
Modeling Neurotransmission: Computational Tools to Investigate Neurological Disorders



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Modeling Neurotransmission
2. Compartmental Modeling
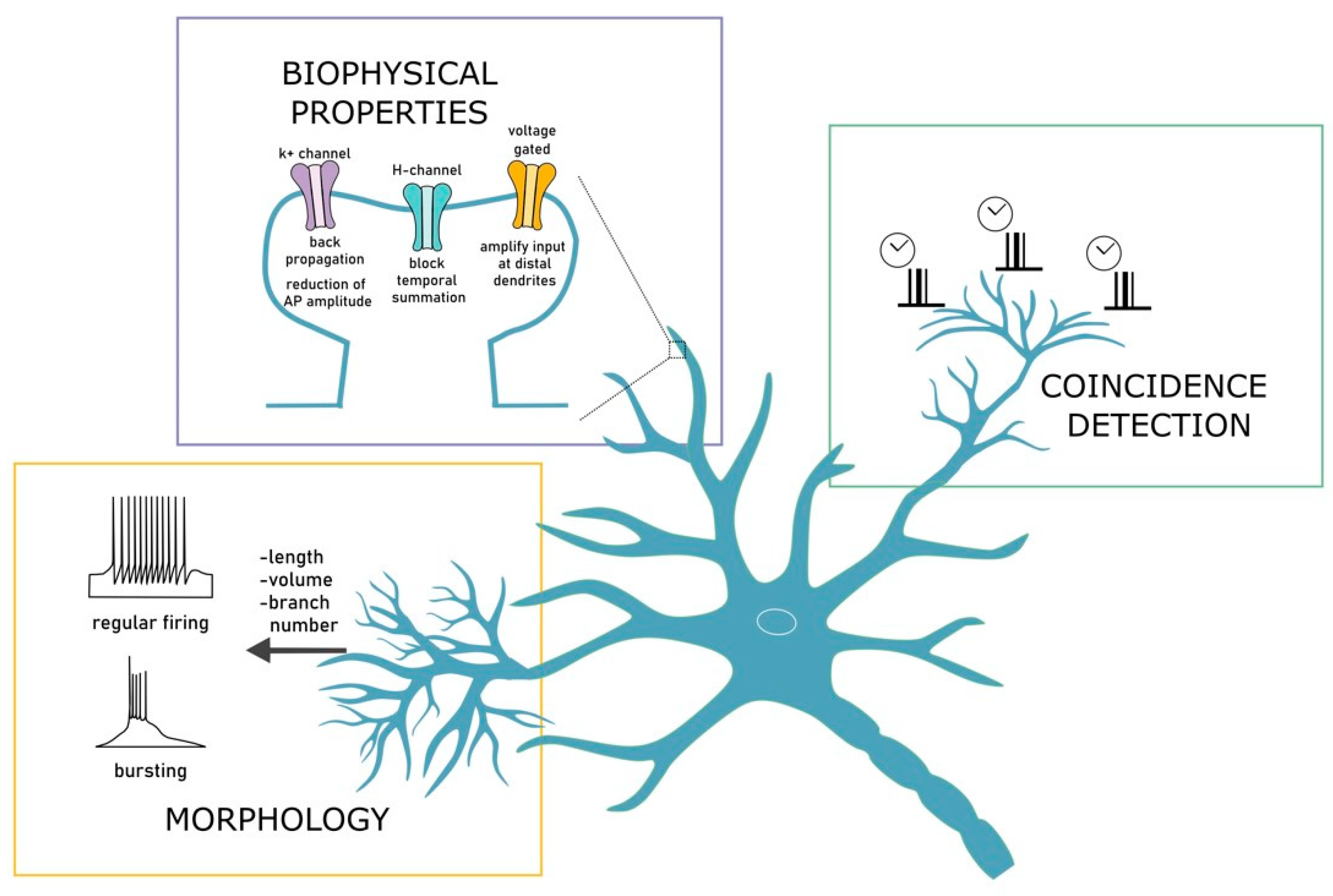
2.1. Dendrites
2.2. Synapses
3. Modeling Diseases
3.1. Epilepsy
3.1.1. Realistic Models
3.1.2. Abstract Models
3.2. Alzheimer’s Disease
3.2.1. Realistic Models
3.2.2. Abstract Models
3.3. Parkinson
3.3.1. Realistic Models
3.3.2. Abstract Models
3.4. Schizophrenia
3.4.1. Realistic Models
3.4.2. Abstract Models
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bornholdt, S. Less Is More in Modeling Large Genetic Networks. Science 2005, 310, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Prinz, A.A.; Bucher, D.; Marder, E. Similar Network Activity from Disparate Circuit Parameters. Nat. Neurosci. 2004, 7, 1345–1352. [Google Scholar] [CrossRef]
- Valton, V.; Romaniuk, L.; Steele, J.D.; Lawrie, S.; Seriès, P. Comprehensive Review: Computational Modelling of Schizophrenia. Neurosci. Biobehav. Rev. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayan, P.; Abbott, L.F. Theoretical Neuroscience: Computational and Mathematical Modeling of Neural Systems; MIT Press: Cambridge, MA, USA, 2001; ISBN 978-0-262-54185-5. [Google Scholar]
- Marr, D. Vision: A Computational Investigation into the Human Representation and Processing of Visual Information; W.H. Freeman and Company: New York, NY, USA, 2010. [Google Scholar] [CrossRef]
- Rall, W. Theory of Physiological Properties of Dendrites. Ann. N. Y. Acad. Sci. 1962. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, T.; Cathala, L.; Matsui, K.; Shigemoto, R.; DiGregorio, D.A. Thin Dendrites of Cerebellar Interneurons Confer Sublinear Synaptic Integration and a Gradient of Short-Term Plasticity. Neuron 2012, 73, 1159–1172. [Google Scholar] [CrossRef] [Green Version]
- Palmer, L.M.; Stuart, G.J. Membrane Potential Changes in Dendritic Spines during Action Potentials and Synaptic Input. J. Neurosci. 2009, 29, 6897–6903. [Google Scholar] [CrossRef] [Green Version]
- Herreras, O. Propagating Dendritic Action Potential Mediates Synaptic Transmission in CA1 Pyramidal Cells in Situ. J. Neurophysiol. 1990, 64, 1429–1441. [Google Scholar] [CrossRef]
- Takahashi, N.; Oertner, T.G.; Hegemann, P.; Larkum, M.E. Active Cortical Dendrites Modulate Perception. Science 2016, 354, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.E.; Whitney, D.E.; Scholl, B.; Fitzpatrick, D. Orientation Selectivity and the Functional Clustering of Synaptic Inputs in Primary Visual Cortex. Nat. Neurosci. 2016, 19, 1003–1009. [Google Scholar] [CrossRef]
- Lavzin, M.; Rapoport, S.; Polsky, A.; Garion, L.; Schiller, J. Nonlinear Dendritic Processing Determines Angular Tuning of Barrel Cortex Neurons in Vivo. Nature 2012, 490, 397–401. [Google Scholar] [CrossRef]
- Lee, D.; Lin, B.-J.; Lee, A.K. Hippocampal Place Fields Emerge upon Single-Cell Manipulation of Excitability during Behavior. Science 2012, 337, 849–853. [Google Scholar] [CrossRef] [Green Version]
- Bittner, K.C.; Milstein, A.D.; Grienberger, C.; Romani, S.; Magee, J.C. Behavioral Time Scale Synaptic Plasticity Underlies CA1 Place Fields. Science 2017, 357, 1033–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheffield, M.E.; Dombeck, D.A. Dendritic Mechanisms of Hippocampal Place Field Formation. Curr. Opin. Neurobiol. 2019, 54, 1–11. [Google Scholar] [CrossRef]
- Poirazi, P.; Papoutsi, A. Illuminating Dendritic Function with Computational Models. Nat. Rev. Neurosci. 2020, 21, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Vetter, P.; Roth, A.; Häusser, M. Propagation of Action Potentials in Dendrites Depends on Dendritic Morphology. J. Neurophysiol. 2001, 85, 926–937. [Google Scholar] [CrossRef]
- Krichmar, J.L.; Nasuto, S.J.; Scorcioni, R.; Washington, S.D.; Ascoli, G.A. Effects of Dendritic Morphology on CA3 Pyramidal Cell Electrophysiology: A Simulation Study. Brain Res. 2002, 941, 11–28. [Google Scholar] [CrossRef]
- Komendantov, A.O.; Ascoli, G.A. Dendritic Excitability and Neuronal Morphology as Determinants of Synaptic Efficacy. J. Neurophysiol. 2009, 101, 1847–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zador, A.M.; Agmon-Snir, H.; Segev, I. The Morphoelectrotonic Transform: A Graphical Approach to Dendritic Function. J. Neurosci. 1995, 15, 1669–1682. [Google Scholar] [CrossRef]
- Van Elburg, R.A.J.; van Ooyen, A. Impact of Dendritic Size and Dendritic Topology on Burst Firing in Pyramidal Cells. PLoS Comput. Biol. 2010, 6, e1000781. [Google Scholar] [CrossRef] [Green Version]
