
Transcriptome and MiRNAomics Analyses Identify Genes Associated with Cytoplasmic Male Sterility in Cotton (Gossypium hirsutum L.)

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Result
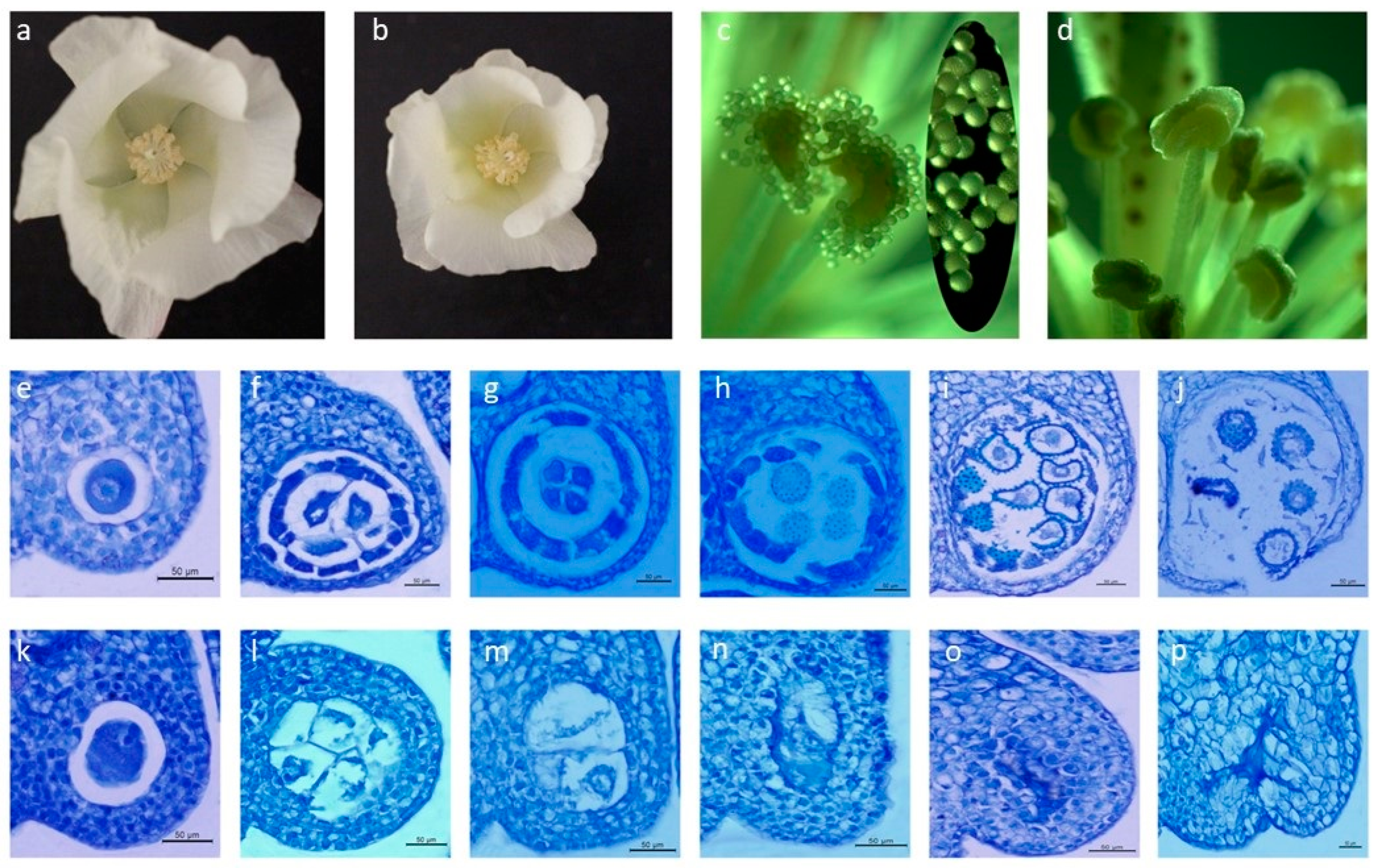
2.1. Morphological and Cytological Observations
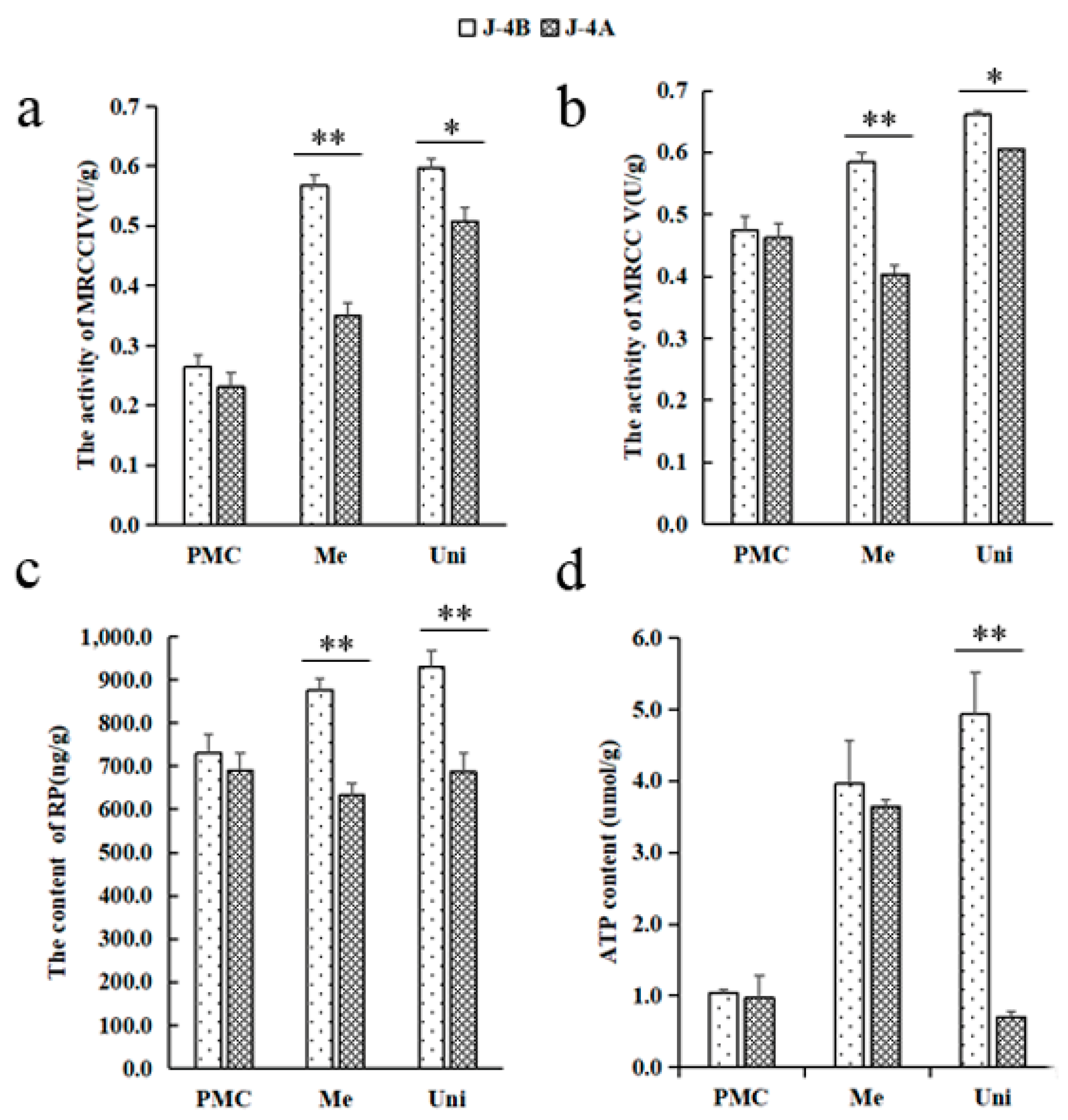
2.2. Mitochondria-Related Indexes
2.3. Overview of the mRNA and miRNA Sequencing Data for J4A and J4B
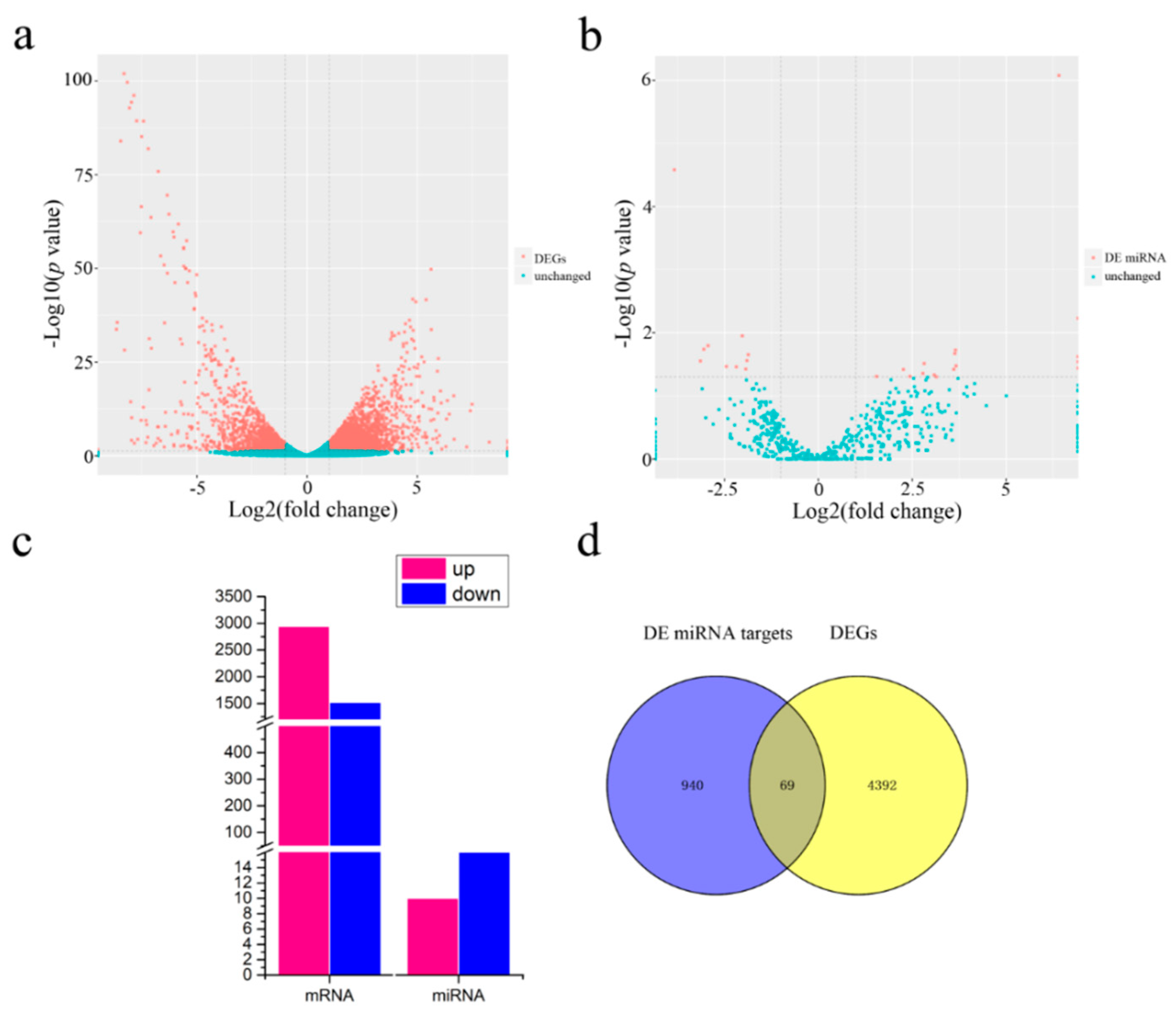
2.4. Differentially Expressed (DE) miRNAs and mRNAs
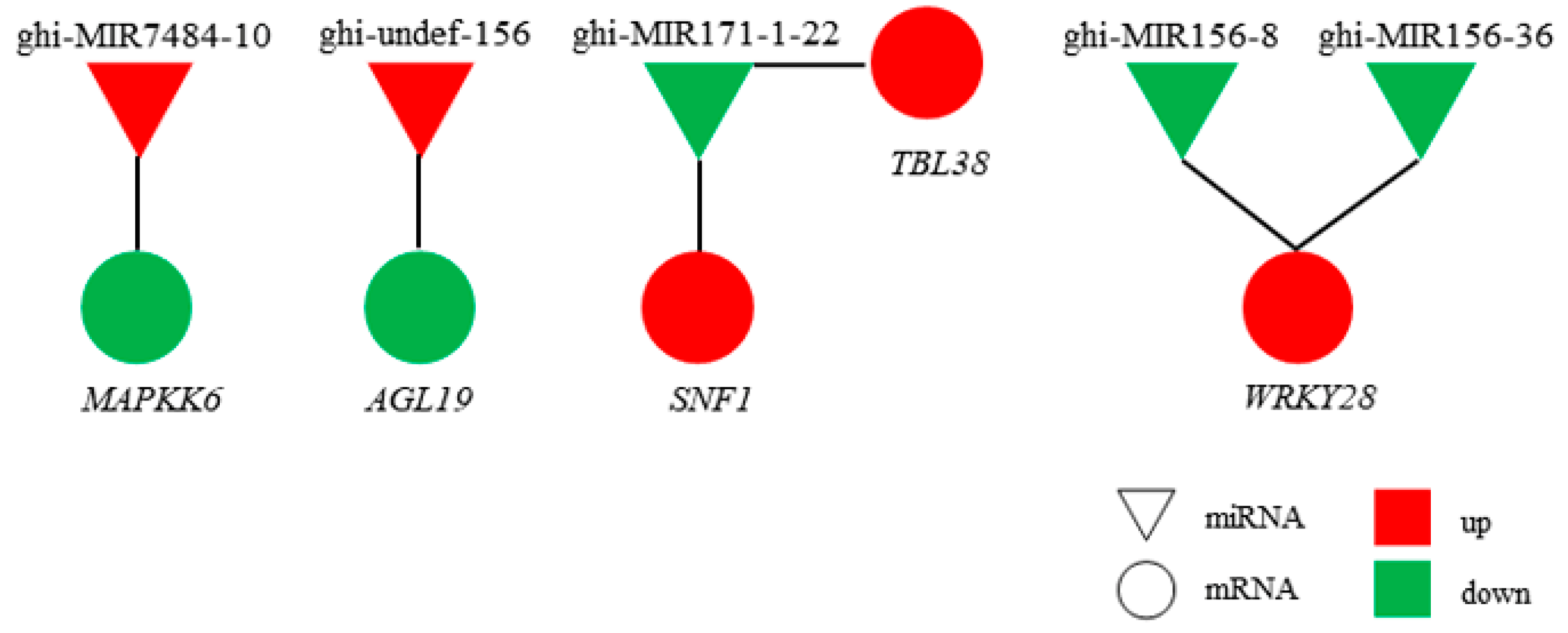
2.5. Regulatory Network of miRNAs and mRNAs
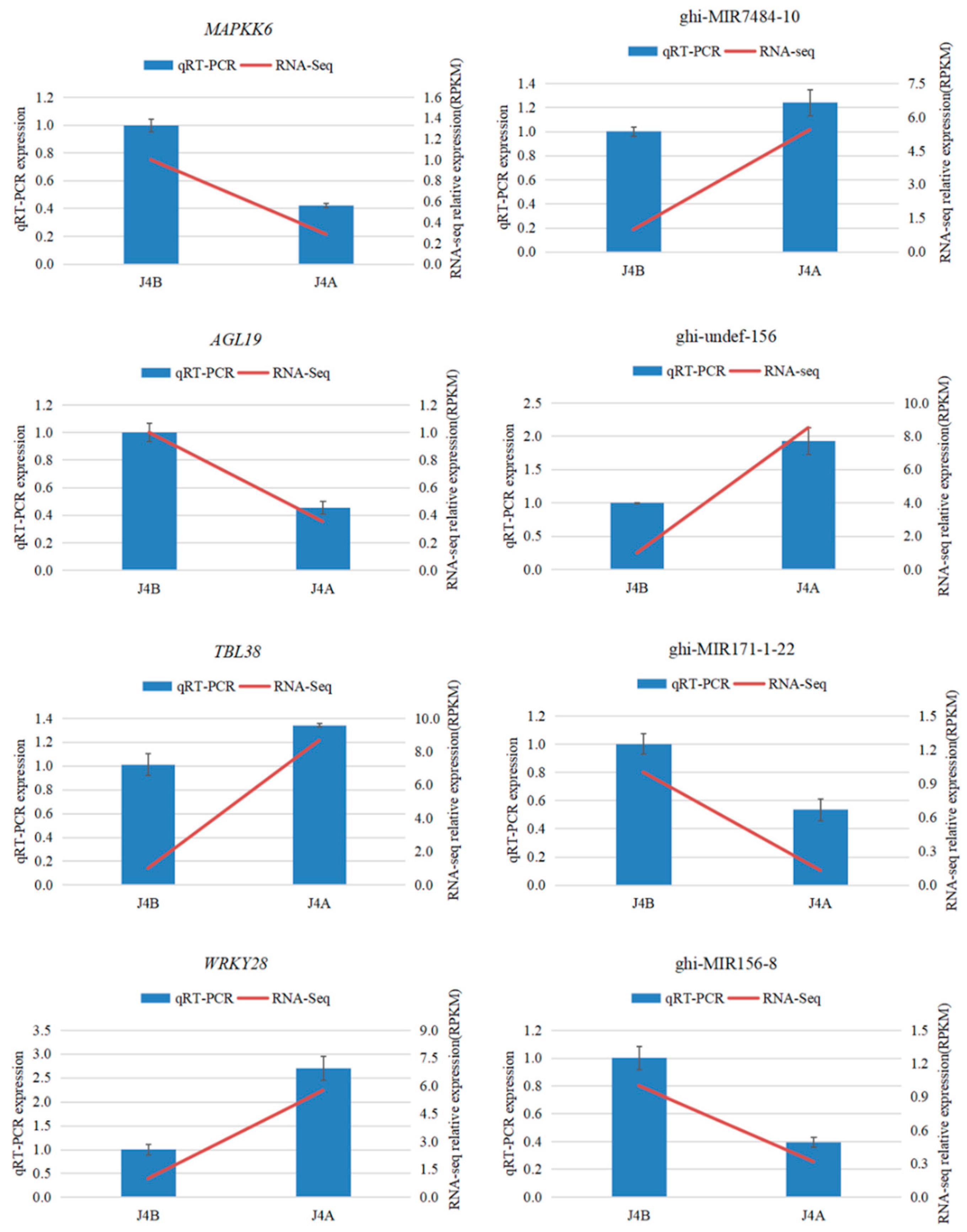
2.6. QRT-PCR Validation of miRNA and mRNA Expression
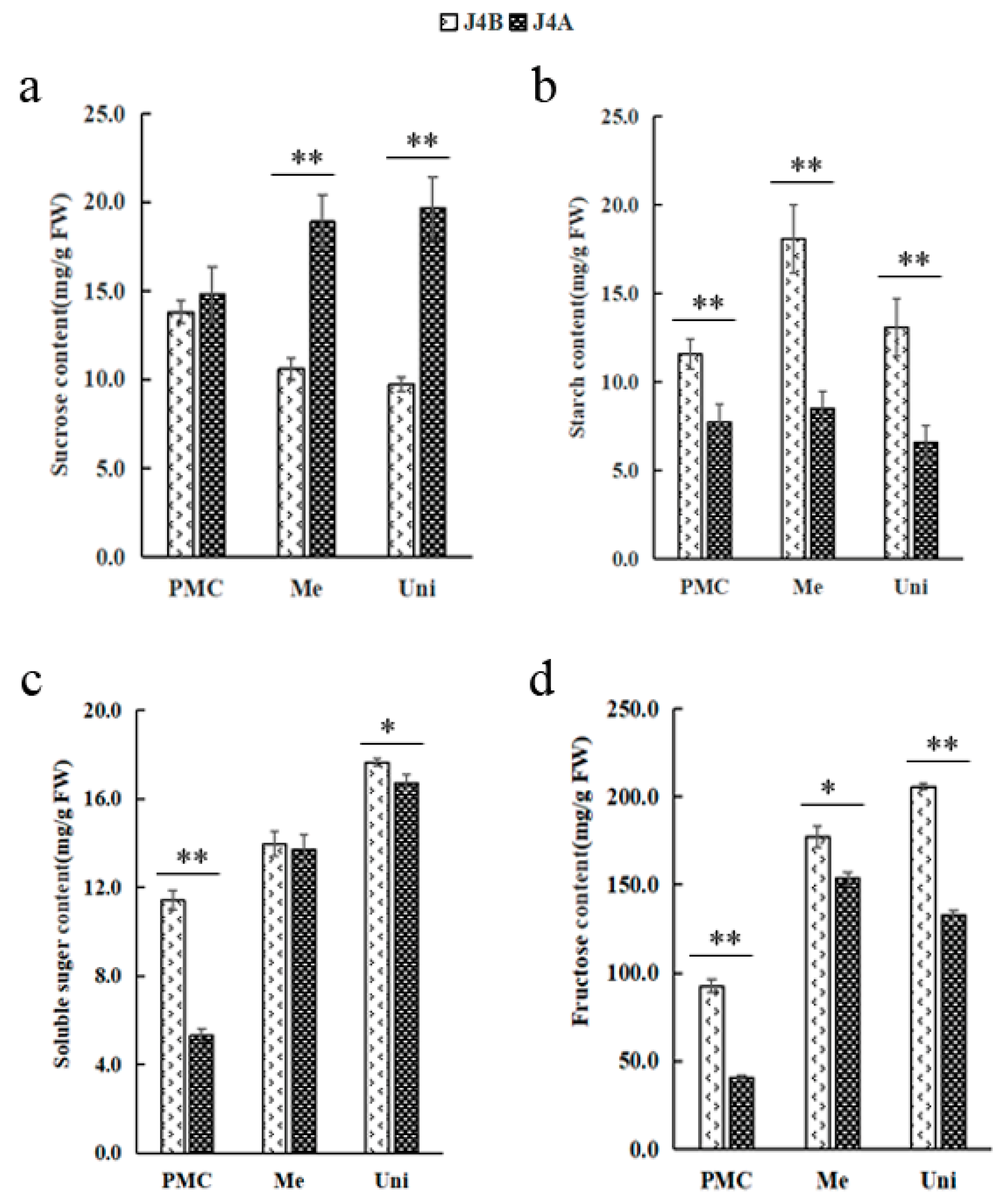
2.7. Determination of Physiological and Biochemical Indexes Related to the Glucose Metabolism Pathway
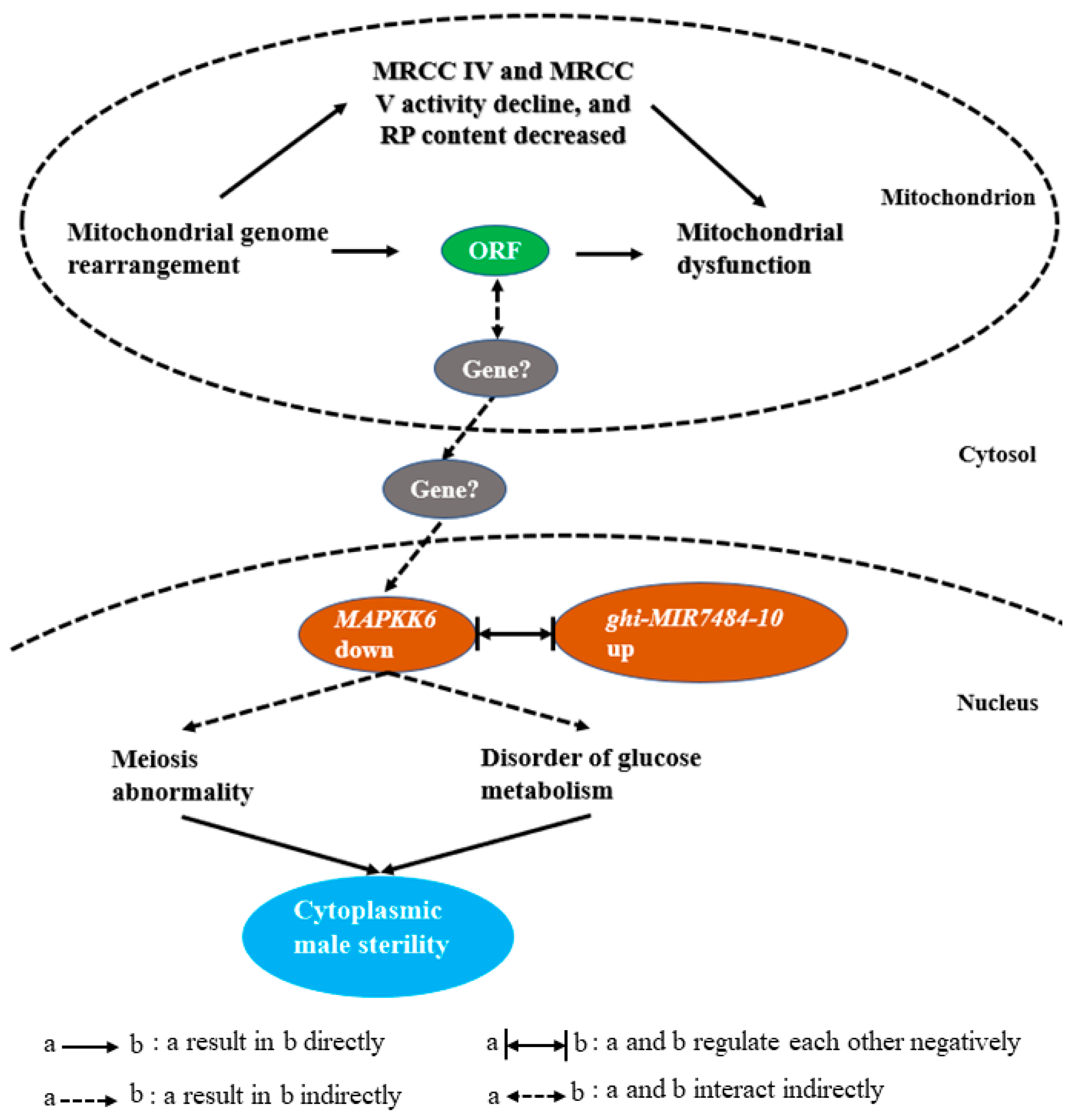
3. Discussion
3.1. Period of Abortion in Cotton
3.2. Disorder of Glucose Metabolism Is Related to the Occurrence of Male Sterility
3.3. The MAPK Signaling Pathway Regulates the Process of Proliferation and Meiosis
4. Materials and Methods
4.1. Plant Materials
4.2. Morphological and Cytological Observations
4.3. Detection of Mitochondria and Glucose Metabolism Related Indexes
4.4. RNA Sequencing and Data Processing
4.5. Construction of miRNA–mRNA Regulatory Network
4.6. Verification of the Transcriptome and miRNA Sequencing Results by Quantitative Real-Time Fluorescence PCR (qRT-PCR)
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferreira Júnior, J.; Ramos, A.; Chambergo, F.; Stambuk, B.; Muschellack, L.; Schumacher, R.; El-Dorry, H. Functional expression of the maize mitochondrial URF13 down-regulates galactose-induced GAL1 gene expression in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2006, 1, 30–36. [Google Scholar] [CrossRef]
- Horn, R.; Gupta, K.J.; Colombo, N. Mitochondrion role in molecular basis of cytoplasmic male sterility. Mitochondrion 2014, 19, 198–205. [Google Scholar] [CrossRef]
