
Extracellular Vesicles and Antiphospholipid Syndrome: State-of-the-Art and Future Challenges


Abstract
:1. Introduction
2. The Molecular Mechanisms of EVs Contributing to Vascular Disorders
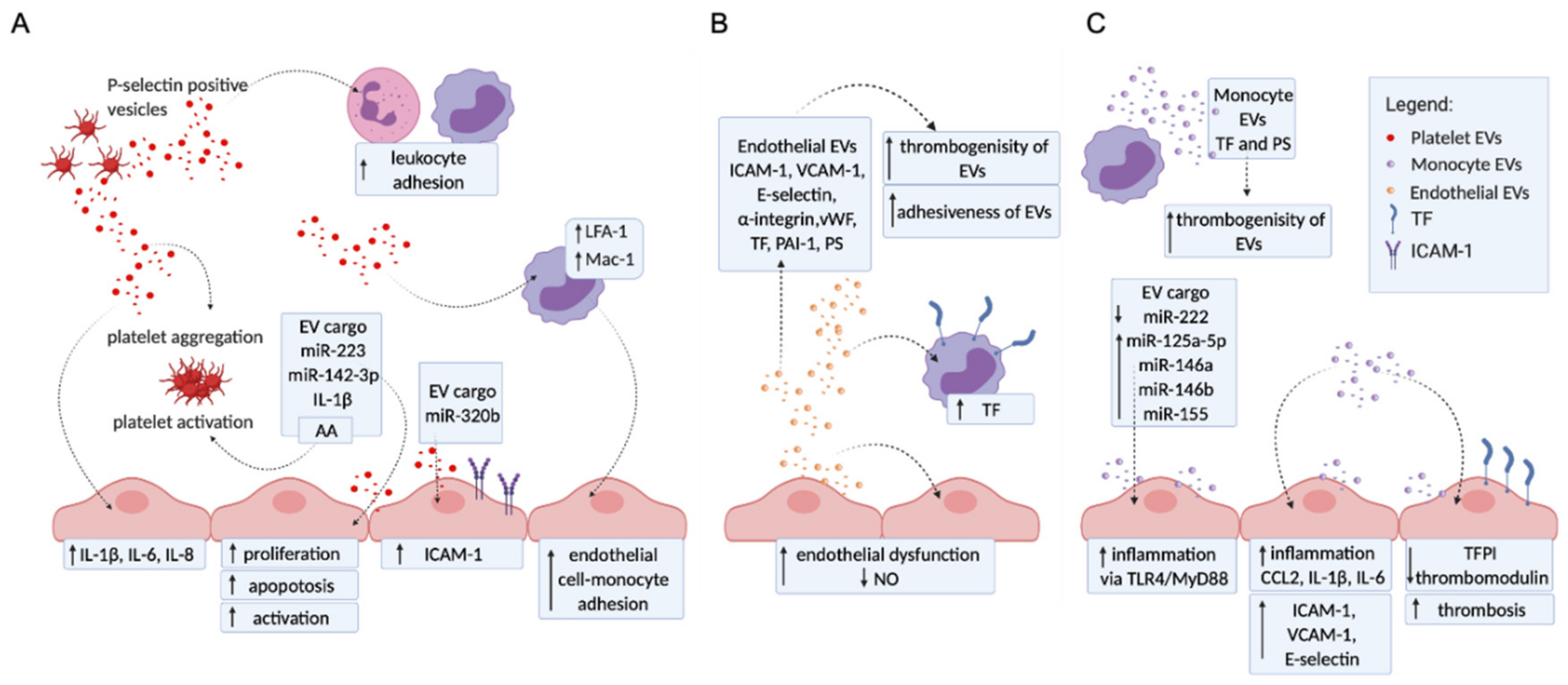
2.1. Platelet EVs Change the Adhesion Profile of Endothelial Cells and Monocytes, Activate Other Platelets, and Influence Cytokine Production
2.2. Endothelial EVs Carry a Proadhesive and Procoagulant Profile
2.3. Monocyte EVs Modulate Adhesion and Coagulation Profile of Endothelial Cells
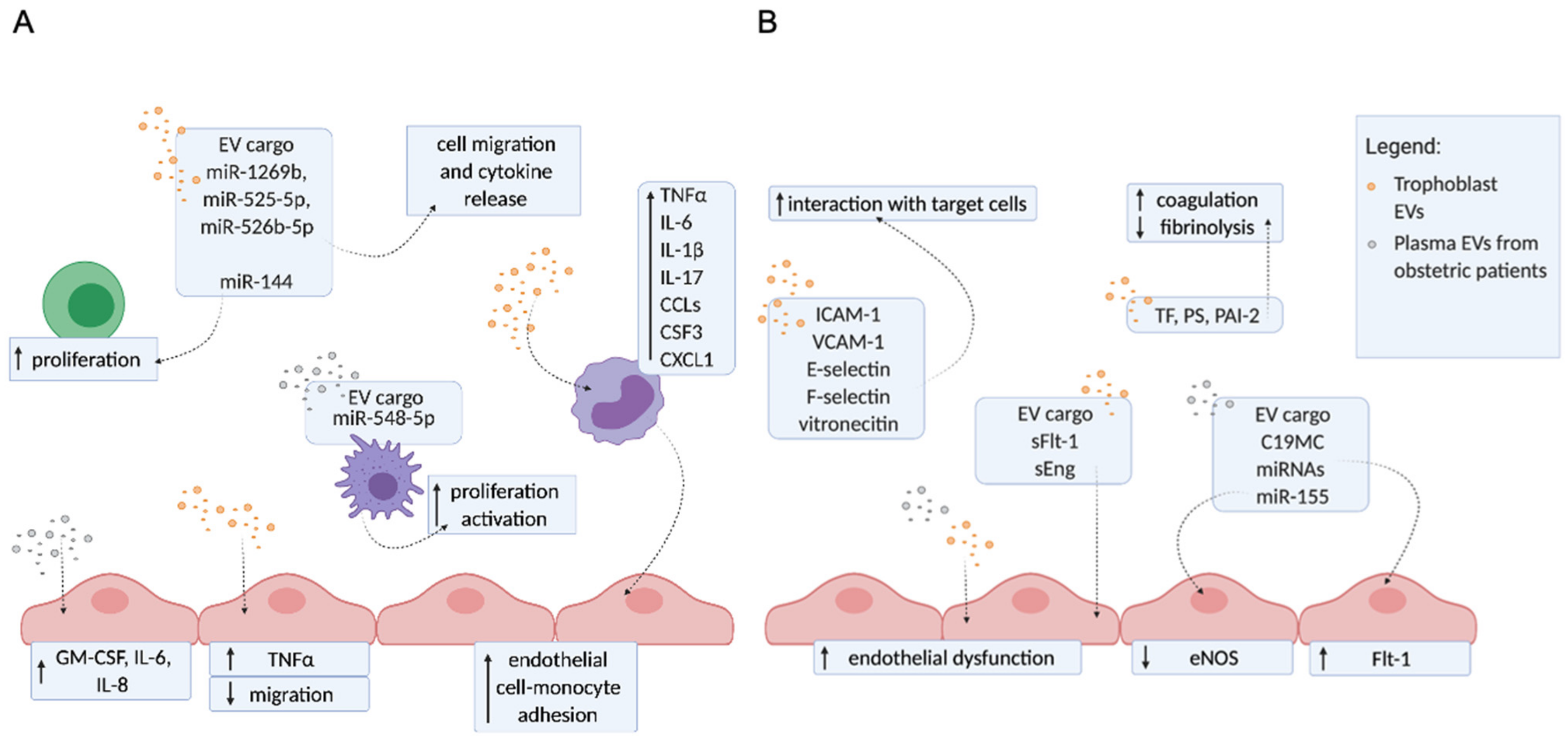
3. The Molecular Mechanisms of EVs Contributing to Pregnancy Disorders
Trophoblast EVs and EVs from Plasma of Patients with Obstetric Complications Enhance Inflammation, Endothelial Dysfunction, and Hypercoagulation
4. Extracellular Vesicles in Antiphospholipid Syndrome
4.1. Literature Search Strategy and Results
4.2. Thrombotic APS
4.2.1. Thrombotic APS In Vivo (Clinical) Studies

{kind=link}
{kind=link}
| Reference | Patient Group | Control Group | Isolation Method | Characterization Method | Type of EVs | Main Findings |
|---|---|---|---|---|---|---|
| Combes et al., 1999 [31] | 5 APS, 8 APS + SLE | 17 asympt. aPL+ (6 SLE or other autoimmune, 4 infections, 5 malignancies, 2 undefined) 30 HBDs | Sodium citrate 2 × 1500× g (15 min), 13,000× g (1 min). Temperature not specified. | FC: Positive for annexin V, CD51. < 1.5 µm (using latex beads). Renumeration beads not specified. | endothelial (CD51+) | ↑ levels of endothelial EVs in aPL + pts. vs. HBDs. ↑ levels of endothelial EVs in aPL + pts. vs. asympt. aPL+. |
| Joseph et al., 2001 [73] | 20 APS 14 APS + SLE | 16 SLE 20 HBDs | Sodium citrate 2 × 1500× g (15 min), 13,000× g (1 min). Temperature not specified. | FC: Positive for GPIIb-IIIa. < 0.8 µm (beads not specified). Renumeration beads not specified. | platelet (GPIIb-IIIa+) | No difference in levels of platelet EVs between APS pts., SLE pts., and HBDs. |
| Nagahama et al., 2003 [72] | 24 APS 13 APS + SLE | 25 SLE 30 HBDs | Sodium citrate 200× g (10 min, RT), 1000× g (15 min, RT). | FC: Positive for annexin V, CD14, CD42a. | platelet (CD42a+) monocyte (annexin V+/CD14+) | ↑ levels of monocyte EVs in APS pts. vs. APS + SLE pts. and vs. HBDs. ↑levels of P-selectin+ platelets and platelet EVs in APS pts. vs. SLE pts. and HBDs. |
| Dignat-George et al., 2004 [65] | 23 APS 14 APS + SLE | 28 SLE aPL+ no thrombosis 23 SLE aPL− no thrombosis 25 aPL− with thrombosis 25 HBDs | Sodium citrate 2 × 1500× g (15 min), 13,000× g (2 min). Temperature not specified. | FC: Positive for CD51. <0.8 µm (using latex beads). Renumeration beads (FlowCount). | endothelial (CD51+) | ↑ levels of endothelial EVs in APS pts. vs. HBDs and vs. aPL− thrombosis. No difference between primary or secondary APS. ↑ levels of endothelial EVs SLE aPL + pts. vs. HBDs. No difference between SLE aPL− pts. and aPL− thrombosis pts. vs. HBDs. |
| Jy et al., 2007 [66] | 60 APS | 28 asympt. aPL+ 39 HBDs | Sodium citrate 160× g (10 min), 1500× g (6 min). Temperature not specified. | FC: Positive for CD31, CD42. < 1.5 µm (beads not specified). Renumeration beads (Standard beads). | endothelial (CD31+/CD42-) platelet (CD31+/CD42+) | ↑ levels of platelet and endothelial EVs in APS pts. vs. HBDs. ↑ levels of endothelial EVs in asympt. aPL+ vs. HBDs. ↑ levels of platelet EVs in APS pts. vs. asympt. aPL+. No difference in levels of endothelial EVs in APS pts. vs. asympt. aPL+. No difference in levels of platelet EVs in asympt. aPL+ vs. HBDs. |
| Flores-Nascimento et al., 2009 [71] | 11 APS 9 DVT pts. at diagnosis 10 DVT pts. after 1–3 years of warfarin withdrawal 7 FVL pts. | 37 HBDs | Sodium citrate 3000× g (20 min), 13,000× g (30 min). Temperature not specified. | FC: Positive for annexin V, CD14, CD31, CD45, CD61, CD142, CD235. | total (annexin V+) platelet (CD61+) erythrocyte (CD235+) monocyte (CD14+) endothelial (CD31+) leukocyte (CD45+) TF (CD142+) | No difference in total EVs in DVT pts. at diagnosis, FVL pts., APS pts. vs. HBDs. ↑ levels of total EVs in DVT 1-3 years vs. HBDs. No difference in platelet, erythrocyte, monocyte, endothelial, and leukocyte EVs in all pts. groups vs. HBDs. |
| Vikerfors et al., 2012 [67] | 40 APS 12 APS + SLE | 52 HBDs | Blood collection and centrifugation not described. | FC: Negative for phalloidin, positive for lacadherin, CD14, CD42a, CD142, CD144. <1 µm (using MegaMix beads). Renumeration beads not specified. | total (lacadherin+) endothelial (CD144+) platelet (CD42a+) monocyte (CD14+) endothelial TF (CD144+/CD142+) | ↑ levels of total EVs in APS pts. vs. HBDs. ↑ levels of endothelial, endothelial TF+, and monocyte EVs in APS pts. vs HBDs. No difference in levels of platelet EVs in APS pts. vs. HBDs. |
| Willemze et al., 2014 [74] | 11 APS 19 APS + SLE | 72 asympt. aPL+ no HBDs | Sodium citrate 1500× g (10 min, 4 °C), 2000× g (5 min, 4 °C), 20,000× g (30 min, 4 °C). | TF activity assay | TF + EVs | ↑ EV-TF activity in APS pts. vs. asympt. aPL+. No difference in EV-TF activity in the presence or absence of underlying SLE. No difference between different APS clinical complications. No correlation between EV-TF activity and aPL subtype. |
| Breen et al., 2015 [69] | 37 APS | 18 asymptomatic aPL+, 18 HBDs | Sodium citrate 2 × 2000× g (15 min, 4 °C), for procoagulant activity additional 12,000× g (2 min, 4 °C). | FC: Positive for CD51, CD41, CD61, CD105. Renumeration beads (flow count fluoroshperes) | endothelial (CD51+ or CD105+) platelet (CD41+ or CD61+) | ↑ levels of endothelial and platelet EVs in APS pts. vs. HBDs. No difference in levels of endothelial and platelet EVs in asymptomatic aPL+ vs. HBDs. No difference in the EVs procoagulant activity between all groups and HBDs. |