- Psarrou, M.; Stefanou, S.S.; Papoutsi, A.; Tzilivaki, A.; Cutsuridis, V.; Poirazi, P. A Simulation Study on the Effects of Dendritic Morphology on Layer V Prefrontal Pyramidal Cell Firing Behavior. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, M.; Migliore, M.; Ascoli, G.A. Functional Impact of Dendritic Branch-Point Morphology. J. Neurosci. 2013, 33, 2156–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, S.; Nikolic, K.; Schultz, S.R. Neuronal Gain Modulability Is Determined by Dendritic Morphology: A Computational Optogenetic Study. PLoS Comput. Biol. 2018, 14, e1006027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connors, B.W.; Regehr, W.G. Neuronal Firing: Does Function Follow Form? Curr. Biol. 1996, 6, 1560–1562. [Google Scholar] [CrossRef] [Green Version]
- Johnston, D.; Magee, J.C.; Colbert, C.M.; Cristie, B.R. Active Properties of Neuronal Dendrites. Annu. Rev. Neurosci. 1996, 19, 165–186. [Google Scholar] [CrossRef]
- Branco, T.; Häusser, M. Synaptic Integration Gradients in Single Cortical Pyramidal Cell Dendrites. Neuron 2011, 69, 885–892. [Google Scholar] [CrossRef] [Green Version]
- Archie, K.A.; Mel, B.W. A Model for Intradendritic Computation of Binocular Disparity. Nat. Neurosci. 2000, 3, 54–63. [Google Scholar] [CrossRef]
- Segev, I.; London, M. Untangling Dendrites with Quantitative Models. Science 2000, 290, 744–750. [Google Scholar] [CrossRef]
- Stuart, G.; Spruston, N. Determinants of Voltage Attenuation in Neocortical Pyramidal Neuron Dendrites. J. Neurosci. 1998, 18, 3501–3510. [Google Scholar] [CrossRef]
- Magee, J.C.; Johnston, D. A Synaptically Controlled, Associative Signal for Hebbian Plasticity in Hippocampal Neurons. Science 1997, 275, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, D.A.; Magee, J.C.; Colbert, C.M.; Johnston, D. K+ Channel Regulation of Signal Propagation in Dendrites of Hippocampal Pyramidal Neurons. Nature 1997, 387, 869–875. [Google Scholar] [CrossRef]
- Migliore, M.; Hoffman, D.A.; Magee, J.C.; Johnston, D. Role of an A-Type K+ Conductance in the Back-Propagation of Action Potentials in the Dendrites of Hippocampal Pyramidal Neurons. J. Comput. Neurosci. 1999, 7, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Ascoli, G.A.; Gasparini, S.; Medinilla, V.; Migliore, M. Local Control of Postinhibitory Rebound Spiking in CA1 Pyramidal Neuron Dendrites. J. Neurosci. 2010, 30, 6434–6442. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, I.; Scimemi, A.; Savtchenko, L.; Kullmann, D.M.; Walker, M.C. Ih-Mediated Depolarization Enhances the Temporal Precision of Neuronal Integration. Nat. Commun. 2011, 2, 199. [Google Scholar] [CrossRef]
- Ferrarese, L.; Jouhanneau, J.-S.; Remme, M.W.H.; Kremkow, J.; Katona, G.; Rózsa, B.; Schreiber, S.; Poulet, J.F.A. Dendrite-Specific Amplification of Weak Synaptic Input during Network Activity In Vivo. Cell Rep. 2018, 24, 3455–3465.e5. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, S.; Migliore, M.; Magee, J.C. On the Initiation and Propagation of Dendritic Spikes in CA1 Pyramidal Neurons. J. Neurosci. 2004, 24, 11046–11056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shai, A.S.; Anastassiou, C.A.; Larkum, M.E.; Koch, C. Physiology of Layer 5 Pyramidal Neurons in Mouse Primary Visual Cortex: Coincidence Detection through Bursting. PLoS Comput. Biol. 2015, 11, e1004090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariav, G.; Polsky, A.; Schiller, J. Submillisecond Precision of the Input-Output Transformation Function Mediated by Fast Sodium Dendritic Spikes in Basal Dendrites of CA1 Pyramidal Neurons. J. Neurosci. 2003, 23, 7750–7758. [Google Scholar] [CrossRef]
- Mainen, Z.F.; Malinow, R.; Svoboda, K. Synaptic Calcium Transients in Single Spines Indicate That NMDA Receptors Are Not Saturated. Nature 1999, 399, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Larkum, M. A Cellular Mechanism for Cortical Associations: An Organizing Principle for the Cerebral Cortex. Trends Neurosci. 2013, 36, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, D.; Bigiani, A.; Porro, C.A.; Mapelli, J. Inhibitory Plasticity: From Molecules to Computation and Beyond. Int. J. Mol. Sci. 2020, 21, 1805. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, D.; Mapelli, J.; D’Angelo, E. Long-Term Spatiotemporal Reconfiguration of Neuronal Activity Revealed by Voltage-Sensitive Dye Imaging in the Cerebellar Granular Layer. Neural Plast. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapelli, J.; Gandolfi, D.; Vilella, A.; Zoli, M.; Bigiani, A. Heterosynaptic GABAergic Plasticity Bidirectionally Driven by the Activity of Pre- and Postsynaptic NMDA Receptors. Proc. Natl. Acad. Sci. USA 2016, 113, 9898–9903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelfsema, P.R.; Holtmaat, A. Control of Synaptic Plasticity in Deep Cortical Networks. Nat. Rev. Neurosci. 2018, 19, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Koch, G.; Di Lorenzo, F.; Bonnì, S.; Giacobbe, V.; Bozzali, M.; Caltagirone, C.; Martorana, A. Dopaminergic Modulation of Cortical Plasticity in Alzheimer’s Disease Patients. Neuropsychopharmacology 2014, 39, 2654–2661. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.S.; Huntsman, M.M. Pathological Plasticity in Fragile X Syndrome. Available online: https://www.hindawi.com/journals/np/2012/275630/ (accessed on 11 February 2021).