- Levings, C.S., III; Pring, D.R. Restriction endonuclease analysis of mitochondrial DNA from normal and texas cytoplasmic male-sterile maize. Science 1976, 193, 158–160. [Google Scholar] [CrossRef]
- Ding, B.; Hao, M.; Mei, D.; Zaman, Q.; Sang, S.; Wang, H.; Wang, W.; Fu, L.; Cheng, H.; Hu, Q. Transcriptome and hormone comparison of three cytoplasmic male sterile systems in Brassica napus. Int. J. Mol. Sci. 2018, 19, 4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, Y.D.; Lee, H.; Ro, N.; Kim, S.H.; Kim, J.; Kang, B.; Kang, S. Mitotypes based on structural variation of mitochondrial genomes imply relationships with morphological phenotypes and cytoplasmic male sterility in peppers. Front. Plant Sci. 2019, 10, 1343. [Google Scholar] [CrossRef] [PubMed]
- Bohra, A.; Jha, U.C.; Adhimoolam, P.; Bisht, D.; Singh, N. Cytoplasmic male sterility (CMS) in hybrid breeding in field crops. Plant Cell Rep. 2016, 35, 967–993. [Google Scholar] [CrossRef]
- Chase, C. Cytoplasmic male sterility: A window to the world of plant mitochondrial-nuclear interactions. Trends. Genet. 2007, 23, 81–90. [Google Scholar] [CrossRef]
- Okazaki, M.; Kazama, T.; Murata, H.; Motomura, K.; Toriyama, K. Whole mitochondrial genome sequencing and transcriptional analysis to uncover an RT102-Type cytoplasmic male sterility-associated candidate gene derived from Oryza rufipogon. Plant Cell Physiol. 2013, 54, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Chen, L.; Tang, D.; Liao, X.; Kong, X.; Li, B.; You, J.; Zhou, R. Discovery of four novel ORFs responsible for cytoplasmic male sterility (CMS) in cotton (Gossypium hirsutum L.) through comparative analysis of the mitochondrial genomes of four isoplasmic lines. Agronomy 2020, 10, 765. [Google Scholar] [CrossRef]
- Li, S.; Chen, Z.; Zhao, N.; Wang, Y.; Nie, H.; Hua, J. The comparison of four mitochondrial genomes reveals cytoplasmic male sterility candidate genes in cotton. BMC Genom. 2018, 19, 775. [Google Scholar] [CrossRef] [PubMed]