| Chaturvedi et al., 2015 [68] | 47 aPL+ patients (38 APS, 2 APS + SLE, 6 asympt. aPL+, 1 aPL+ migraine with aura) | 144 HBDs | Sodium citrate 2 × 1500× g (15 min), 13,000× g (2 min). Temperature not specified. | FC: Positive for annexin V or CD14, CD41, CD105, CD142, CD144. < 1µm (using latex beads). Renumeration beads not specified. | total (annexin V+) endothelial (CD105+ or CD144+) platelet (CD41+) monocyte (CD14+) TF (CD142+) | ↑ levels of total EVs in aPL+ vs. HBDs. ↑ levels of endothelial, platelet, and TF + EVs in aPL+ vs. HBDs. No difference in levels of monocyte EVs in aPL+ vs. HBDs. |
| Niccolai et al., 2015 [70] | 16 APS | 16 asympt. aPL+ 16 HBDs | Sodium citrate Serial centrifugation: 1500× g (15 min), 3000× g (3 min). Temperature not specified. | FC: Positive for VPD450 or CD31, CD41a, CD45. < 0.9 µm (using Megamix beads). Renumeration beads (Trucount). | total (VPD450+/7AAD-) endothelial (CD31+) platelet (CD41a+) leukocyte (CD45+) | ↑ levels of total, endothelial, platelet, and leukocyte EVs between APS pts. and HBDs, between APS pts. and asympt. aPL+ and between asympt. aPL + pts. and HBDs. ↑ levels of total EVs in APS triple positivity vs. single positivity. ↑ levels of endothelial EVs in asympt. aPL+ triple positivity vs. single positivity. |
| Hell et al., 2018 [75] | 64 APS 18 APS + SLE 12 APS+LLD | 30 HBDs | Sodium citrate 2500× g (15 min, 15 °C) | TF activity assay | TF + EVs | No difference in EV-TF activity in APS pts. vs. HBDs. No difference in EV-TF activity in single, double, or triple aPL + pts. No correlation between different aPL and EV-TF activity. No difference in EV-TF activity in aPL + pts. with arterial thrombosis vs. venous thrombosis vs. combination of both. No difference in EV-TF activity and the number of thromboses. |
| Štok et al., 2020 [76] | 14 APS | 5 aPL− with thrombosis 7 HBD | Sodium citrate 820× g (10 min, RT), 2500× g (10 min, RT), 10,000× g, (45 min, RT), 100,000× g (2 h 15 min, 4 °C ). | NTA | < 200 nm. multiplex flow cytometry for 38 markers (detection via tetraspanins) | Small EVs were investigated. ↑ levels of sEVs in plasma of APS pts. vs. HBD. Platelet (CD41b+, CD42a+), lymphocyte (CD8+), leukocyte (CD45+), and endothelial (CD31+) sEVs were detected. ↑ levels of P-selectin on sEVs from APS pts. vs. HBDs. ↑levels of CD133/1 on sEVs from APS pts. vs. aPL− pts. with thrombosis. |
4.2.2. Thrombotic APS In Vitro (Translational) Studies
| Reference | Cell Type | Stimulation | Isolation Method | Characterization Method | Main Findings |
|---|---|---|---|---|---|
| Ford et al., 1998 [81] | Platelets | Serum from aPL + pts. and HBDs. | No isolation. | FC: Platelet EVs (CD61+). | No difference in levels of CD61+ vesicles after stimulation of platelets with aPL + pts. serum vs. HBDs. |
| Dignat-George et al., 2004 [65] | HUVEC | Plasma from APS pts. and HBDs. | None for flow cytometry. 14,000× g (2 h 30 min) for procoagulant activity. | FC: Total EVs (annexin V+). <0.8 µm (using latex beads). Renumeration beads (Flowcount). | ↑ levels and procoagulant activity of EVs after stimulation with APS pts. plasma vs. HBDs. |
| Wu et al., 2015 [82] | HUVEC | Anti-β2GPI from APS pts. and rabbits immunized with β2GPI. Control IgG. | 2500× g (15 min), 13,000× g (2 min), 100,000× g (90 min). | qPCR, immunoblotting. | ↑ activation of endothelial cells after stimulation with endothelial EVs released in response to aPL possibly through presentation of ssRNA (miRNA) to TLR7. ↑ active IL-1β in endothelial EVs. |
| Pericleous et al., 2012 [79] | HUVEC | Polyclonal IgG from APS pts. and HBDs. | 3000× g (5 min), 12,000× g (60 min). | FC: Total EVs (annexin V+) Specific endothelial EVs: CD62E+ (E-selectin), CD106+ (VCAM-1), CD54+ (ICAM-1), CD142+ (TF), CD105+ (endoglin), CD144+ (VE-cadherin). <1 µm (using latex beads). | ↑ levels of total endothelial and E-selectin+ EVs after APS IgG stimulation vs. HBD IgG. No difference in levels of ICAM-1+, endoglin+, and VE-cadherin+ EVs after APS IgG stimulation vs. HBD IgG. VCAM-1+ and TF + EVs could not be detected. |
| Betapudi et al., 2013 [80] | HUVEC | Anti-β2GPI purified from APS pts. and rabbits immunized with β2GPI. Control IgG. | 1500× g, (30 min), 13,000× g (2 min). | FC: <1 µm (using latex beads ). Endothelial EVs (CD144+). | ↑ levels of endothelial EVs after stimulation with aPL compared to control IgG. Anti-β2GPI antibodies stimulate EVs release via a nonmuscle myosin II motor protein-dependent pathway. |
4.3. Obstetric APS
4.3.1. Obstetric APS In Vivo (Clinical) Studies
| Reference | Patient Group | Control Group | Isolation Method | Characterization Method | Type of EVs | Main Findings |
|---|---|---|---|---|---|---|
| Alijotas-Reig et al., 2011 [83] | 9 pregnant obstetric APS | 40 aPL− pregnant women with a history of pregnancy loss | Sodium citrate. 1500× g (15 min), 13,000× g (2 min). | FC: Positive for annexin V, CD41, CD45, CD144. <1 µm (calibrated latex beads). Renumeration beads (FlowCount). | total (annexin V+) platelet (CD41+) endothelial (CD144+) leukocyte (CD45+) | No difference in levels of total, platelet, leukocyte, and endothelial EVs between pregnant APS pts. and pregnant aPL− pts. with a history of pregnancy loss. |