- Abbott, L.F.; Regehr, W.G. Synaptic Computation. Nature 2004, 431, 796–803. [Google Scholar] [CrossRef]
- Mapelli, J.; D’Angelo, E. The Spatial Organization of Long-Term Synaptic Plasticity at the Input Stage of Cerebellum. J. Neurosci. 2007, 27, 1285–1296. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.P.; Padamsey, Z.; D’Amour, J.A.; Emptage, N.J.; Froemke, R.C.; Vogels, T.P. Synaptic Transmission Optimization Predicts Expression Loci of Long-Term Plasticity. Neuron 2017, 96, 177–189.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennig, M.H. Theoretical Models of Synaptic Short Term Plasticity. Front. Comput. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Liaw, J.S.; Berger, T.W. Dynamic Synapse: A New Concept of Neural Representation and Computation. Hippocampus 1996, 6, 591–600. [Google Scholar] [CrossRef]
- Maass, W.; Zador, A.M. Dynamic Stochastic Synapses as Computational Units. Neural Comput. 1999, 11, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Tong, R.; Emptage, N.J.; Padamsey, Z. A Two-Compartment Model of Synaptic Computation and Plasticity. Mol. Brain 2020, 13, 79. [Google Scholar] [CrossRef]
- Tsodyks, M.V.; Markram, H. The Neural Code between Neocortical Pyramidal Neurons Depends on Neurotransmitter Release Probability. Proc. Natl. Acad. Sci. USA 1997, 94, 719–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liaw, J.-S.; Berger, T.W. Dynamic Synapse: Harnessing the Computing Power of Synaptic Dynamics. Neurocomputing 1999, 26–27, 199–206. [Google Scholar] [CrossRef]
- Goda, Y.; Stevens, C.F. Two Components of Transmitter Release at a Central Synapse. Proc. Natl. Acad. Sci. USA 1994, 91, 12942–12946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, E.R. How Does Synaptotagmin Trigger Neurotransmitter Release? Annu. Rev. Biochem. 2008, 77, 615–641. [Google Scholar] [CrossRef] [Green Version]
- Daw, M.I.; Tricoire, L.; Erdelyi, F.; Szabo, G.; McBain, C.J. Asynchronous Transmitter Release from Cholecystokinin-Containing Inhibitory Interneurons Is Widespread and Target-Cell Independent. J. Neurosci. 2009, 29, 11112–11122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hefft, S.; Jonas, P. Asynchronous GABA Release Generates Long-Lasting Inhibition at a Hippocampal Interneuron–Principal Neuron Synapse. Nat. Neurosci. 2005, 8, 1319–1328. [Google Scholar] [CrossRef]
- D’Angelo, E.; Solinas, S.; Garrido, J.; Casellato, C.; Pedrocchi, A.; Mapelli, J.; Gandolfi, D.; Prestori, F. Realistic Modeling of Neurons and Networks: Towards Brain Simulation. Funct. Neurol. 2013, 28, 153–166. [Google Scholar] [CrossRef]
- Meier-Schellersheim, M.; Fraser, I.D.C.; Klauschen, F. Multiscale Modeling for Biologists. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Forti, L.; Cesana, E.; Mapelli, J.; D’Angelo, E. Ionic Mechanisms of Autorhythmic Firing in Rat Cerebellar Golgi Cells. J. Physiol. 2006, 574, 711–729. [Google Scholar] [CrossRef]
- Solinas, S.; Forti, L.; Cesana, E.; Mapelli, J.; De Schutter, E.; D’Angelo, E. Computational Reconstruction of Pacemaking and Intrinsic Electroresponsiveness in Cerebellar Golgi Cells. Front. Cell. Neurosci. 2007, 1. [Google Scholar] [CrossRef] [Green Version]
- Lytton, W. Computer Modelling of Epilepsy. Nat. Rev. Neurosci. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasscock, E.; Qian, J.; Yoo, J.W.; Noebels, J.L. Masking Epilepsy by Combining Two Epilepsy Genes. Nat. Neurosci. 2007, 10, 1554–1558. [Google Scholar] [CrossRef] [PubMed]
- Mulley, J.C.; Scheffer, I.E.; Petrou, S.; Berkovic, S.F. Channelopathies as a Genetic Cause of Epilepsy. Curr. Opin. Neurol. 2003, 16, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Franks, K.M.; Bartol, T.M.; Sejnowski, T.J. A Monte Carlo Model Reveals Independent Signaling at Central Glutamatergic Synapses. Biophys. J. 2002, 83, 2333–2348. [Google Scholar] [CrossRef] [Green Version]
- Lytton, W.; Omurtag, A. Tonic-Clonic Transitions in Computer Simulation. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, R.C.; Howell, F.W.; Goddard, N.H.; Schutter, E.D. Non-Curated Distributed Databases for Experimental Data and Models in Neuroscience. Netw. Comput. Neural Syst. 2002, 13, 415–428. [Google Scholar] [CrossRef]
- Strogatz, S.H. Exploring Complex Networks. Nature 2001, 410, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.J.; Santhakumar, V.; Soltesz, I. Modeling the dentate gyrus. In Progress in Brain Research; Scharfman, H.E., Ed.; The Dentate Gyrus: A Comprehensive Guide to Structure, Function, and Clinical Implications; Elsevier: Amsterdam, The Netherlands, 2007; Volume 163, pp. 639–658. [Google Scholar]
- Sunderam, S.; Osorio, I.; Frei, M.; Watkins, J.F. Stochastic Modeling and Prediction of Experimental Seizures in Sprague-Dawley Rats. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 2001. [Google Scholar] [CrossRef]
- Wendling, F.; Bartolomei, F.; Bellanger, J.J.; Chauvel, P. Epileptic Fast Activity Can Be Explained by a Model of Impaired GABAergic Dendritic Inhibition. Eur. J. Neurosci. 2002, 15, 1499–1508. [Google Scholar] [CrossRef]
- LeMasson, G.; Marder, E.; Abbott, L.F. Activity-Dependent Regulation of Conductances in Model Neurons. Science 1993, 259, 1915–1917. [Google Scholar] [CrossRef] [Green Version]
- Mantegazza, M.; Cestèle, S.; Catterall, W. Sodium Channelopathies of Skeletal Muscle and Brain. Physiol. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lytton, W.; Sejnowski, T. Computer Model of Ethosuximide’s Effect on a Thalamic Neuron. Ann. Neurol. 1992. [Google Scholar] [CrossRef]