- L’Homme, Y.; Stahl, R.; Li, X.-Q.; Hameed, A.; Brown, G.G. Brassica nap cytoplasmic male sterility is associated with expression of a mtDNA region containing a chimeric gene similar to the pol CMS associated orf224 gene. Curr. Genet. 1997, 31, 325–335. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, M.; Yu, J. Mitochondrial nad2 gene is co-transcripted with CMS-associated orfB gene in cytoplasmic male-sterile stem mustard (Brassica juncea). Mol. Biol. Rep. 2009, 36, 345–351. [Google Scholar] [CrossRef]
- Yamamoto, M.; Shinada, H.; Onodera, Y.; Komaki, C.; Mikami, T.; Kubo, T. A male sterility-associated mitochondrial protein in wild beets causes pollen disruption in transgenic plants. Plant J. 2008, 54, 1027–1036. [Google Scholar] [CrossRef]
- Igarashi, K.; Kazama, T.; Motomura, K.; Toriyama, K. Whole genomic sequencing of RT98 mitochondria derived from Oryza rufipogon and Northern Blot analysis to uncover a cytoplasmic male sterility-associated gene. Plant Cell Physiol. 2013, 54, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewey, R.; Timothy, D.; Levings, C. Chimeric mitochondrial genes expressed in the C male-sterile cytoplasm of maize. Curr. Genet. 1991, 20, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Ducos, E.; Touzet, P.; Boutry, M. The male sterile G cytoplasm of wild beet displays modified mitochondrial respiratory complexes. Plant J. 2001, 26, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, Y.; Lee, J.; Choi, B.; Kim, S.; Yang, T. Complete mitochondrial genome sequence and identification of a candidate gene responsible for cytoplasmic male sterility in radish (Raphanus sativus L.) containing DCGMS cytoplasm. Theor. Appl. Genet. 2013, 126, 1763–1774. [Google Scholar] [CrossRef]
- Gulyas, G.; Shin, Y.; Kim, H.; Lee, J.; Hirata, Y. Altered transcript reveals an orf507 sterility-related gene in chili pepper (Capsicum annuum L.). Plant Mol. Biol. Rep. 2010, 28, 605–612. [Google Scholar] [CrossRef]
- Itabashi, E.; Kazama, T.; Toriyama, K. Characterization of cytoplasmic male sterility of rice with Lead Rice cytoplasm in comparison with that with Chinsurah Boro II cytoplasm. Plant Cell Rep. 2009, 28, 233–239. [Google Scholar] [CrossRef]