| Martinez-Zamora et al., 2015 [86] | 50 non-pregnant obstetric APS | 50 non-pregnant aPL− patients with a history of unexplained RM. 50 HBDs. | Sodium citrate. 2000× g (10 min), RT 5000× g (10 min), 4 °C. | Functional assay (ZYMUPHEN MP-activity). | total EVs | ↑ levels of total EVs in non-pregnant APS pts. vs. HBDs ↑ levels of total EVs in non-pregnant aPL− RM pts. vs. HBDs. No differences in levels of total EVs between non-pregnant APS pts. and non-pregnant aPL− RM pts. |
| Breen et al., 2015 [69] | 11 non-pregnant obstetric APS. | 18 HBDs | Sodium citrate 2x 2000× g (15 min, 4 °C), for procoagulant activity additional 12,000× g (2 min, 4 °C). | FC: Positive for CD51, CD41, CD61, or CD105. Renumeration beads (flow count fluoroshperes). | endothelial (CD51+ or CD105+) platelet (CD41+ or CD61+) | No difference in levels of endothelial and platelet EVs in non-pregnant APS pts. vs. HBDs. No difference in the EVs procoagulant activity between non-pregnant APS pts. and HBDs. |
| Campello et al., 2018 [84] | 11 pregnant obstetric APS | 15 pregnant HBDs. | Not described | FC: Positive for PS, TF, Endoglin. Detection beads: 0.5, 0.9, 3 µm (MegaMix) Renumeration beads not defined. | endothelial and platelet (markers not defined) TF+ endoglin+ | ↑ levels of PS+, endoglin+, and endothelial EVs in first and second trimester obstetric APS pts. vs. pregnant HBDs. ↑ levels of PS+, endoglin+, TF+, endothelial, and platelet EVs in third trimester obstetric APS pts. vs. pregnant HBDs. ↑ levels of endoglin+, TF+, and platelet EVs in high risk (triple aPL positive) pts. compared to low risk (single aPL positive) pts. |
| Zhou et al., 2019 [85] | 25 pregnant obstetric APS | 17 pregnant HBDs. | Sodium citrate. 1500× g (15 min) 13,000× g (2 min) | FACS sorting (CytoFlex). Positive for annexin V, CD41. Detection beads: 0.1 µm, 0.3 µm, 0.5 µm and 0.9 µm (MegaMix) Renumeration (Flow count fluorospheres). | platelet (annexin V+/CD41+) | No difference in levels of platelet EVs in first trimester APS pts. vs. pregnant HBDs. |
4.3.2. Obstetric APS In Vitro (Translational) Studies
The Release of Different EVs Populations from aPL Stimulated Placenta
EVs Derived from aPL-Exposed Placentae or from Plasma of APS Patients Activate Endothelial Cells
EVs Derived from aPL-Exposed Placentae Reflect ER Dysfunction and Carry Different Danger Signals
| Reference | Cell Type | Stimulation | Isolation of EVs | Characterization of EVs | Main Findings |
|---|---|---|---|---|---|
| Chen et al. 2012 [90] | 1st trimester human placental explants, HMEC-1 (stimulated with aPL and trophoblastic debris), human U937 monocytes. | Murine monoclonal anti-β2GPI (ID2, IIC5), isotype IgG, trophoblastic debris from placental explants. | Trophoblastic debris: CD45+ leukocyte depletion using magnetic beads Red blood cells removed by incubation with MilliQ water, 1300× g. | NA | aPL did not increase ICAM-1 expression or monocyte adhesion to HMEC-1 in the absence of trophoblastic debris. ↑ surface ICAM-1 and E-selectin expression and monocyte adhesion to HMEC-1 by trophoblastic debris is prolonged following the stimulation with aPL. |
| Viall et al. 2013 [91] | 1st trimester human placental explants (stimulated with aPL), HMEC-1 (stimulated with trophoblastic debris). | Murine monoclonal anti-β2GPI (ID2, IIC5), isotype IgG, trophoblastic debris from placental explants. | Not described. | NA | ↑ surface ICAM-1 expression on HMEC-1 after stimulation with trophoblast debris extruded from ID2 and IIC5 stimulated placental explants compared to isotype controls. ↓ levels of ICAM-1 on HMEC-1 when inhibition of aPL internalization was used. |
| Gysler et al., 2016 [95] | 1st trimester human extravillous trophoblast cell line (HTR8). | Murine monoclonal anti-β2GPI (IIC5), control IgG. | ExoQuick | Taqman MicroRNA Assay. | ↑ of mIR-146a-5p, miR-146a-3p, and miR-210 in exosomes isolated from trophoblasts after treatment with aPL compared to isotype control. |
| Shao et al., 2016 [94] | 1st trimester human placental explants (stimulated with aPL). | Murine monoclonal anti-β2GPI (IDT2), control IgG, serum of preeclamptic pts. and healthy pregnant women. | Trophoblastic debris: 300× g (10 min) CD45+ leukocyte depletion using magnetic beads Red blood cells removed by incubation with MilliQ water. | Immunohistochemistry and western blotting. | ↑ HMGB1 in trophoblastic debris derived from placental explants treated with aPL or patient sera compared to controls. |
| Tong et al., 2017 [89] | 1st trimester human placental explants (stimulated with aPL), HMEC-1 (stimulated with macro-, micro-, and nanovesicles), human U937 monocytes. | Murine monoclonal anti-β2GPI (ID2), control IgG, aPL derived from 5 APS pts. and controls, macro-, micro-, and nanovesicles from placental explants. | Macrovesicles: 2000× g, (5 min, 4 °C.), CD45+ leukocyte depletion using magnetic beads Red blood cells removed by incubation with MilliQ water. Microvesicles: 20,000× g (60 min, 4 °C.), Nanovesicles: 100,000× g (60 min, 4 °C). | NTA, PCR | Levels of nano- and microvesicles extruded from aPL stimulated placental explants were not increased compared to controls. ↑ mean and modal size of nanovesicles extruded from human serum-derived aPL stimulation of placental explants compared to control. ↑ surface ICAM-1 expression and monocyte adhesion to HMEC-1 after stimulation with macro-, micro-, and nanovesicles extruded from ID2 stimulated placental explants compared to isotype control. ↑ of mtDNA but not nucleolar DNA in micro- and nanovesicles extruded from ID2 stimulated placental explants compared to isotype control. Micro- and nanovesicles extruded from ID2 stimulated placental explants activated HMEC-1 through TLR-9 receptor signaling. |