- Spampanato, J.; Aradi, I.; Soltesz, I.; Goldin, A. Increased Neuronal Firing in Computer Simulations of Sodium Channel Mutations That Cause Generalized Epilepsy with Febrile Seizures Plus. J. Neurophysiol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, J.V.; Adams, P.R. Voltage-Clamp Analysis of Muscarinic Excitation in Hippocampal Neurons. Brain Res. 1982, 250, 71–92. [Google Scholar] [CrossRef]
- Maccaferri, G.; McBain, C.J. The Hyperpolarization-Activated Current (Ih) and Its Contribution to Pacemaker Activity in Rat CA1 Hippocampal Stratum Oriens-Alveus Interneurones. J. Physiol. 1996, 497, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Pape, H.C. Queer Current and Pacemaker: The Hyperpolarization-Activated Cation Current in Neurons. Annu. Rev. Physiol. 1996, 58, 299–327. [Google Scholar] [CrossRef]
- Chen, K.; Aradi, I.; Thon, N.; Eghbal-Ahmadi, M.; Baram, T.; Soltesz, I. Persistently Modified H-Channels after Complex Febrile Seizures Convert the Seizure-Induced Enhancement of Inhibition to Hyperexcitability. Nat. Med. 2001. [Google Scholar] [CrossRef]
- Cressman, J.R.; Ullah, G.; Ziburkus, J.; Schiff, S.; Barreto, E. The Influence of Sodium and Potassium Dynamics on Excitability, Seizures, and the Stability of Persistent States: I. Single Neuron Dynamics. J. Comput. Neurosci. 2008. [Google Scholar] [CrossRef] [Green Version]
- Rutecki, P.A.; Lebeda, F.J.; Johnston, D. Epileptiform Activity Induced by Changes in Extracellular Potassium in Hippocampus. J. Neurophysiol. 1985, 54, 1363–1374. [Google Scholar] [CrossRef]
- Traub, R.D.; Dingledine, R. Model of Synchronized Epileptiform Bursts Induced by High Potassium in CA3 Region of Rat Hippocampal Slice. Role of Spontaneous EPSPs in Initiation. J. Neurophysiol. 1990, 64, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Ullah, G.; Cressman, J.R.; Barreto, E.; Schiff, S. The Influence of Sodium and Potassium Dynamics on Excitability, Seizures, and the Stability of Persistent States: II. Network and Glial Dynamics. J. Comput. Neurosci. 2008. [Google Scholar] [CrossRef] [PubMed]
- Beck, H. Plasticity of Antiepileptic Drug Targets. Epilepsia 2007, 48, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, B.H.; Schmitt, N.; Calloe, K.; Dalby Brown, W.; Grunnet, M.; Olesen, S.-P. The Acrylamide (S)-1 Differentially Affects Kv7 (KCNQ) Potassium Channels. Neuropharmacology 2006, 51, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic Seizures and Epilepsy: Definitions Proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef]
- Lyman, K.A.; Chetkovich, D.M. CRAFTing a New Approach to Antiepileptic Drug Discovery. Epilepsy Curr. 2019, 19, 182–183. [Google Scholar] [CrossRef]
- Goldwyn, J.H.; Shea-Brown, E. The What and Where of Adding Channel Noise to the Hodgkin-Huxley Equations. PLoS Comput. Biol. 2011, 7, e1002247. [Google Scholar] [CrossRef] [Green Version]
- Destexhe, A.; Contreras, D.; Steriade, M. LTS Cells in Cerebral Cortex and Their Role in Generating Spike-and-Wave Oscillations. Neurocomputing 2001, 38–40, 555–563. [Google Scholar] [CrossRef]
- Traub, R.D.; Wong, R.K. Cellular Mechanism of Neuronal Synchronization in Epilepsy. Science 1982, 216, 745–747. [Google Scholar] [CrossRef]
- Traub, R.D.; Jefferys, J.G.; Whittington, M.A. Enhanced NMDA Conductance Can Account for Epileptiform Activity Induced by Low Mg2+ in the Rat Hippocampal Slice. J. Physiol. 1994, 478 Pt 3, 379–393. [Google Scholar] [CrossRef]
- Traub, R.D. Cellular Mechanisms Underlying the Inhibitory Surround of Penicillin Epileptogenic Foci. Brain Res. 1983, 261, 277–284. [Google Scholar] [CrossRef]
- Traub, R.D.; Contreras, D.; Cunningham, M.O.; Murray, H.; LeBeau, F.E.N.; Roopun, A.; Bibbig, A.; Wilent, W.B.; Higley, M.J.; Whittington, M.A. Single-Column Thalamocortical Network Model Exhibiting Gamma Oscillations, Sleep Spindles, and Epileptogenic Bursts. J. Neurophysiol. 2005, 93, 2194–2232. [Google Scholar] [CrossRef]
- Volman, V.; Perc, M.; Bazhenov, M. Gap Junctions and Epileptic Seizures—Two Sides of the Same Coin? PLoS ONE 2011, 6, e20572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C. Sleeping and Dreaming. Science 1991, 251, 326–327. [Google Scholar] [CrossRef]
- Kiesmann, M.; Marescaux, C.; Vergnes, M.; Micheletti, G.; Depaulis, A.; Warter, J.M. Audiogenic Seizures in Wistar Rats before and after Repeated Auditory Stimuli: Clinical, Pharmacological, and Electroencephalographic Studies. J. Neural Transm. 1988, 72, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Lytton, W.W. Computer Model of Antiepileptic Effects Mediated by Alterations in GABA(A)-Mediated Inhibition. Neuroreport 1998, 9, 691–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, C.; Lecar, H. Voltage Oscillations in the Barnacle Giant Muscle Fiber. Biophys. J. 1981, 35, 193–213. [Google Scholar] [CrossRef] [Green Version]
- Beverlin, B.; Kakalios, J.; Nykamp, D.; Netoff, T.I. Dynamical Changes in Neurons during Seizures Determine Tonic to Clonic Shift. J. Comput. Neurosci. 2012, 33, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Naze, S.; Bernard, C.; Jirsa, V. Computational Modeling of Seizure Dynamics Using Coupled Neuronal Networks: Factors Shaping Epileptiform Activity. PLoS Comput. Biol. 2015, 11, e1004209. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, G.P.; Bazhenov, M. Ionic Dynamics Mediate Spontaneous Termination of Seizures and Postictal Depression State. J. Neurosci. 2011, 31, 8870–8882. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhu, J.; Liu, Y.; Yang, M.; Tian, C.; Jiang, S.; Wang, Y.; Guo, H.; Wang, K.; Shu, Y. Enhancement of Asynchronous Release from Fast-Spiking Interneuron in Human and Rat Epileptic Neocortex. PLoS Biol. 2012, 10, e1001324. [Google Scholar] [CrossRef] [Green Version]
- Lavi, A.; Perez, O.; Ashery, U. Shaping Neuronal Network Activity by Presynaptic Mechanisms. PLoS Comput. Biol. 2015, 11, e1004438. [Google Scholar] [CrossRef] [Green Version]