- Green, D.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 5381, 1309–1312. [Google Scholar] [CrossRef]
- Wallace, D. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Liu, D.; Liao, X.; Zheng, J.; Diao, Y.; Liu, Y.; Zhou, R. Comparative analysis of the cytology and transcriptomes of the cytoplasmic male sterility line H276A and its maintainer line H276B of cotton (Gossypium barbadense L.). Int. J. Mol. Sci. 2017, 18, 2240. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhao, Y.; Chen, P.; Zhou, B.; Diao, Y.; Yu, M.; Huang, Z.; Zhou, R. A comparative analysis of the atp8 gene between a cytoplasmic male sterile line and its maintainer and further development of a molecular marker specific to male sterile cytoplasm in kenaf (Hibiscus cannabinus L.). Plant Mol. Biol. Rep. 2016, 34, 29–36. [Google Scholar] [CrossRef]
- Chen, G.; Ye, X.; Zhang, S.; Zhu, S.; Yuan, L.; Hou, J.; Wang, C. Comparative transcriptome analysis between fertile and CMS flower buds in Wucai (Brassica campestris L.). BMC Genom. 2018, 19, 908. [Google Scholar] [CrossRef]
- Zheng, J.; Kong, X.; Li, B.; Khan, A.; Li, Z.; Liu, Y.; Kang, H.; Ullah Dawar, F.; Zhou, R. Comparative transcriptome analysis between a novel allohexaploid cotton progeny CMS line LD6A and its maintainer line LD6B. Int. J. Mol. Sci. 2019, 20, 6127. [Google Scholar] [CrossRef] [Green Version]
- Mei, S.; Liu, T.; Wang, Z. Comparative transcriptome profile of the cytoplasmic male sterile and fertile floral buds of radish (Raphanus sativus L.). Int. J. Mol. Sci. 2016, 17, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Qin, T.; Wei, C.; Sun, J.; Dong, T.; Zhou, R.; Chen, Q.; Wang, Q. Using transcriptome analysis to screen for key genes and pathways related to cytoplasmic male sterility in cotton (Gossypium hirsutum L.). Int. J. Mol. Sci. 2019, 20, 5120. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Cheng, J.; Qin, C.; Hu, Z.; Yin, C.; Hu, K. Differential proteomic analysis of anthers between cytoplasmic male sterile and maintainer lines in Capsicum annuum L. Int. J. Mol. Sci. 2013, 14, 22982–22996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, L.; Wang, H.; Li, D.; Lin, Z.; Li, Y.; Zhao, W.; Chao, H.; Miao, L.; Li, M. Transcriptomic and proteomic analysis of Shaan2A cytoplasmic male sterility and its maintainer line in Brassica napus. Front. Plant Sci. 2019, 10, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Wei, F.; Kashif, M.; Khan, A.; Li, Z.; Shi, Q.; Jia, R.; Xie, H.; Zhang, L.; Li, B.; et al. Analysis of chloroplast differences in leaves of rice isonuclear alloplasmic lines. Protoplasma 2018, 255, 863–871. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, Q.; Zhang, L.; Li, Z.; Wang, C.; Zhang, G. Comparative proteomic analysis of developmental changes in P-type cytoplasmic male sterile and maintainer anthers in wheat. Int. J. Mol. Sci. 2021, 22, 2012. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.; Bartel, D.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Li, Z.; An, X.; Zhu, T.; Yan, T.; Wu, S.; Tian, Y.; Li, J.; Wan, X. Discovering and constructing ceRNA-miRNA target gene regulatory networks during anther development in maize. Int. J. Mol. Sci. 2019, 20, 3480. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zheng, B.; Wang, L.; Yang, W.; Wu, X.; Xu, Q.; Guo, W. High-throughput sequencing and degradome analysis reveal altered expression of miRNAs and their targets in a male-sterile cybrid pummelo (Citrus grandis). BMC Genom. 2016, 17, 591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, M.; Li, N.; Wang, H.; Qiu, P.; Pei, L.; Xu, Z.; Wang, T.; Gao, E.; Liu, J.; et al. Long noncoding RNAs involve in resistance to Verticillium dahliae, a fungal disease in cotton. Plant Biotechnol. J. 2018, 16, 1172–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, V.; Meyer, J. Cytoplasmically controlled male sterility in cotton. Crop. Sci. 1965, 5, 444–448. [Google Scholar] [CrossRef]
