forse, P.A.; van Rooijen, N.; Liblau, R.S.; Perry, V.H. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J. Exp. Med. 2007, 204, 2023–2030. [Google Scholar] [CrossRef] [Green Version]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespel, A.; Coubes, P.; Rousset, M.C.; Brana, C.; Rougier, A.; Rondouin, G.; Bockaert, J.; Baldy-Moulinier, M.; Lerner-Natoli, M. Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res. 2002, 952, 159–169. [Google Scholar] [CrossRef]
- Iyer, A.; Zurolo, E.; Spliet, W.G.; van Rijen, P.C.; Baayen, J.C.; Gorter, J.A.; Aronica, E. Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 2010, 51, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, H.; Konishi, Y.; Beach, T.G.; Yamada, N.; Makino, S.; Tooyama, I. Infiltration of T lymphocytes and expression of icam-1 in the hippocampus of patients with hippocampal sclerosis. Acta Histochem. Cytochem. 2010, 43, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steve, T.A.; Jirsch, J.D.; Gross, D.W. Quantification of subfield pathology in hippocampal sclerosis: A systematic review and meta-analysis. Epilepsy Res. 2014, 108, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Kohm, A.P.; McMahon, J.S.; Podojil, J.R.; Begolka, W.S.; DeGutes, M.; Kasprowicz, D.J.; Ziegler, S.F.; Miller, S.D. Cutting Edge: Anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J. Immunol. 2006, 176, 3301–3305. [Google Scholar] [CrossRef] [Green Version]
- Setiady, Y.Y.; Coccia, J.A.; Park, P.U. In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII + phagocytes. Eur. J. Immunol. 2010, 40, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Rasmussen, J.P.; Rudensky, A.Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007, 8, 191–197. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Cao, C.; Chen, Z.; Bankaitis, V.; Tzima, E.; Sheibani, N.; Burridge, K. Pericytes regulate vascular basement membrane remodeling and govern neutrophil extravasation during inflammation. PLoS ONE 2012, 7, e45499. [Google Scholar] [CrossRef] [Green Version]
- Stark, K.; Eckart, A.; Haidari, S.; Tirniceriu, A.; Lorenz, M.; von Brühl, M.L.; Gärtner, F.; Khandoga, A.G.; Legate, K.R.; Pless, R.; et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat. Immunol. 2013, 14, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Muñoz, I.; Compte, M.; Álvarez-Cienfuegos, A.; Álvarez-Vallina, L.; Sanz, L. Lipopolysaccharide activates Toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway and proinflammatory response in human pericytes. J. Biol. Chem. 2014, 289, 2457–2468. [Google Scholar] [CrossRef] [Green Version]
- Pieper, C.; Marek, J.J.; Unterberg, M.; Schwerdtle, T.; Galla, H.J. Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Res. 2014, 1550, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jansson, D.; Rustenhoven, J.; Feng, S.; Hurley, D.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Faull, R.L.; Dragunow, M. A role for human brain pericytes in neuroinflammation. J. Neuroinflamm. 2014, 11, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, J.; Takata, F.; Machida, T.; Takahashi, H.; Soejima, Y.M.; Funakoshi, M.; Futagami, K.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Tumor necrosis factor-α-stimulated brain pericytes possess a unique cytokine and chemokine release profile and enhance microglial activation. Neurosci. Lett. 2014, 578, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Kovac, A.; Erickson, M.A.; Banks, W.A. Brain microvascular pericytes are immunoactive in culture: Cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J. Neuroinflamm. 2011, 8, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Alcendor, D.J.; Charest, A.M.; Zhu, W.Q.; Vigil, H.E.; Knobel, S.M. Infection and upregulation of proinflammatory cytokines in human brain vascular pericytes by human cytomegalovirus. J. Neuroinflamm. 2012, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Takata, F.; Dohgu, S.; Matsumoto, J.; Takahashi, H.; Machida, T.; Wakigawa, T.; Harada, E.; Miyaji, H.; Koga, M.; Nishioku, T.; et al. Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J. Neuroinflamm. 2011, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Machida, T.; Takata, F.; Matsumoto, J.; Takenoshita, H.; Kimura, I.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Brain pericytes are the most thrombin-sensitive matrix metalloproteinase-9-releasing cell type constituting the blood-brain barrier in vitro. Neurosci. Lett. 2015, 599, 109–114. [Google Scholar] [CrossRef]