- Destexhe, A.; Rudolph, M.; Fellous, J.M.; Sejnowski, T.J. Fluctuating Synaptic Conductances Recreate in Vivo-like Activity in Neocortical Neurons. Neuroscience 2001, 107, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Marder, E.; Prinz, A.A. Modeling Stability in Neuron and Network Function: The Role of Activity in Homeostasis. Bioessays 2002, 24, 1145–1154. [Google Scholar] [CrossRef]
- Compte, A.; Brunel, N.; Goldman-Rakic, P.S.; Wang, X.J. Synaptic Mechanisms and Network Dynamics Underlying Spatial Working Memory in a Cortical Network Model. Cereb. Cortex 2000, 10, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Schiff, S.J.; Sauer, T.; Kumar, R.; Weinstein, S.L. Neuronal Spatiotemporal Pattern Discrimination: The Dynamical Evolution of Seizures. Neuroimage 2005, 28, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Park, E.-H.; Barreto, E.; Gluckman, B.J.; Schiff, S.J.; So, P. A Model of the Effects of Applied Electric Fields on Neuronal Synchronization. J. Comput. Neurosci. 2005, 19, 53–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirsa, V.K.; Stacey, W.C.; Quilichini, P.P.; Ivanov, A.I.; Bernard, C. On the nature of seizure dynamics. Brain 2014, 137, 2210–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chizhov, A.V.; Zefirov, A.V.; Amakhin, D.V.; Smirnova, E.Y.; Zaitsev, A.V. Minimal model of interictal and ictal discharges “epileptor-2”. PLoS Comput. Biol. 2018, 14, e1006186. [Google Scholar] [CrossRef]
- Netoff, T.I.; Schiff, S.J. Decreased Neuronal Synchronization during Experimental Seizures. J. Neurosci. 2002, 22, 7297–7307. [Google Scholar] [CrossRef] [Green Version]
- Feldt, S.; Wang, J.X.; Shtrahman, E.; Dzakpasu, R.; Olariu, E.; Zochowski, M. Functional Clustering in Hippocampal Cultures: Relating Network Structure and Dynamics. Phys. Biol. 2010, 7, 046004. [Google Scholar] [CrossRef]
- Casali, S.; Tognolina, M.; Gandolfi, D.; Mapelli, J.; D’Angelo, E. Cellular-Resolution Mapping Uncovers Spatial Adaptive Filtering at the Rat Cerebellum Input Stage. Commun. Biol. 2020, 3, 635. [Google Scholar] [CrossRef]
- Pozzi, P.; Gandolfi, D.; Porro, C.A.; Bigiani, A.; Mapelli, J. Scattering Compensation for Deep Brain Microscopy: The Long Road to Get Proper Images. Front. Phys. 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Sombati, S.; Delorenzo, R.J. Recurrent Spontaneous Seizure Activity in Hippocampal Neuronal Networks in Culture. J. Neurophysiol. 1995, 73, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Beggs, J.M.; Plenz, D. Neuronal Avalanches in Neocortical Circuits. J. Neurosci. 2003, 23, 11167–11177. [Google Scholar] [CrossRef] [Green Version]
- Achard, S.; Bullmore, E. Efficiency and Cost of Economical Brain Functional Networks. PLoS Comput. Biol. 2007, 3, e17. [Google Scholar] [CrossRef] [PubMed]
- Achard, S.; Salvador, R.; Whitcher, B.; Suckling, J.; Bullmore, E. A Resilient, Low-Frequency, Small-World Human Brain Functional Network with Highly Connected Association Cortical Hubs. J. Neurosci. 2006, 26, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.J.; Soltesz, I. Nonrandom Connectivity of the Epileptic Dentate Gyrus Predicts a Major Role for Neuronal Hubs in Seizures. Proc. Natl. Acad. Sci. USA 2008, 105, 6179–6184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, M.; Soltesz, I. Computational Modeling of Epilepsy. Epilepsia 2011, 52, 12–15. [Google Scholar] [CrossRef]
- Kaiser, M.; Hilgetag, C.C. Nonoptimal Component Placement, but Short Processing Paths, Due to Long-Distance Projections in Neural Systems. PLoS Comput. Biol. 2006, 2, e95. [Google Scholar] [CrossRef]
- Kaiser, M.; Varier, S. Evolution and Development of Brain Networks: From Caenorhabditis Elegans to Homo Sapiens. Network 2011, 22, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Wilke, C.; Worrell, G.; He, B. Graph Analysis of Epileptogenic Networks in Human Partial Epilepsy. Epilepsia 2011, 52, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, G.K.; Esiri, M.M. Plaques, Tangles and Dementia. A Quantitative Study. J. Neurol. Sci. 1982, 56, 343–356. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The Cholinergic Hypothesis of Alzheimer’s Disease: A Review of Progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutajangout, A.; Wisniewski, T. Tau-Based Therapeutic Approaches for Alzheimer’s Disease—A Mini-Review. Gerontology 2014, 60, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 Hypothesis of Alzheimer’s Disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhikav, V.; Anand, K. Potential Predictors of Hippocampal Atrophy in Alzheimer’s Disease. Drugs Aging 2011, 28, 1–11. [Google Scholar] [CrossRef]
- Joshi, A.; Wang, D.H.; Watterson, S.; McCleand, P.L.; Behera, C.K.; Sharp, T.; Wong-Lina, K.F. Opportunities for multiscale computational modelling of serotonergic drug effects in Alzheimer’s disease. Neuropharmacology 2020, 174, 108118. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Murphy, R.M.; Pallitto, M.M. Probing the Kinetics of Beta-Amyloid Self-Association. J. Struct. Biol. 2000, 130, 109–122. [Google Scholar] [CrossRef]
- Lomakin, A.; Chung, D.S.; Benedek, G.B.; Kirschner, D.A.; Teplow, D.B. On the Nucleation and Growth of Amyloid Beta-Protein Fibrils: Detection of Nuclei and Quantitation of Rate Constants. Proc. Natl. Acad. Sci. USA 1996, 93, 1125–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomakin, A.; Kirschner, D.; Benedek, G. Kinetic Theory of Fibrillogenesis of Amyloid B-Protein (Alzheimer Disease y Light Scattering). Available online: /paper/Kinetic-theory-of-fibrillogenesis-of-amyloid-(-y-)-Lomakin-Kirschner/6ab1dbdfa474fc16dc3474c499aef50dd3012bda (accessed on 5 February 2021).