| Zhao et al., 2017 [92] | 1st trimester human placental explants (stimulated with aPL), HMEC-1. | Murine monoclonal anti-β2GPI (ID2), control IgG, sera of preeclamptic pts., and healthy pregnant women. | Trophoblastic debris: 300× g (10 min) CD45+ leukocyte depletion using magnetic beads Red blood cells removed by incubation with MilliQ water | NA | ↑ ICAM-1 on HMEC-1 after stimulation with trophoblastic debris extruded from ID2 and pts. sera stimulated placental explants compared to controls. Melatonin prevented this increase. |
| Zhou et al., 2019 [85] | HUVEC, HTR-8/SVneo, THP-1. | Platelet microparticles from pregnant APS RM pts. and healthy pregnant women. | NA | NA | ↑ HUVEC expression levels of TNFα, ICAM-1, VCAM-1 after stimulation with platelet microparticles from APS RM pts. compared to healthy pregnant group. ↑ THP-1 adherence to HUVEC and inhibition of HUVEC tube formation after stimulation with platelet microparticles from APS RM pts. compared to healthy pregnant group. ↑ HUVEC apoptosis (via p38 MAP kinase pathway) after stimulation with platelet microparticles from APS RM pts. compared to healthy pregnant group. ↑ apoptosis and inhibition of invasion and migration of HTR-8/SVneo after stimulation with platelet microparticles from APS RM pts. compared to healthy pregnant group. |
| Tang et al., 2020 [93] | 1st trimester human healthy term and APS placental explants (stimulated with aPL). | Murine monoclonal anti-β2GPI (ID2, IIC5), isotype control IgG. | Macrovesicles: 2000× g, (5 min), Microvesicles: 20,000× g (60 min), Nanovesicles: 100,000× g (60 min). | Western blotting. | ↑ HSP 70 (ER stress sensor) in microvesicles and nanovesicles derived from placental explants treated with aPL compared to isotype control. ↑ levels of misfolded proteins in microvesicles and nanovesicles derived from placental explants treated with aPL compared to isotype control. ↑ MLKL in microvesicles and nanovesicles derived from placental explants treated with aPL compared to isotype control. |
5. Conclusions and Future Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; DE Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Radic, M.; Pattanaik, D. Cellular and Molecular Mechanisms of Anti-Phospholipid Syndrome. Front. Immunol. 2018, 9, 969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Žigon, P.; Čučnik, S.; Ambrožič, A.; Kveder, T.; Šemrl, S.S.; Rozman, B.; Božič, B. Detection of antiphosphatidylserine/prothrombin antibodies and their potential diagnostic value. Clin. Dev. Immunol. 2013, 2013, 724592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Žigon, P.; Podovšovnik, A.; Ambrožič, A.; Tomšič, M.; Hočevar, A.; Gašperšič, N.; Rotar, Ž.; Praprotnik, S.; Šemrl, S.S.; Čučnik, S. Added value of non-criteria antiphospholipid antibodies for antiphospholipid syndrome: Lessons learned from year-long routine measurements. Clin. Rheumatol. 2019, 38, 371–378. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Gudbergsson, J.M.; Andresen, T.L.; Simonsen, J.B. What is the blood concentration of extracellular vesicles? Implications for the use of extracellular vesicles as blood-borne biomarkers of cancer. Biochim. Biophys. Acta Rev. Cancer. 2019, 1871, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Berezin, A.E.; Berezin, A.A. Endothelial cell-derived extracellular vesicles in atherosclerosis: The emerging value for diagnosis, risk stratification and prognostication. Vessel Plus 2020, 4. [Google Scholar] [CrossRef]
- Oggero, S.; Austin-Williams, S.; Norling, L.V. The Contrasting Role of Extracellular Vesicles in Vascular Inflammation and Tissue Repair. Front. Pharmacol. 2019, 10, 1479. [Google Scholar] [CrossRef]
- Zara, M.; Guidetti, G.F.; Camera, M.; Canobbio, I.; Amadio, P.; Torti, M.; Tremoli, E.; Barbieri, S.S. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. Int. J. Mol. Sci. 2019, 20, 2840. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Han, L.; Huang, P.; Chen, Y.; Wang, Y.; Xue, F. Syncytiotrophoblast-Derived Extracellular Vesicles in Pathophysiology of Preeclampsia. Front. Physiol. 2019, 10, 1236. [Google Scholar] [CrossRef]
- Shetty, S.; Patil, R.; Ghosh, K. Role of microparticles in recurrent miscarriages and other adverse pregnancies: A review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 169, 123–129. [Google Scholar] [CrossRef]
- Buca, D.; Bologna, G.; D’Amico, A.; Cugini, S.; Musca, F.; Febbo, M.; D’Arcangelo, D.; Buca, D.; Simeone, P.; Liberati, M.; et al. Extracellular Vesicles in Feto-Maternal Crosstalk and Pregnancy Disorders. Int. J. Mol. Sci. 2020, 21, 2120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, H.; Fan, B.; Xu, W.; Zhang, X. Extracellular vesicles in normal pregnancy and pregnancy-related diseases. J. Cell Mol. Med. 2020, 24, 4377–4388. [Google Scholar] [CrossRef] [Green Version]
- Campello, E.; Radu, C.M.; Spiezia, L.; Simioni, P. Modulating thrombotic diathesis in hereditary thrombophilia and antiphospholipid antibody syndrome: A role for circulating microparticles? Clin. Chem. Lab. Med. 2017, 55, 934–943. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Alluri, R.; McCrae, K.R. Extracellular Vesicles in the Antiphospholipid Syndrome. Semin. Thromb. Hemost. 2018, 44, 493–504. [Google Scholar]
- Tong, M.; Tsai, B.W.; Chamley, L.W. Antiphospholipid antibodies and extracellular vesicles in pregnancy. Am. J. Reprod. Immunol. 2020, 85, e13312. [Google Scholar]
- Gidlöf, O.; van der Brug, M.; Ohman, J.; Gilje, P.; Olde, B.; Wahlestedt, C.; Erlinge, D. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood 2013, 121, 3908–3917. [Google Scholar] [CrossRef] [Green Version]