- Garbelli, R.; de Bock, F.; Medici, V.; Rousset, M.C.; Villani, F.; Boussadia, B.; Arango-Lievano, M.; Jeanneteau, F.; Daneman, R.; Bartolomei, F.; et al. PDGFRβ(+) cells in human and experimental neuro-vascular dysplasia and seizures. Neuroscience 2015, 306, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Jansson, D.; Scotter, E.L.; Rustenhoven, J.; Coppieters, N.; Smyth, L.C.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Graham, E.S.; Faull, R.L.; et al. Interferon-γ blocks signalling through PDGFRβ in human brain pericytes. J. Neuroinflamm. 2016, 13, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoni, P.; Badaut, J.; Dargazanli, C.; De Maudave, A.F.; Klement, W.; Costalat, V.; Marchi, N. The pericyte-glia interface at the blood-brain barrier. Clin. Sci. 2018, 132, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klement, W.; Blaquiere, M.; Zub, E.; deBock, F.; Boux, F.; Barbier, E.; Audinat, E.; Lerner-Natoli, M.; Marchi, N. A pericyte-glia scarring develops at the leaky capillaries in the hippocampus during seizure activity. Epilepsia 2019, 60, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Takata, F.; Yamanaka, G.; Yasunaga, M.; Hashiguchi, K.; Tominaga, K.; Itoh, K.; Kataoka, Y.; Yamauchi, A.; Dohgu, S. Reactive pericytes in early phase are involved in glial activation and late-onset hypersusceptibility to pilocarpine-induced seizures in traumatic brain injury model mice. J. Pharmacol. Sci. 2021, 145, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Klement, W.; Garbelli, R.; Zub, E.; Rossini, L.; Tassi, L.; Girard, B.; Blaquiere, M.; Bertaso, F.; Perroy, J.; de Bock, F.; et al. Seizure progression and inflammatory mediators promote pericytosis and pericyte-microglia clustering at the cerebrovasculature. Neurobiol. Dis. 2018, 113, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Aalderink, M.; Scotter, E.L.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Graham, E.S.; Faull, R.L.; Curtis, M.A.; Park, T.I.; et al. TGF-beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function. J. Neuroinflamm. 2016, 13, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morganti, J.M.; Jopson, T.D.; Liu, S.; Riparip, L.K.; Guandique, C.K.; Gupta, N.; Ferguson, A.R.; Rosi, S. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J. Neurosci. 2015, 35, 748–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biber, K.; Möller, T.; Boddeke, E.; Prinz, M. Central nervous system myeloid cells as drug targets: Current status and translational challenges. Nat. Rev. Drug Discov. 2016, 15, 110–124. [Google Scholar] [CrossRef]
- Shechter, R.; London, A.; Varol, C.; Raposo, C.; Cusimano, M.; Yovel, G.; Rolls, A.; Mack, M.; Pluchino, S.; Martino, G.; et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009, 6, e1000113. [Google Scholar] [CrossRef] [PubMed]
- London, A.; Itskovich, E.; Benhar, I.; Kalchenko, V.; Mack, M.; Jung, S.; Schwartz, M. Neuroprotection and progenitor cell renewal in the injured adult murine retina requires healing monocyte-derived macrophages. J. Exp. Med. 2011, 208, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Planas, R.; Martin, R.; Sospedra, M. Long-term safety and efficacy of natalizumab in relapsing-remitting multiple sclerosis: Impact on quality of life. Patient Relat. Outcome Meas. 2014, 5, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappos, L.; O’Connor, P.; Radue, E.W.; Polman, C.; Hohlfeld, R.; Selmaj, K.; Ritter, S.; Schlosshauer, R.; von Rosenstiel, P.; Zhang-Auberson, L.; et al. Long-term effects of fingolimod in multiple sclerosis: The randomized FREEDOMS extension trial. Neurology 2015, 84, 1582–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotgiu, S.; Murrighile, M.R.; Constantin, G. Treatment of refractory epilepsy with natalizumab in a patient with multiple sclerosis. Case report. BMC Neurol. 2010, 10, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider-Hohendorf, T.; Mohan, H.; Bien, C.G.; Breuer, J.; Becker, A.; Görlich, D.; Kuhlmann, T.; Widman, G.; Herich, S.; Elpers, C.; et al. CD8(+) T-cell pathogenicity in Rasmussen encephalitis elucidated by large-scale T-cell receptor sequencing. Nat. Commun. 2016, 7, 11153. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Wise, L.E.; Allegood, J.C.; O’Brien, M.; Avni, D.; Reeves, T.M.; Knapp, P.E.; Lu, J.; Luo, C.; Miles, M.F.; et al. Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat. Neurosci. 2014, 17, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Gnatkovsky, V.; Othman, I.; Shaikh, M.F. From the Molecular Mechanism to Pre-clinical Results: Anti-epileptic Effects of Fingolimod. Curr. Neuropharmacol. 2020, 18, 1126–1137. [Google Scholar] [CrossRef]
- Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O’Connor, P.W. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2003, 348, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Schneider-Hohendorf, T.; Rossaint, J.; Mohan, H.; Böning, D.; Breuer, J.; Kuhlmann, T.; Gross, C.C.; Flanagan, K.; Sorokin, L.; Vestweber, D.; et al. VLA-4 blockade promotes differential routes into human CNS involving PSGL-1 rolling of T cells and MCAM-adhesion of TH17 cells. J. Exp. Med. 2014, 211, 1833–1846. [Google Scholar] [CrossRef]
- Steinman, L. Blocking adhesion molecules as therapy for multiple sclerosis: Natalizumab. Nat. Rev. Drug Discov. 2005, 4, 510–518. [Google Scholar] [CrossRef]
- Abkur, T.M.; Kearney, H.; Hennessy, M.J. Refractory epilepsy following natalizumab associated PML. Mult. Scler. Relat. Disord. 2018, 20, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.J.; Card, D.; Keohane, C.; et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Yanagawa, Y.; Masubuchi, Y.; Kataoka, H.; Kawaguchi, T.; Ohtsuki, M.; Hoshino, Y. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J. Immunol. 1998, 160, 5037–5044. [Google Scholar] [PubMed]
- Chun, J.; Hartung, H.P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebbins, M.J.; Gastfriend, B.D.; Canfield, S.G.; Lee, M.S.; Richards, D.; Faubion, M.G.; Li, W.J.; Daneman, R.; Palecek, S.P.; Shusta, E.V. Human pluripotent stem cell-derived brain pericyte-like cells induce blood-brain barrier properties. Sci. Adv. 2019, 5, eaau7375. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Monocytes | Microglia |
|---|---|
| CCR2-red fluorescent protein (RFP) [11,12,14] | Identified the binding adaptor molecule 1 (IBA1) P2Y12 [45] Transmembrane protein 119 (TMEM119) [50] |
| Mouse Model | Genetic Mouse Model | mRNA Upregulation in Hippocampus | Significant Points | |
|---|---|---|---|---|
| Varvel et al., 2016 [11] | Pilocarpine Kainic acid | CCR2 RFP/+ | IL-1β, IL-6 TNF-α, CCL2, iNOS | Blocking monocyte entry using the CCR2 KO model mouse did not alter iNOS, CCL2, TNF-α, or IL-6 mRNA levels in hippocampal tissues, but reduced IL-1β by 50%. |
| Tian et al. 2017 [12] | Kainic acid | CX3CR1 GFP/+:CCR2 RFP/+ | IL-1 a, IL-1β, IL-1RA, CCL2, CCL3, CCL5, CCL12, CXCL10 | Knocking out CCR2R can virtually abolish KA-induced IL-1β upregulation |
| Monocytes | CD4+ | CD8+ | |
|---|---|---|---|
| Kainic acid-treated mice | Appears from day 1, peaks in 3 days, disappears in 7–14 days [11,12,14] | Initially appears within blood vessels but mainly within the neuropils at 2–4 weeks [7] | Appears, peaks 2 weeks after injection and persists at 4 weeks [7] |
| Pilocarpine-treated | Appears after 96 h [79] | Not described | |
| Electrical stimulation | Not described | Appears after 24–48 h and disappears after 7 days [8] | |
| Not identified [19] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamanaka, G.; Morichi, S.; Takamatsu, T.; Watanabe, Y.; Suzuki, S.; Ishida, Y.; Oana, S.; Yamazaki, T.; Takata, F.; Kawashima, H. Links between Immune Cells from the Periphery and the Brain in the Pathogenesis of Epilepsy: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 4395. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094395
Yamanaka G, Morichi S, Takamatsu T, Watanabe Y, Suzuki S, Ishida Y, Oana S, Yamazaki T, Takata F, Kawashima H. Links between Immune Cells from the Periphery and the Brain in the Pathogenesis of Epilepsy: A Narrative Review. International Journal of Molecular Sciences. 2021; 22(9):4395. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094395
Chicago/Turabian StyleYamanaka, Gaku, Shinichiro Morichi, Tomoko Takamatsu, Yusuke Watanabe, Shinji Suzuki, Yu Ishida, Shingo Oana, Takashi Yamazaki, Fuyuko Takata, and Hisashi Kawashima. 2021. "Links between Immune Cells from the Periphery and the Brain in the Pathogenesis of Epilepsy: A Narrative Review" International Journal of Molecular Sciences 22, no. 9: 4395. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094395