- Pallitto, M.; Murphy, R. A Mathematical Model of the Kinetics of Beta-Amyloid Fibril Growth from the Denatured State. Biophys. J. 2001. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.R.; Mureșan, A.; Lee, K.; Murphy, R. Urea Modulation of Β-amyloid Fibril Growth: Experimental Studies and Kinetic Models. Protein Sci. 2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, L.; Urbanc, B.; Buldyrev, S.; Christie, R.; Gómez-Isla, T.; Havlin, S.; McNamara, M.; Stanley, H.; Hyman, B. Aggregation and Disaggregation of Senile Plaques in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, F.; Stott, J.; Visser, S.A.G.; Bendtsen, C. Interplay between α-, β-, and γ-Secretases Determines Biphasic Amyloid-β Protein Level in the Presence of a γ-Secretase Inhibitor. J. Biol. Chem. 2013, 288, 785–792. [Google Scholar] [CrossRef] [Green Version]
- De Caluwé, D.; Dupont, G. The Progression towards Alzheimer’s Disease Described as a Bistable Switch Arising from the Positive Loop between Amyloids and Ca2+. J. Theor. Biol. 2013. [Google Scholar] [CrossRef]
- Anastasio, T. Exploring the Contribution of Estrogen to Amyloid-Beta Regulation: A Novel Multifactorial Computational Modeling Approach. Front. Pharm. 2013. [Google Scholar] [CrossRef] [Green Version]
- Sasidharakurup, H.; Melethadathil, N.; Nair, B.; Diwakar, S. A Systems Model of Parkinson’s Disease Using Biochemical Systems Theory. OMICS J. Integr. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Petrella, J.R.; Hao, W.; Rao, A.; Doraiswamy, P.M. Computational Causal Modeling of the Dynamic Biomarker Cascade in Alzheimer’s Disease. Comput. Math. Methods Med. 2019, 2019, 6216530. [Google Scholar] [CrossRef] [Green Version]
- Romani, A.; Marchetti, C.; Bianchi, D.; Leinekugel, X.; Poirazi, P.; Migliore, M.; Marie, H. Computational Modeling of the Effects of Amyloid-Beta on Release Probability at Hippocampal Synapses. Front. Comput. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [Green Version]
- Culmone, V.; Migliore, M. Progressive Effect of Beta Amyloid Peptides Accumulation on CA1 Pyramidal Neurons: A Model Study Suggesting Possible Treatments. Front. Comput. Neurosci. 2012, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Coyle, D.; Wong-Lin, K.; Maguire, L. Computational Study of Hippocampal-Septal Theta Rhythm Changes Due to Beta-Amyloid-Altered Ionic Channels. PLoS ONE 2011, 6, e21579. [Google Scholar] [CrossRef] [PubMed]
- Abuhassan, K.; Coyle, D.; Maguire, L.P. Investigating the Neural Correlates of Pathological Cortical Networks in Alzheimer’s Disease Using Heterogeneous Neuronal Models. IEEE Trans. Biomed. Eng. 2012, 59, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.; Spiros, A.; Geerts, H. Simulations of Symptomatic Treatments for Alzheimer’s Disease: Computational Analysis of Pathology and Mechanisms of Drug Action. Alzheimer’s Res. Ther. 2012. [Google Scholar] [CrossRef] [Green Version]
- Menschik, E.D.; Finkel, L.H. Neuromodulatory Control of Hippocampal Function: Towards a Model of Alzheimer’s Disease. Artif. Intell. Med. 1998, 13, 99–121. [Google Scholar] [CrossRef]
- Buzsáki, G. Two-Stage Model of Memory Trace Formation: A Role for “Noisy” Brain States. Neuroscience 1989, 31, 551–570. [Google Scholar] [CrossRef]
- Bianchi, D.; Michele, P.D.; Marchetti, C.; Tirozzi, B.; Cuomo, S.; Marie, H.; Migliore, M. Effects of Increasing CREB-dependent Transcription on the Storage and Recall Processes in a Hippocampal CA1 Microcircuit. Hippocampus 2014. [Google Scholar] [CrossRef]
- Rowan, M.; Neymotin, S.; Lytton, W. Electrostimulation to Reduce Synaptic Scaling Driven Progression of Alzheimer’s Disease. Front. Comput. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Coyle, D.; Maguire, L. A Thalamo-Cortico-Thalamic Neural Mass Model to Study Alpha Rhythms in Alzheimer’s Disease. Neural Netw. 2011. [Google Scholar] [CrossRef]
- Meeter, M.; Murre, J. Tracelink: A Model of Consolidation and Amnesia. Cogn. Neuropsychol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Kéri, S.; Herzallah, M.M.; Myers, C.; Gluck, M. A Neural Model of Hippocampal–Striatal Interactions in Associative Learning and Transfer Generalization in Various Neurological and Psychiatric Patients. Brain Cogn. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAuley, M.T.; Kenny, R.; Kirkwood, T.; Wilkinson, D.; Jones, J.J.; Miller, V. A Mathematical Model of Aging-Related and Cortisol Induced Hippocampal Dysfunction. BMC Neurosci. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, D.; Ruppin, E.; Usher, M.; Herrmann, M. Neural Network Modeling of Memory Deterioration in Alzheimer’s Disease. Neural Comput. 1993, 5, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Tippett, L.J.; Farah, M.J. A Computational Model of Naming in Alzheimer’s Disease: Unitary or Multiple Impairments? Neuropsychology 1994, 8, 3–13. [Google Scholar] [CrossRef]
- Ruppin, E.; Reggia, J. A Neural Model of Memory Impairment in Diffuse Cerebral Atrophy. Br. J. Psychiatry J. Ment. Sci. 1995. [Google Scholar] [CrossRef]
- Bullmore, E.; Sporns, O. Complex Brain Networks: Graph Theoretical Analysis of Structural and Functional Systems. Nat. Rev. Neurosci. 2009, 10, 186–198. [Google Scholar] [CrossRef]
- Sporns, O. The Non-Random Brain: Efficiency, Economy, and Complex Dynamics. Front. Comput. Neurosci. 2011, 5. [Google Scholar] [CrossRef] [Green Version]