- Nomura, S.; Tandon, N.N.; Nakamura, T.; Cone, J.; Fukuhara, S.; Kambayashi, J. High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis 2001, 158, 277–287. [Google Scholar] [CrossRef]
- Barry, O.P.; Praticò, D.; Savani, R.C.; FitzGerald, G.A. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J. Clin. Invest 1998, 102, 136–144. [Google Scholar] [CrossRef]
- Forlow, S.B.; McEver, R.P.; Nollert, M.U. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood 2000, 95, 1317–1323. [Google Scholar] [CrossRef]
- Kuravi, S.J.; Harrison, P.; Rainger, G.E.; Nash, G.B. Ability of Platelet-Derived Extracellular Vesicles to Promote Neutrophil-Endothelial Cell Interactions. Inflammation 2019, 42, 290–305. [Google Scholar] [CrossRef] [Green Version]
- Suades, R.; Padró, T.; Vilahur, G.; Badimon, L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb. Haemost. 2012, 108, 1208–1219. [Google Scholar] [CrossRef]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Invest 1997, 99, 2118–2127. [Google Scholar] [CrossRef] [Green Version]
- Bissels, U.; Wild, S.; Tomiuk, S.; Hafner, M.; Scheel, H.; Mihailovic, A.; Choi, Y.H.; Tuschl, T.; Bosio, A. Combined characterization of microRNA and mRNA profiles delineates early differentiation pathways of CD133+ and CD34+ hematopoietic stem and progenitor cells. Stem Cells 2011, 29, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fordham, J.B.; Naqvi, A.R.; Nares, S. Regulation of miR-24, miR-30b, and miR-142-3p during macrophage and dendritic cell differentiation potentiates innate immunity. J. Leukoc. Biol. 2015, 98, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, H.; Chen, Y.X.; Huang, K.; Zhuang, F.; Bao, M.; Han, Y.; Chen, X.H.; Shi, Q.; Yao, Q.P.; Qi, Y.X. Platelet-derived microparticles promote endothelial cell proliferation in hypertension via miR-142-3p. FASEB J. 2018, 32, 3912–3923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Liang, H.; Liu, H.; Li, D.; Chen, X.; Li, L.; Zhang, C.Y.; Zen, K. Platelet-secreted microRNA-223 promotes endothelial cell apoptosis induced by advanced glycation end products via targeting the insulin-like growth factor 1 receptor. J. Immunol. 2014, 192, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Tan, M.; Xiang, Q.; Zhou, Z.; Yan, H. Thrombin-activated platelet-derived exosomes regulate endothelial cell expression of ICAM-1 via microRNA-223 during the thrombosis-inflammation response. Thromb. Res. 2017, 154, 96–105. [Google Scholar] [CrossRef]
- Somajo, S.; Koshiar, R.L.; Norström, E.; Dahlbäck, B. Protein S and factor V in regulation of coagulation on platelet microparticles by activated protein C. Thromb. Res. 2014, 134, 144–152. [Google Scholar] [CrossRef]
- Srikanthan, S.; Li, W.; Silverstein, R.L.; McIntyre, T.M. Exosome poly-ubiquitin inhibits platelet activation, downregulates CD36 and inhibits pro-atherothombotic cellular functions. J. Thromb. Haemost. 2014, 12, 1906–1917. [Google Scholar] [CrossRef] [Green Version]
- Combes, V.; Simon, A.C.; Grau, G.E.; Arnoux, D.; Camoin, L.; Sabatier, F.; Mutin, M.; Sanmarco, M.; Sampol, J.; Dignat-George, F. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J. Clin. Invest 1999, 104, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Abid Hussein, M.N.; Böing, A.N.; Biró, E.; Hoek, F.J.; Vogel, G.M.; Meuleman, D.G.; Sturk, A.; Nieuwland, R. Phospholipid composition of in vitro endothelial microparticles and their in vivo thrombogenic properties. Thromb. Res. 2008, 121, 865–871. [Google Scholar] [CrossRef]
- Dignat-George, F.; Boulanger, C.M. The many faces of endothelial microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatier, F.; Roux, V.; Anfosso, F.; Camoin, L.; Sampol, J.; Dignat-George, F. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood 2002, 99, 3962–3970. [Google Scholar] [CrossRef]
- Brodsky, S.V.; Zhang, F.; Nasjletti, A.; Goligorsky, M.S. Endothelium-derived microparticles impair endothelial function in vitro. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1910–H1915. [Google Scholar] [CrossRef] [Green Version]
- Hromada, C.; Muhleder, S.; Grillari, J.; Redl, H.; Holnthoner, W. Endothelial Extracellular Vesicles-Promises and Challenges. Front. Physiol. 2017, 8, 275. [Google Scholar] [CrossRef]
- Aharon, A.; Tamari, T.; Brenner, B. Monocyte-derived microparticles and exosomes induce procoagulant and apoptotic effects on endothelial cells. Thromb. Haemost. 2008, 100, 878–885. [Google Scholar]
- Wang, J.G.; Williams, J.C.; Davis, B.K.; Jacobson, K.; Doerschuk, C.M.; Ting, J.P.; Mackman, N. Monocytic microparticles activate endothelial cells in an IL-1beta-dependent manner. Blood 2011, 118, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Sun, B.; Gupta, A.; Rempel, H.; Pulliam, L. Monocyte exosomes induce adhesion molecules and cytokines via activation of NF-kappaB in endothelial cells. FASEB J. 2016, 30, 3097–3106. [Google Scholar] [CrossRef] [Green Version]
- Dalvi, P.; Sun, B.; Tang, N.; Pulliam, L. Immune activated monocyte exosomes alter microRNAs in brain endothelial cells and initiate an inflammatory response through the TLR4/MyD88 pathway. Sci. Rep. 2017, 7, 9954. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Huang, H.; Xu, Y.; Zhu, H.; Zhong, C. MiR-222 in Cardiovascular Diseases: Physiology and Pathology. Biomed. Res. Int. 2017, 2017, 4962426. [Google Scholar] [CrossRef] [PubMed]