- Meyer, V. Male sterility from Gossypium harknessii. Heredity 1975, 66, 23–27. [Google Scholar] [CrossRef]
- Stewart, J. A new cytoplasmic male sterile and restorer for cotton. Proc. Beltwide Cotton Conf. Natl. Cotton Counc. Memphis 1992, 610, 50–53. [Google Scholar]
- Wei, Z.; Liu, S.; Su, J. A preliminary study on male sterile lines with Zhongmian cytoplasm and upland cotton nucleus. Chin. Cotton 1987, 2, 15–17. [Google Scholar]
- Jia, Z. Selection of a male-sterile line 104-7A of cotton and its complete set of three lines. Chin. Cotton 1990, 17, 13. [Google Scholar]
- Zhu, C.; Zhou, D.; Wu, J.; Li, X.; Peng, F.; Gong, Y.; Zhou, S. Three-line combination of the hybrid cotton Xiangyuan A and its utilization. Hunan Agric. Sci. 2013, 10, 1–3. [Google Scholar]
- Li, M.; Liu, D.; Liao, X.; Tang, D.; Chen, P.; Zhou, R. Cloning and expression analysis of a novel transcription factor GhbZIP1 discovered in upland cotton. J. Chin. Agricul. Univ. 2015, 20, 61–67. [Google Scholar]
- Zhou, R.; Li, M. A method of creating cytoplasmic male sterile line by transgenic cotton. CN Patent ZL201711061141.8, 11 May 2020. [Google Scholar]
- Huang, J.; Yang, P.; Li, B.; An, Z.; Sun, D. Microsporogenesis and ultrastructure of cytoplasmic male sterile line Jin A in cotton. J. Cotton Sci. 2001, 13, 259–263. [Google Scholar]
- Wang, X.; Zhang, T.; Pan, J. Cytological observation of microsporogenesis and RAPD analysis of mitochondrial DNA in cytoplasmic male sterile cotton. Agr. Sci. Chin 1998, 2, 70–75. [Google Scholar]
- Clément, C.; Chavant, L.; Burrus, M.; Audran, J. Anther starch variations in lilium during pollen development. Sex. Plant Reprod. 1994, 7, 347–356. [Google Scholar] [CrossRef]
- Oliver, S.; Dongen, J.; Alfred, S.; Mamun, E.; Dolferus, R. Cold-induced repression of the rice anther-specific cell wall invertase gene OSINV4 is correlated with sucrose accumulation and pollen sterility. Plant Cell Environ. 2010, 12, 1534–1551. [Google Scholar] [CrossRef]