- Rembach, A.; Stingo, F.; Peterson, C.; Vannucci, M.; Do, K.A.; Wilson, W.J.; Macaulay, S.L.; Ryan, T.M.; Martins, R.N.; Ames, D.; et al. Bayesian Graphical Network Analyses Reveal Complex Biological Interactions Specific to Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 44, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wang, X.; Wang, Q.; Wang, Q. A Review of Computational Modeling and Deep Brain Stimulation: Applications to Parkinson’s Disease. Appl. Math. Mech. 2020, 1–22. [Google Scholar] [CrossRef]
- Caligiore, D.; Mannella, F.; Baldassarre, G. Different Dopaminergic Dysfunctions Underlying Parkinsonian Akinesia and Tremor. Front. Neurosci. 2019. [Google Scholar] [CrossRef] [Green Version]
- Muddapu, V.R.; Chakravarthy, V.S. Influence of energy deficiency on the subcellular processes of Substantia Nigra Pars Compacta cell for understanding Parkinsonian neurodegeneration. Neurol. Neurosurg. Psychiatry 2018, 89, 1181–1188. [Google Scholar]
- Wightman, R.M.; Zimmerman, J.B. Control of Dopamine Extracellular Concentration in Rat Striatum by Impulse Flow and Uptake. Brain Res. Brain Res. Rev. 1990, 15, 135–144. [Google Scholar] [CrossRef]
- John, C.E.; Jones, S.R. Voltammetric Characterization of the Effect of Monoamine Uptake Inhibitors and Releasers on Dopamine and Serotonin Uptake in Mouse Caudate-Putamen and Substantia Nigra Slices. Neuropharmacology 2007, 52, 1596–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieus, T.; Sola, E.; Mapelli, J.; Saftenku, E.; Rossi, P.; D’Angelo, E. LTP Regulates Burst Initiation and Frequency at Mossy Fiber–Granule Cell Synapses of Rat Cerebellum: Experimental Observations and Theoretical Predictions. J. Neurophysiol. 2006, 95, 686–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiencke, K.; Horstmann, A.; Mathar, D.; Villringer, A.; Neumann, J. Dopamine Release, Diffusion and Uptake: A Computational Model for Synaptic and Volume Transmission. PLoS Comput. Biol. 2020, 16, e1008410. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Pendyam, S.; Kalivas, P.W.; Nair, S.S. Molecular Diffusion Model of Neurotransmitter Homeostasis Around Synapses Supporting Gradients. Neural Comput. 2011, 23, 984–1014. [Google Scholar] [CrossRef]
- Moyer, J.T.; Wolf, J.; Finkel, L. Effects of Dopaminergic Modulation on the Integrative Properties of the Ventral Striatal Medium Spiny Neuron. J. Neurophysiol. 2007. [Google Scholar] [CrossRef]
- Nair, A.; Gutierrez-Arenas, O.; Eriksson, O.; Vincent, P.; Kotaleski, J.H. Sensing Positive versus Negative Reward Signals through Adenylyl Cyclase-Coupled GPCRs in Direct and Indirect Pathway Striatal Medium Spiny Neurons. J. Neurosci. 2015. [Google Scholar] [CrossRef] [Green Version]
- Yapo, C.; Nair, A.; Clement, L.; Castro, L.R.V.; Kotaleski, J.H.; Vincent, P. Detection of Phasic Dopamine by D1 and D2 Striatal Medium Spiny Neurons. J. Physiol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Albin, R.L.; Young, A.B.; Penney, J.B. The Functional Anatomy of Basal Ganglia Disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- DeLong, M.R. Primate Models of Movement Disorders of Basal Ganglia Origin. Trends Neurosci. 1990, 13, 281–285. [Google Scholar] [CrossRef]
- Rubin, J.; McIntyre, C.; Turner, R.S.; Wichmann, T. Basal Ganglia Activity Patterns in Parkinsonism and Computational Modeling of Their Downstream Effects. Eur. J. Neurosci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.; Wood, R.; Gurney, K. Dopamine-Modulated Dynamic Cell Assemblies Generated by the GABAergic Striatal Microcircuit. Neural Netw. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damodaran, S.; Evans, R.; Blackwell, K. Synchronized Firing of Fast-Spiking Interneurons Is Critical to Maintain Balanced Firing between Direct and Indirect Pathway Neurons of the Striatum. J. Neurophysiol. 2014. [Google Scholar] [CrossRef]
- Humphries, M.; Stewart, R.; Gurney, K. A Physiologically Plausible Model of Action Selection and Oscillatory Activity in the Basal Ganglia. J. Neurosci. 2006. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, M.; Kotaleski, J.H. Untangling Basal Ganglia Network Dynamics and Function: Role of Dopamine Depletion and Inhibition Investigated in a Spiking Network Model. eNeuro 2016. [Google Scholar] [CrossRef] [Green Version]
- Leblois, A.; Boraud, T.; Meissner, W.; Bergman, H.; Hansel, D. Competition between Feedback Loops Underlies Normal and Pathological Dynamics in the Basal Ganglia. J. Neurosci. 2006. [Google Scholar] [CrossRef] [Green Version]
- Humphries, M.D.; Obeso, J.; Dreyer, J.K. Insights into Parkinson’s disease from computational models of the basal ganglia. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Romano, M.R.; Moioli, R.C.; Elias, L.A. Evaluation of Frequency-Dependent Effects of Deep Brain Stimulation in a Cortex-Basal Ganglia-Thalamus Network Model of Parkinson’s Disease. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2020, 2020, 3638–3641. [Google Scholar] [CrossRef] [PubMed]
- Valverde, S.; Vandecasteele, M.; Piette, C.; Derousseaux, W.; Gangarossa, G.; Arbelaiz, A.A.; Touboul, J.; Degos, B.; Venance, L. Deep brain stimulation-guided optogenetic rescue of parkinsonian symptoms. Nat. Commun. 2020, 11, 2388. [Google Scholar] [CrossRef] [PubMed]
- Bardakjian, B.L.; Diamant, N.E. A Mapped Clock Oscillator Model for Transmembrane Electrical Rhythmic Activity in Excitable Cells. J. Theor. Biol. 1994, 166, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Hitti, F.L.; Chang, S.-Y.; Lee, D.C.; Roberts, D.W.; McIntyre, C.C.; Leiter, J.C. High Frequency Stimulation Abolishes Thalamic Network Oscillations: An Electrophysiological and Computational Analysis. J. Neural Eng. 2011, 8, 046001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.J.; Beverlin, B.; Netoff, T. Chaotic Desynchronization as the Therapeutic Mechanism of Deep Brain Stimulation. Front. Syst. Neurosci. 2011, 5. [Google Scholar] [CrossRef] [Green Version]