- Celic, T.; Metzinger-Le Meuth, V.; Six, I.; Massy, Z.A.; Metzinger, L. The mir-221/222 Cluster is a Key Player in Vascular Biology via the Fine-Tuning of Endothelial Cell Physiology. Curr. Vasc. Pharmacol. 2017, 15, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Holland, O.; Kroneis, T.; El-Heliebi, A.; McDowell-Hook, M.; Stone, P.; Sedlmayr, P.; Chamley, L. Detection of Fetal Sex, Aneuploidy and a Microdeletion from Single Placental Syncytial Nuclear Aggregates. Fetal. Diagn. Ther. 2017, 41, 32–40. [Google Scholar] [CrossRef]
- Tong, M.; Chamley, L.W. Placental extracellular vesicles and feto-maternal communication. Cold Spring. Harb. Perspect. Med. 2015, 5, a023028. [Google Scholar] [CrossRef] [Green Version]
- Kovács, Á.F.; Láng, O.; Turiák, L.; Ács, A.; Kőhidai, L.; Fekete, N.; Alasztics, B.; Mészáros, T.; Buzás, E.I.; Rigó Jr, J.; et al. The impact of circulating preeclampsia-associated extracellular vesicles on the migratory activity and phenotype of THP-1 monocytic cells. Sci. Rep. 2018, 8, 5426. [Google Scholar] [CrossRef]
- Holder, B.S.; Tower, C.L.; Jones, C.J.; Aplin, J.D.; Abrahams, V.M. Heightened pro-inflammatory effect of preeclamptic placental microvesicles on peripheral blood immune cells in humans. Biol. Reprod. 2012, 86, 103. [Google Scholar] [CrossRef]
- Salomon, C.; Scholz-Romero, K.; Sarker, S.; Sweeney, E.; Kobayashi, M.; Correa, P.; Longo, S.; Duncombe, G.; Mitchell, M.D.; Rice, G.E.; et al. Gestational Diabetes Mellitus Is Associated With Changes in the Concentration and Bioactivity of Placenta-Derived Exosomes in Maternal Circulation Across Gestation. Diabetes 2016, 65, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.E.; Scholz-Romero, K.; Sweeney, E.; Peiris, H.; Kobayashi, M.; Duncombe, G.; Mitchell, M.D.; Salomon, C. The Effect of Glucose on the Release and Bioactivity of Exosomes From First Trimester Trophoblast Cells. J. Clin. Endocrinol. Metab. 2015, 100, E1280–E1288. [Google Scholar] [CrossRef]
- Truong, G.; Guanzon, D.; Kinhal, V.; Elfeky, O.; Lai, A.; Longo, S.; Nuzhat, Z.; Palma, C.; Scholz-Romero, K.; Menon, R.; et al. Oxygen tension regulates the miRNA profile and bioactivity of exosomes released from extravillous trophoblast cells-Liquid biopsies for monitoring complications of pregnancy. PLoS ONE 2017, 12, e0174514. [Google Scholar] [CrossRef] [Green Version]
- Ospina-Prieto, S.; Chaiwangyen, W.; Herrmann, J.; Groten, T.; Schleussner, E.; Markert, U.R.; Morales-Prieto, D.M. MicroRNA-141 is upregulated in preeclamptic placentae and regulates trophoblast invasion and intercellular communication. Transl. Res. 2016, 172, 61–72. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, P.; Wang, Z.; Qin, Z.; Xiu, X.; Xu, D.; Zhang, X.; Wang, Y. MiRNA-548c-5p downregulates inflammatory response in preeclampsia via targeting PTPRO. J. Cell Physiol. 2019, 234, 11149–11155. [Google Scholar] [CrossRef]
- Lyall, F.; Robson, S.C.; Bulmer, J.N. Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction: Relationship to clinical outcome. Hypertension. 2013, 62, 1046–1054. [Google Scholar] [CrossRef] [Green Version]
- Goulopoulou, S.; Davidge, S.T. Molecular mechanisms of maternal vascular dysfunction in preeclampsia. Trends. Mol. Med. 2015, 21, 88–97. [Google Scholar] [CrossRef]
- Tannetta, D.S.; Dragovic, R.A.; Gardiner, C.; Redman, C.W.; Sargent, I.L. Characterisation of syncytiotrophoblast vesicles in normal pregnancy and pre-eclampsia: Expression of Flt-1 and endoglin. PLoS ONE 2013, 8, e56754. [Google Scholar] [CrossRef]
- Lok, C.A.; Böing, A.N.; Sargent, I.L.; Sooranna, S.R.; van der Post, J.A.; Nieuwland, R.; Sturk, A. Circulating platelet-derived and placenta-derived microparticles expose Flt-1 in preeclampsia. Reprod. Sci. 2008, 15, 1002–1010. [Google Scholar] [CrossRef]
- Chang, X.; Yao, J.; He, Q.; Liu, M.; Duan, T.; Wang, K. Exosomes From Women With Preeclampsia Induced Vascular Dysfunction by Delivering sFlt (Soluble Fms-Like Tyrosine Kinase)-1 and sEng (Soluble Endoglin) to Endothelial Cells. Hypertension 2018, 72, 1381–1390. [Google Scholar] [CrossRef]
- Cronqvist, T.; Tannetta, D.; Mörgelin, M.; Belting, M.; Sargent, I.; Familari, M.; Hansson, S.R. Syncytiotrophoblast derived extracellular vesicles transfer functional placental miRNAs to primary human endothelial cells. Sci. Rep. 2017, 7, 4558. [Google Scholar] [CrossRef] [Green Version]
- Motta-Mejia, C.; Kandzija, N.; Zhang, W.; Mhlomi, V.; Cerdeira, A.S.; Burdujan, A.; Tannetta, D.; Dragovic, R.; Sargent, I.L.; Redman, C.W.; et al. Placental Vesicles Carry Active Endothelial Nitric Oxide Synthase and Their Activity is Reduced in Preeclampsia. Hypertension 2017, 70, 372–381. [Google Scholar] [CrossRef]
- Shen, L.; Li, Y.; Li, R.; Diao, Z.; Yany, M.; Wu, M.; Sun, H.; Yan, G.; Hu, Y. Placenta-associated serum exosomal miR-155 derived from patients with preeclampsia inhibits eNOS expression in human umbilical vein endothelial cells. Int. J. Mol. Med. 2018, 41, 1731–1739. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, F.G.; Nelson, D.B. Disseminated Intravascular Coagulation Syndromes in Obstetrics. Obstet. Gynecol. 2015, 126, 999–1011. [Google Scholar] [CrossRef]
- Gardiner, C.; Tannetta, D.S.; Simms, C.A.; Harrison, P.; Redman, C.W.; Sargent, I.L. Syncytiotrophoblast microvesicles released from pre-eclampsia placentae exhibit increased tissue factor activity. PLoS ONE 2011, 6, e26313. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Guller, S.; Tang, Z.; Ma, Y.Y.; Di Santo, S.; Sager, R.; Schneider, H. Protein composition of microparticles shed from human placenta during placental perfusion: Potential role in angiogenesis and fibrinolysis in preeclampsia. Placenta 2011, 32, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dignat-George, F.; Camoin-Jau, L.; Sabatier, F.; Arnoux, D.; Anfosso, F.; Bardin, N.; Veit, V.; Combes, V.; Gentile, S.; Moal, V.; et al. Endothelial microparticles: A potential contribution to the thrombotic complications of the antiphospholipid syndrome. Thromb. Haemost. 2004, 91, 667–673. [Google Scholar] [PubMed] [Green Version]