- Koch, K. Carbohydrate-modulated gene expression in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 509–540. [Google Scholar] [CrossRef] [Green Version]
- Rolland, F.; Baena-Gonzalez, E.; Sheen, J. Sugar sensing and signaling in plants: Conserved and novel mechanisms. Annu. Rev. Plant Biol. 2006, 57, 675–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeekens, S.; Ma, J.; Hanson, J.; Rolland, F. Sugar signals and molecular networks controlling plant growth. Curr. Opin. Plant Biol. 2010, 3, 274–279. [Google Scholar] [CrossRef]
- Pacini, E. Types and meaning of pollen carbohydrate reserves. Sex. Plant Reprod. 1996, 6, 362–366. [Google Scholar] [CrossRef]
- Goetz, M.; Godt, D.E.; Guivarc’h, A.; Kahmann, U.; Chriqui, D.; Roitsch, T. Induction of male sterility in plants by metabolic engineering of the carbohydrate supply. Proc. Natl. Acad. Sci USA. 2001, 98, 6522–6527. [Google Scholar] [CrossRef] [Green Version]
- Dorion, S.; Lalonde, S.; Saini, H. Induction of male sterility in wheat by meiotic-stage water deficit is preceded by a decline in invertase activity and changes in carbohydrate metabolism in anthers. Plant Physiol. 1996, 111, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Datta, R.; Chamusco, K.; Chourey, P. Starch biosynthesis during pollen maturation is associated with altered patterns of gene expression in maize. Plant Physiol. 2002, 130, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z. Construction of regulatory model for pollen development and functional analysis of fertility related gene TaUGP1-6A in isogenic and heterogenic male sterile wheat. Degree of Doctor, Northwest A & F University, Xianyang, China, May 2019. [Google Scholar]
- Gumienny, T.; Lambie, E.; Hartwieg, E.; Horvitz, H.; Hengartner, M. Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 1999, 126, 1011–1022. [Google Scholar] [CrossRef]
- Church, D.; Guan, K.; Lambie, E. Three genes of the MAP kinase cascade, mek-2, mpk-1/sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development 1995, 121, 2525–2535. [Google Scholar] [CrossRef]