- Yousif, N.; Bain, P.G.; Nandi, D.; Borisyuk, R. A Population Model of Deep Brain Stimulation in Movement Disorders From Circuits to Cells. Front. Hum. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhugra, D. The Global Prevalence of Schizophrenia. PLoS Med. 2005, 2, e151. [Google Scholar] [CrossRef] [Green Version]
- Howes, O.D.; Kapur, S. The Dopamine hypothesis of schizophrenia: Version III—The final common pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef] [Green Version]
- van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef]
- Lewis, D.; Hashimoto, T. Cortical inhibitory neurons and schizophrenia. Nat. Rev. 2005, 6, 312–324. [Google Scholar] [CrossRef]
- Friston, K. A Theory of Cortical Responses. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 815–836. [Google Scholar] [CrossRef]
- Durstewitz, D.; Kelc, M.; Güntürkün, O. A Neurocomputational Theory of the Dopaminergic Modulation of Working Memory Functions. J. Neurosci. 1999. [Google Scholar] [CrossRef] [Green Version]
- Durstewitz, D.; Seamans, J.; Sejnowski, T. Dopamine-Mediated Stabilization of Delay-Period Activity in a Network Model of Prefrontal Cortex. J. Neurophysiol. 2000. [Google Scholar] [CrossRef] [PubMed]
- Seamans, J.; Yang, C. The Principal Features and Mechanisms of Dopamine Modulation in the Prefrontal Cortex. Prog. Neurobiol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J. Synaptic Basis of Cortical Persistent Activity: The Importance of NMDA Receptors to Working Memory. J. Neurosci. 1999. [Google Scholar] [CrossRef]
- Wang, X. Synaptic Reverberation Underlying Mnemonic Persistent Activity. Trends Neurosci. 2001. [Google Scholar] [CrossRef]
- Rolls, E.; Loh, M.; Deco, G.; Winterer, G. Computational Models of Schizophrenia and Dopamine Modulation in the Prefrontal Cortex. Nat. Rev. Neurosci. 2008. [Google Scholar] [CrossRef]
- Loh, M.; Rolls, E.; Deco, G. A Dynamical Systems Hypothesis of Schizophrenia. PLoS Comput. Biol. 2007. [Google Scholar] [CrossRef]
- Diwadkar, V.A.; Flaugher, B.; Jones, T.; Zalányi, L.; Ujfalussy, B.; Keshavan, M.S.; Erdi, P. Impaired associative learning in schizophrenia: Behavioral and computational studies. Cogn. Neurodyn. 2008, 2, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Siekmeier, P.J.; Hasselmo, M.E.; Howard, M.W.; Coyle, J. Modeling of context-dependent retrieval in hippocampal region CA1: Implications for cognitive function in schizophrenia. Schizophr. Res. 2007, 89, 177–190. [Google Scholar] [CrossRef]
- Spencer, K.M. The functional consequences of cortical circuit abnormalities on gamma oscillations in schizophrenia: Insights from computational modeling. Front. Hum. Neurosci. 2009, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.; Coombes, S.; Liddle, P.F. A Neural Mass Model for Abnormal Beta-Rebound in Schizophrenia. Multiscale Models Brain Disord. 2019, 13, 21–27. [Google Scholar]
- Friston, K.J. The Labile Brain. I. Neuronal Transients and Nonlinear Coupling. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fries, P.; Roelfsema, P.; Engel, A.; König, P.; Singer, W. Synchronization of Oscillatory Responses in Visual Cortex Correlates with Perception in Interocular Rivalry. Proc. Natl. Acad. Sci. USA 1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabuffo, G.; Fousek, J.; Bernard, C.; Jirsa, V. Neuronal cascades shape whole-brain functional dynamics at rest. Biorxiv 2020. [Google Scholar] [CrossRef]
- Knill, D.C.; Pouget, A. The Bayesian Brain: The Role of Uncertainty in Neural Coding and Computation. Trends Neurosci. 2004, 27, 712–719. [Google Scholar] [CrossRef]
- Mirza, M.B.; Adams, R.A.; Friston, K.; Parr, T. Introducing a Bayesian Model of Selective Attention Based on Active Inference. Sci. Rep. 2019, 9, 13915. [Google Scholar] [CrossRef] [Green Version]
- Friston, K.J.; Harrison, L.; Penny, W. Dynamic Causal Modelling. Neuroimage 2003, 19, 1273–1302. [Google Scholar] [CrossRef]
- Cooray, G.K.; Sengupta, B.; Douglas, P.K.; Friston, K. Dynamic Causal Modelling of Electrographic Seizure Activity Using Bayesian Belief Updating. Neuroimage 2016, 125, 1142–1154. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, R. Computer Simulations of Neural Information Processing and the Schizophrenia-Mania Dichotomy. Arch. Gen. Psychiatry 1987. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Sandoval, C.; Portillo, V.; Fresán, A.; Apiquian, R. Replication of a computer model of auditory hallucinations in schizophrenia. Actas Esp. Psiquiatr. 2005, 33, 141–146. [Google Scholar]
- Corlett, P.R.; Frith, C.D.; Fletcher, P.C. From Drugs to Deprivation: A Bayesian Framework for Understanding Models of Psychosis. Psychopharmacology 2009, 206, 515–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandolfi, D.; Boiani, G.M.; Bigiani, A.; Mapelli, J. Modeling Neurotransmission: Computational Tools to Investigate Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4565. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094565
Gandolfi D, Boiani GM, Bigiani A, Mapelli J. Modeling Neurotransmission: Computational Tools to Investigate Neurological Disorders. International Journal of Molecular Sciences. 2021; 22(9):4565. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094565
Chicago/Turabian StyleGandolfi, Daniela, Giulia Maria Boiani, Albertino Bigiani, and Jonathan Mapelli. 2021. "Modeling Neurotransmission: Computational Tools to Investigate Neurological Disorders" International Journal of Molecular Sciences 22, no. 9: 4565. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094565