- Jy, W.; Tiede, M.; Bidot, C.J.; Horstman, L.L.; Jimenez, J.J.; Chirinos, J.; Ahn, Y.S. Platelet activation rather than endothelial injury identifies risk of thrombosis in subjects positive for antiphospholipid antibodies. Thromb. Res. 2007, 121, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Vikerfors, A.; Mobarrez, F.; Bremme, K.; Holmström, M.; Ågren, A.; Eelde, A.; Bruzelius, M.; Antovic, A.; Wallén, H.; Svenungsson, E. Studies of microparticles in patients with the antiphospholipid syndrome (APS). Lupus 2012, 21, 802–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, S.; Cockrell, E.; Espinola, R.; Hsi, L.; Fulton, S.; Khan, M.; Li, L.; Fonseca, F.; Kundu, S.; McCrae, K.R. Circulating microparticles in patients with antiphospholipid antibodies: Characterization and associations. Thromb. Res. 2015, 135, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Breen, K.A.; Sanchez, K.; Kirkman, N.; Seed, P.T.; Parmar, K.; Moore, G.W.; Hunt, B.J. Endothelial and platelet microparticles in patients with antiphospholipid antibodies. Thromb. Res. 2015, 135, 368–374. [Google Scholar] [CrossRef]
- Niccolai, E.; Squatrito, D.; Emmi, G.; Silvestri, E.; Emmi, L.; Ciucciarelli, L.; Ricci, F.; Manganaro, D.; Amedei, A.; Prisco, D. A new cytofluorimetric approach to evaluate the circulating microparticles in subjects with antiphospholipid antibodies. Thromb. Res. 2015, 136, 1252–1258. [Google Scholar] [CrossRef]
- Flores-Nascimento, M.C.; Beltrame, M.P.; De Paula, E.V.; Montalvao, S.L.; Pereira, F.G.; Orsi, F.L.; Lorand-Metze, I.; Annichino-Bizzacchi, J.M. Microparticles in deep venous thrombosis, antiphospholipid syndrome and Factor V Leiden. Platelets 2009, 20, 367–375. [Google Scholar] [CrossRef]
- Nagahama, M.; Nomura, S.; Kanazawa, S.; Ozaki, Y.; Kagawa, H.; Fukuhara, S. Significance of anti-oxidized LDL antibody and monocyte-derived microparticles in anti-phospholipid antibody syndrome. Autoimmunity 2003, 36, 125–131. [Google Scholar] [CrossRef]
- Joseph, J.E.; Harrison, P.; Mackie, I.J.; Isenberg, D.A.; Machin, S.J. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br. J. Haematol. 2001, 115, 451–459. [Google Scholar] [CrossRef]
- Willemze, R.; Bradford, R.L.; Mooberry, M.J.; Roubey, R.A.; Key, N.S. Plasma microparticle tissue factor activity in patients with antiphospholipid antibodies with and without clinical complications. Thromb. Res. 2014, 133, 187–189. [Google Scholar] [CrossRef] [Green Version]
- Hell, L.; Ay, C.; Posch, F.; Gebhart, J.; Koder, S.; Mackman, N.; Pabinger, I.; Thaler, J. Low extracellular vesicle-associated tissue factor activity in patients with persistent lupus anticoagulant and a history of thrombosis. Ann. Hematol. 2019, 98, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Stok, U.; Blokar, E.; Lenassi, M.; Holcar, M.; Frank-Bertoncelj, M.; Erman, A.; Resnik, N.; Sodin-Semrl, S.; Cucnik, S.; Pirkmajer, K.P.; et al. Characterization of Plasma-Derived Small Extracellular Vesicles Indicates Ongoing Endothelial and Platelet Activation in Patients with Thrombotic Antiphospholipid Syndrome. Cells 2020, 9, 1211. [Google Scholar] [CrossRef]
- Velásquez, M.; Rojas, M.; Abrahams, V.M.; Escudero, C.; Cadavid, Á.P. Mechanisms of Endothelial Dysfunction in Antiphospholipid Syndrome: Association With Clinical Manifestations. Front. Physiol. 2018, 9, 1840. [Google Scholar] [CrossRef] [Green Version]
- Lackner, K.J.; Muller-Calleja, N. Pathogenesis of antiphospholipid syndrome: Recent insights and emerging concepts. Expert. Rev. Clin. Immunol. 2018, 15, 199–209. [Google Scholar] [CrossRef]
- Pericleous, C.; Clarke, L.A.; Brogan, P.A.; Latchman, D.S.; Isenberg, D.A.; Ioannou, Y.; Giles, I.P.; Rahman, A. Endothelial microparticle release is stimulated in vitro by purified IgG from patients with the antiphospholipid syndrome. Thromb. Haemost. 2013, 109, 72–78. [Google Scholar]
- Betapudi, V.; Lominadze, G.; Hsi, L.; Willard, B.; Wu, M.; McCrae, K.R. Anti-beta2GPI antibodies stimulate endothelial cell microparticle release via a nonmuscle myosin II motor protein-dependent pathway. Blood 2013, 122, 3808–3817. [Google Scholar] [CrossRef] [Green Version]
- Ford, I.; Urbaniak, S.; Greaves, M. IgG from patients with antiphospholipid syndrome binds to platelets without induction of platelet activation. Br. J. Haematol. 1998, 102, 841–849. [Google Scholar] [CrossRef]
- Wu, M.; Barnard, J.; Kundu, S.; McCrae, K.R. A novel pathway of cellular activation mediated by antiphospholipid antibody-induced extracellular vesicles. J. Thromb. Haemost. 2015, 13, 1928–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alijotas-Reig, J.; Palacio-Garcia, C.; Farran-Codina, I.; Zarzoso, C.; Cabero-Roura, L.; Vilardell-Tarres, M. Circulating cell-derived microparticles in women with pregnancy loss. Am. J. Reprod. Immunol. 2011, 66, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Campello, E.; Radu, C.M.; Tonello, M.; Kuzenko, A.; Bulato, C.; Hoxha, A.; Mattia, E.; Spiezia, L.; Ruffatti, A.; Simioni, P. Circulating microparticles in pregnant patients with primary anti-phospholipid syndrome: An exploratory study. Scand. J. Rheumatol. 2018, 47, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Lian, Y.; Zhang, Y.; Li, L.; Li, H.; Shen, D.; Zhou, Y.; Zhang, M.; Lu, Y.; Liu, J.; et al. Platelet-derived microparticles from recurrent miscarriage associated with antiphospholipid antibody syndrome influence behaviours of trophoblast and endothelial cells. Mol. Hum. Reprod. 2019, 25, 483–494. [Google Scholar] [CrossRef]