- Yin, Y.; Donlevy, S.; Smolikove, S. Coordination of recombination with meiotic progression in the Caenorhabditis elegans germline by KIN-18, a TAO kinase that regulates the timing of MPK-1 signaling. Genetics 2016, 202, 45–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achache, H.; Laurent, L.; Hecker-Mimoun, Y.; Ishtayeh, H.; Rappaport, Y.; Kroizer, E.; Colaiácovo, M.P.; Tzur, Y.B. Progression of meiosis is coordinated by the level and location of MAPK activation via OGR-2 in Caenorhabditis elegans. Genetics 2019, 1, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhang, K.; Wu, Y.; Li, C. Detection of apple chlorotic leaf spot virus-sensitivity comparison between PCR and ELISA. J. Dalian Univer. 2001, 6, 19–22. [Google Scholar]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic. Acids. Res. 2011, 39, 155–159. [Google Scholar] [CrossRef] [Green Version]
- Kenneth, J.; Thomas, D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2002, 25, 402–408. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Chen, L.; Khan, A.; Kong, X.; Khan, M.R.; Rao, M.J.; Wang, J.; Wang, L.; Zhou, R. Transcriptome and MiRNAomics Analyses Identify Genes Associated with Cytoplasmic Male Sterility in Cotton (Gossypium hirsutum L.). Int. J. Mol. Sci. 2021, 22, 4684. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094684
Li M, Chen L, Khan A, Kong X, Khan MR, Rao MJ, Wang J, Wang L, Zhou R. Transcriptome and MiRNAomics Analyses Identify Genes Associated with Cytoplasmic Male Sterility in Cotton (Gossypium hirsutum L.). International Journal of Molecular Sciences. 2021; 22(9):4684. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094684
Chicago/Turabian StyleLi, Min, Li Chen, Aziz Khan, Xiangjun Kong, Muhammad Rabnawaz Khan, Muhammad Junaid Rao, Jibin Wang, Lingqiang Wang, and Ruiyang Zhou. 2021. "Transcriptome and MiRNAomics Analyses Identify Genes Associated with Cytoplasmic Male Sterility in Cotton (Gossypium hirsutum L.)" International Journal of Molecular Sciences 22, no. 9: 4684. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094684