- Martínez-Zamora, M.A.; Tàssies, D.; Creus, M.; Reverter, J.C.; Puerto, B.; Monteagudo, J.; Carmona, F.; Balasch, J. Higher levels of procoagulant microparticles in women with recurrent miscarriage are not associated with antiphospholipid antibodies. Hum. Reprod. 2016, 31, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Viall, C.; Kang, Y.; Liu, B.; Stone, P.; Chamley, L. Anti-phospholipid antibodies increase non-apoptotic trophoblast shedding: A contribution to the pathogenesis of pre-eclampsia in affected women? Placenta 2009, 30, 767–773. [Google Scholar] [CrossRef]
- Pantham, P.; Viall, C.A.; Chen, Q.; Kleffmann, T.; Print, C.G.; Chamley, L.W. Antiphospholipid antibodies bind syncytiotrophoblast mitochondria and alter the proteome of extruded syncytial nuclear aggregates. Placenta 2015, 36, 1463–1473. [Google Scholar] [CrossRef]
- Tong, M.; Johansson, C.; Xiao, F.; Stone, P.R.; James, J.L.; Chen, Q.; Cree, L.M.; Chamley, L.W. Antiphospholipid antibodies increase the levels of mitochondrial DNA in placental extracellular vesicles: Alarmin-g for preeclampsia. Sci. Rep. 2017, 7, 16556. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Guo, F.; Hensby-Bennett, S.; Stone, P.; Chamley, L. Antiphospholipid antibodies prolong the activation of endothelial cells induced by necrotic trophoblastic debris: Implications for the pathogenesis of preeclampsia. Placenta 2012, 33, 810–815. [Google Scholar] [CrossRef]
- Viall, C.A.; Chen, Q.; Liu, B.; Hickey, A.; Snowise, S.; Salmon, J.E.; Stone, P.R.; Chamley, L.W. Antiphospholipid antibodies internalised by human syncytiotrophoblast cause aberrant cell death and the release of necrotic trophoblast debris. J. Autoimmun. 2013, 47, 45–57. [Google Scholar] [CrossRef]
- Zhao, M.; Li, Y.; Xu, L.; Hickey, A.; Groom, K.; Stone, P.R.; Chamley, L.W.; Chen, Q. Melatonin prevents preeclamptic sera and antiphospholipid antibodies inducing the production of reactive nitrogen species and extrusion of toxic trophoblastic debris from first trimester placentae. Placenta 2017, 58, 17–24. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Y.; Nursalim, Y.; Groom, K.; Hickey, A.; Chamley, L.; Chen, Q. Endoplasmic reticulum stress occurs in association with the extrusion of toxic extracellular vesicles from human placentae treated with antiphospholipid antibodies. Clin. Sci. 2020, 134, 459–472. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Zhao, M.; Tong, M.; Wei, J.; Wise, M.R.; Stone, P.; Chamley, L.; Chen, Q. Increased levels of HMGB1 in trophoblastic debris may contribute to preeclampsia. Reproduction 2016, 152, 775–784. [Google Scholar] [CrossRef]
- Gysler, S.M.; Mulla, M.J.; Guerra, M.; Brosens, J.J.; Salmon, J.E.; Chamley, L.W.; Abrahams, V.M. Antiphospholipid antibody-induced miR-146a-3p drives trophoblast interleukin-8 secretion through activation of Toll-like receptor 8. Mol. Hum. Reprod. 2016, 22, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; DiBernardo, E.; Parks, E.; Fox, H.; Zheng, S.Y.; Wayne, E. The Role of Extracellular Vesicles in the Pathogenesis and Treatment of Autoimmune Disorders. Front. Immunol. 2021, 12, 566299. [Google Scholar] [CrossRef]
- Withrow, J.; Murphy, C.; Liu, Y.; Hunter, M.; Fulzele, S.; Hamrick, M.W. Extracellular vesicles in the pathogenesis of rheumatoid arthritis and osteoarthritis. Arthritis. Res. Ther. 2016, 18, 286. [Google Scholar] [CrossRef] [Green Version]
- Viñuela-Berni, V.; Doníz-Padilla, L.; Figueroa-Vega, N.; Portillo-Salazar, H.; Abud-Mendoza, C.; Baranda, L.; González-Amaro, R. Proportions of several types of plasma and urine microparticles are increased in patients with rheumatoid arthritis with active disease. Clin. Exp. Immunol. 2015, 180, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Negrini, S.; Pappalardo, F.; Murdaca, G.; Indiveri, F.; Puppo, F. The antiphospholipid syndrome: From pathophysiology to treatment. Clin. Exp. Med. 2017, 17, 257–267. [Google Scholar] [CrossRef]
- Riazifar, M.; Mohammadi, M.R.; Pone, E.J.; Yeri, A.; Lässer, C.; Segaliny, A.I.; McIntyre, L.L.; Shelke, G.V.; Hutchins, E.; Hamamoto, A.; et al. Stem Cell-Derived Exosomes as Nanotherapeutics for Autoimmune and Neurodegenerative Disorders. ACS Nano. 2019, 13, 6670–6688. [Google Scholar] [CrossRef]
- Sousa, C.; Pereira, I.; Santos, A.C.; Carbone, C.; Kovačević, A.B.; Silva, A.M.; Souto, E.B. Targeting dendritic cells for the treatment of autoimmune disorders. Colloids. Surf. B Biointerfaces 2017, 158, 237–248. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štok, U.; Čučnik, S.; Sodin-Šemrl, S.; Žigon, P. Extracellular Vesicles and Antiphospholipid Syndrome: State-of-the-Art and Future Challenges. Int. J. Mol. Sci. 2021, 22, 4689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094689
Štok U, Čučnik S, Sodin-Šemrl S, Žigon P. Extracellular Vesicles and Antiphospholipid Syndrome: State-of-the-Art and Future Challenges. International Journal of Molecular Sciences. 2021; 22(9):4689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094689
Chicago/Turabian StyleŠtok, Ula, Saša Čučnik, Snežna Sodin-Šemrl, and Polona Žigon. 2021. "Extracellular Vesicles and Antiphospholipid Syndrome: State-of-the-Art and Future Challenges" International Journal of Molecular Sciences 22, no. 9: 4689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094689